Introduction
Several recombinant protein expression systems have been developed to produce proteins of interest (POI)1,2,3,4,5,6,7,8,9, including mammalian cells3,4, yeast strains8,9, and Escherichia coli (E. coli)4,5,6,7. Among them, E. coli system is particularly favored due to lower cost, higher yield, short expression period, and ease of manufacturing. Meanwhile, special tags have always been fused to the N- or C-terminus of the recombinant proteins for purification, or to enhance protein solubility and expression yield10,11. Among these tags, maltose-binding protein (MBP)12,13,14, glutathione S-transferase (GST)15, and cysteine protease domain (CPD)16 could effectively enhance solubility17. Poly-histidine tag and FLAG tag, which consist of several amino acids, are commonly employed for purification18.
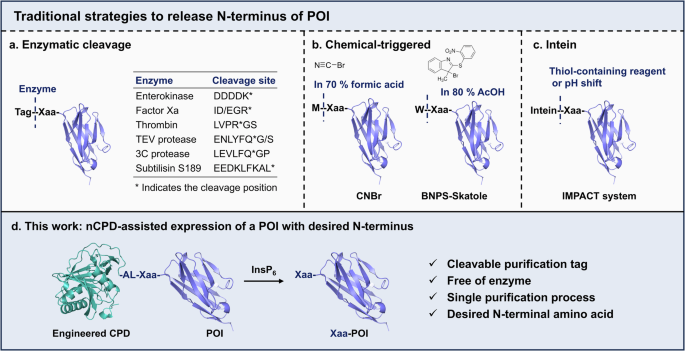
However, the addition of the tag may significantly alter the properties of POI and interfere with subsequent research, especially when fused to the N-terminus17,18,19. To address this issue, various methods have been developed to remove these N-terminal tags after purification18,20. Traditional approaches include enzymatic methods18, chemical induction21,22 and intein-assisted methods20,23,24,25 (Fig. 1a–c). Enzymatic methods typically release the native N-terminus of the POI based on the enzyme recognition of a specific sequence18. Enterokinase is a traditional system to achieve Xaa-POI by cleaving the five-peptide sequence DDDDK26. The Profinity eXact system, developed by Bio-Rad Laboratories Inc., fuses a subtilisin BPN+ pro-domain to the N-terminus of the POI, which could bind to subtilisin S189 crosslinked agarose and be released under F– triggering conditions, enabling the purification of native POI without tags27. In addition, TEV protease12,13, Thrombin28, Factor Xa29, and 3C protease12,30 are favorable alternatives for N-terminal tag removal by cleaving various sequences indicated in Fig. 1a25. Chemical-induced cleavage offers an alternative solution due to better reaction efficiency and lower cost. For example, cyanogen bromide (CNBr) effectively cleaves the peptide bond after Met31, while BNPS-Skatole recognizes and cleaves at the carboxyl terminal of Trp22 (Fig. 1b). However, the traditional cleavage condition of CNBr requires 70% formic acid or trifluoroacetic acid, or 0.1–0.5 M HCl, and 20–100 molar excess of CNBr31. Similarly, BNPS-Skatole often works in acidic conditions like 80% acetic acid20. These harsh conditions can compromise the POI or lead to unnecessary modification20. Additionally, chemical reagents display insufficient sequence specificity, always resulting in undesired hydrolysis on other amino acids.

a–c Traditional strategies to remove the N-terminal tag. d An engineered cleavable CPD tag fused to the N-terminus of a POI enables the customization of the first amino acid.
Intein-based removal of the N-terminal tag offers researchers an alternative option. One such commercial system, developed by New England Biolabs, is known as Intein-Mediated Purification with an Affinity Chitin-binding Tag (IMPACT)23,24. In this system, intein and chitin-binding domain (CBD) affinity tag are fused to the N-terminus of a POI, and the fusion protein is immobilized on a column with chitin matrix. Next, the intein is released by adding a thiol-containing reagent or adjusting the pH, ultimately yielding the native POI (Fig. 1c). This system provides a rapid and straightforward approach to prepare native POI under mild conditions. However, a risk of pre-cleavage exists during in vivo expression, and some inducers may alter the POI structure32.
The cysteine protease domain (CPD), derived from the Vibrio cholerae MARTX toxin, can be activated by eukaryotic-specific small molecule inositol hexakisphosphate (InsP6), and subsequently leads to self-cleavage after a specific leucine residue at its own N-terminus with a notable preference for the leucine residues surrounded by smaller amino acids33,34. Previous studies have shown that CPD enhances the expression and solubility of the POI when used as a C-terminal tag16,35,36. In our previous study, we inactivated the CPD by covalently conjugating the Cys144 with a peptide and found that the fusion protein, Endo-F3 (D165A)-CPDinactive, is stable in the presence of InsP6. However, an interesting phenomenon occurred that Endo-F3 (D165A)-CPDinactive can be cleaved by a purified CPD or by co-incubating with Endo-F3 (D165A)-CPD in the presence of InsP637 (Supplementary Fig. 11 in ref. 37), indicating that CPD possesses the potential for cleaving target proteins in trans processing34. Given the self- or in trans cleavage characteristic of CPD tag, we speculated that the N-terminal CPD tag is applicable for tag-removal to acquire native POIs with specific N-terminus (Fig. 1d). Taking an anti-Her2 nanobody as a model, we designed a series of engineered CPD tags fused to the N-terminus. After evaluating the cleavage rate of these fusion proteins, we ultimately identified Fusion 9 with L207I/L214A dual-mutations as the most effective candidate. We successfully applied this engineered CPD tag-removal strategy to express anti-Her2 nanobody carrying different N-terminal amino acids except Pro residue and Glu residue. In addition, anti-RNF43 nanobody with N-terminal Gln (N-Gln), an EGFR-targeting nanobody with N-Ala, the Sortase A enzyme with N-Gln, the fluorescent protein TurboGFP with N-Glu, and Δ15 Pd2,6ST with N-Cys can also be achieved, validating the feasibility and the broad substrate applicability of the strategy.
Results and discussion
Investigation of fusing the CPD tag to the N-terminus of a POI
Traditionally, the CPD tag is fused to the C-terminus of a protein to increase its soluble expression16,38. It remains unknown if it still works when fusing the CPD at the N-terminus of a POI. To answer it, we firstly fused the CPD (from pET22b-CPDSalI, see Materials section for plasmid information) to the N- or C-terminus of a nanobody against Her2, named as NbHer2, giving CPD-(G4S)3-NbHer2 and NbHer2-CPD respectively (Fig. 2a and Supplementary Fig. 1). After protein expression, we extracted the same amount of E. coli cells for SDS-PAGE analysis. As illustrated in Fig. 2b, fusing CPD at either the C-terminus or the N-terminus of NbHer2 gave the same expression levels, indicating that fusing CPD to the N-terminus of a protein also works well.

a Structure of the CPD-POI fusion and CPD sequence. b SDS-PAGE analysis of the expression levels of the fused NbHer2. CPD-(G4S)3-NbHer2: fusing the CPD tag to the N-terminus of the NbHer2; NbHer2-CPD: fusing the CPD tag to the C-terminus of NbHer2. c Peptide sequence of Fusion 1 and 2. SDS-PAGE (d) and LC-MS (e) analysis of the InsP6-induced cleavage of the fusion proteins. The proteins were incubated with 100 μM of InsP6 in 1 × PBS buffer at 4 °C for 16 h. FL, full length protein; A235-NbHer2 indicates the expected product when hydrolyzed at amino acid L234; E215-NbHer2 indicates the NbHer2-associated product after the hydrolysis at L214; A5-NbHer2 indicates the product after hydrolysis at L4. * indicates a half or third molecular weight of the released products.
To further evaluate the cleavage characterization of the N-terminal CPD, we next constructed another fusion protein CPD5-215-(G4S)3-VDALA-NbHer2 (Fusion 2, Fig. 2c), in which VDALA is the preferred cleavage site of CPD. After incubation with InsP6, the SDS-PAGE analysis illustrated that both fusion proteins exhibited complicated hydrolytic products (Fig. 2d). The LC-MS analysis further revealed that the cleavage of Fusion 1 mainly occurs at sites L4, L207, and L214, with L4 as the favorable cleavage site, which results in the predominant product A5-NbHer2. For Fusion 2, however, three CPD-associated fragments were identified with L234 being the preferred cleavage site (Fig. 2e and Supplementary Fig. 2). These data are consistent with prior data that CPD can recognize and cleave after any Leucine within flexible protein strands33,39. Thus, we sought to optimize the reaction conditions, including pH and temperature, to promote the cleavage efficiency at L234 of Fusion 2. The SDS-PAGE and LC-MS analysis demonstrated that a pH of 7.5 at 37 °C yielded favorable cleavage results (Supplementary Fig. 3). Overall, though with multi-site cleavage, these results demonstrated the feasibility of the N-terminal CPD tag fusion.
Engineering N-terminal CPD to facilitate the desired cleavage at the design site
Based on this intriguing observation, we next chose Fusion 2 as the initial template to engineer the CPD tag, aiming to achieve the neatly hydrolyzed product of Xaa-POI. First, we deleted two amino acids ‘VD’ of the cleavage sequence to give CPD-(G4S)3-ALA-NbHer2 (Fusion 3). Second, to avoid the undesired hydrolysis observed above, we mutated L207 and L214 to Ala, giving CPDL214A-G4S-VDALA-NbHer2 (Fusion 4), CPDL214A-(G4S)3-VDALA-NbHer2 (Fusion 5), and CPDL207A/L214A-(G4S)3-VDALA-NbHer2 (Fusion 6), respectively (Fig. 3a). We subsequently investigated the cleavage capabilities of these fusions and found that either Fusion 3 with ‘VD’ deletion or Fusion 4 with L214A and shorter GS linker impaired the specific hydrolysis at L234. The Fusion 5, with a single mutation at L214 and a longer GS spacer, yielded a greater proportion of the desired product in the presence of InsP6 according to the SDS-PAGE and LC-MS results (Fig. 3b and Supplementary Fig. 4). Notably, Fusion 6, featuring double mutations at L207 and L214, however, almost lost its cleavage activity completely, indicating that L207 plays pivotal role (Fig. 3b and Supplementary Fig. 4). Next, we shortened Fusion 2 and Fusion 5 by removing the ‘ADGK’ sequence at N-terminus, resulting in Fusion 7 and Fusion 8, respectively (Fig. 3a). As expected, this deletion accelerated the cleavage rate induced by InsP6 (Fig. 3c, d), since that Fusion 7 and 8 were completely consumed within 4–8 h. Thereafter, we proceeded to engineer the CPD tag based on Fusion 8.

a The design of the N-terminus CPD engineering. b SDS-PAGE analysis of the InsP6-induced cleavage efficacy of Fusion 2-6. Conditions: 200 μM of InsP6 in 50 mM Tris-HCl, pH 7.5, 37 °C, 3 h. SDS-PAGE (c) and LC-MS (d) analysis of the InsP6-induced cleavage of Fusion 2, 5, 7, and 8, 16 h. SDS-PAGE (e) and LC-MS (f) analysis of the InsP6 induced cleavage of Fusion 8, 9, and 10, 16 h. g SDS-PAGE analysis of Fusion 9 mutants CPD-A/GLH-NbHer2 or CPD-A/GLK-NbHer2, 8 h. Conditions for CPD cleavage: 1 mM of InsP6 in 50 mM Tris-HCl, pH 7.5, 37 °C. * indicates a half or third molecular weight of CPDs.
According to the above-mentioned results that Fusion 6 with L207A/L214A lost the cleavage activity completely, here we further replaced L207 with its isomer Ile to construct Fusion 9, trying to find a suitable substitution to avoid the alternative hydrolysis at L207. Additionally, considering that the smaller amino acids near L234 might promote cleavage, we replaced the tetra-peptide ‘SVDA’ before L234 in Fusion 8 to a single Gly residue to construct Fusion 10 (Fig. 3a). With Fusion 8, 9, and 10 in hand, we then incubated them with InsP6 to determine their cleavage activity. Both SDS-PAGE and LC-MS indicated that, though impaired the cleavage rate, L207I gave pure product that exclusively cleaved at L234 (Fig. 3e, f). Meanwhile, similar to Fusion 3, the replacement of ‘SVDA’ to a single ‘G’ didn’t contribute to the cleavage (Fig. 3e, f). We also conducted a preliminary analysis of the amino acids surrounding L234 by substituting A233 with Gly or Ser, and mutated A235 to His, Lys, or Gly (Fig. 3g and Supplementary Figs. 5, 6). Cleavage evaluation results of the fusions with either H235 or K235 revealed that A233 exhibited greater specificity than G233. Additionally, the cleavage process of G233 was slower compared to that of A233 (Supplementary Fig. 5). However, as to fusions with G235, all gave complete and site-specific cleavage at L234, yielding the expected Gly-NbHer2 (Supplementary Fig. 6). All these data indicated a significant correlation between the InsP6-induced cleavage capability and the amino acids surrounding L234.
Condition optimization of cleavage process induced by InsP6
Since that Fusion 9 gave the best site-specific cleavage, we further optimized the reaction conditions induced by InsP6. First, we tested the reaction temperatures at 4 °C, room temperature, and 37 °C. The SDS-PAGE analysis revealed the nearly complete consumption of Fusion 9 at r.t. or 37 °C, indicating that higher temperature significantly accelerates the cleavage process and increases the yield of Ala-NbHer2 (Fig. 4a). pH is another important study point in our study. Considering that strong alkaline and acidic conditions may affect protein stability, we preferred to conduct the reaction in a near-neutral environment. And we found that pH values ranging from 5.5 to 8.5 are suitable for the cleavage reaction (Fig. 4b). We also tested the cleavage process in the presence of calcium, our results suggested that no obvious influence on the cleavage efficiency was found when the concentration of Ca2+ is less than 5 mM, but the cleavage was dramatically impaired at 10 mM Ca2+ (Supplementary Fig. 7). Finally, we explored the InsP6 concentration and identified 0.25–1 mM as the optimal concentration (Fig. 4c). Based on these inspired results, we proceeded to purify the Ala-NbHer2 following the cleavage of Fusion 9. The SDS-PAGE and LC-MS analysis confirmed the effective acquisition of Ala-NbHer2 with high purity (Fig. 4d, e). Overall, these findings indicated that we successfully identified a suitable N-terminal CPD tag.

The SDS-PAGE analysis of the cleavage activity of Fusion 9 under different temperatures (a), pH (b), and InsP6 concentration (c). SDS-PAGE (d) and LC-MS (e) analysis of the purified A-NbHer2 after cleavage of Fusion 9 induced by InsP6. Lane 1: Marker; Lane 2: A-NbHer2 after purification; Lane 3: reaction system of InsP6-induced cleavage of Fusion 9. The above reactions were performed in 50 mM Tris-HCl.
Removal of N-terminal CPD to create proteins with the desired N-terminal amino acid
Further, we mutated A235, an amino acid before the NbHer2, of Fusion 9 into 19 other amino acid via site-directed mutagenesis to evaluate the universality of the screened N-terminal CPD. Though the residue changes of nCPD-NbHer2 caused a change in mobility in some cases, all fusion proteins were successfully expressed as full-length as determined by LC-MS (Fig. 5a, b and Supplementary Figs. 8–27). We then assessed the cleavage capability of these mutants to generate the desired POI with a distinct N-terminus. After incubation with InsP6, all groups were detected by SDS-PAGE and LC-MS. As illustrated in Fig. 5c, the fusion proteins with Ala, His, Lys, Cys, Gly, Gln, Thr, Val, Ser, Tyr, Leu, Asn, and Arg showed remarkable cleavage activity in yielding the NbHer2 with designed N-terminal amino acid. However, the ones with Phe, Trp, Met, and Ile, performed poorly in achieving the desired products. And the Pro-NbHer2 could not be detected by LC-MS (Supplementary Fig. 22). In addition, the engineered N-terminal CPD tag acted to be compatible with either small size or alkalescent amino acids, while the acidic residues like Asp and Glu resulted in alternative cleavage sites (Fig. 5c and Supplementary Figs. 8–27). Consequently, our strategy facilitates the systemic generation of POIs with diverse N-terminus, serving as a valuable tool for N-terminal modification and function research.

a Schematic representation of the InsP6-induced cleavage process of the nCPD fusion proteins. b SDS-PAGE profiles of all expressed nCPD-Xaa-NbHer2. c SDS-PAGE analysis of Xaa-POI from InsP6-induced cleavage of nCPD-Xaa-NbHer2, 20 h. SDS-PAGE and LC-MS determination of the InsP6-induced cleavage process of all nCPD-POIs: NbRNF43 with N-Gln (d), Δ15Pd2,6ST with N-Cys (e), NbEGFR with N-Ala (f), Sortase A with N-Gln (g), TurboGFP with N-Glu (h). All reactions were conducted under 1 mM InsP6 in 50 mM Tris-HCl, pH 7.5 at 37 °C.
With the above encouraging results, we then sought to apply our strategy to other POIs featuring different N-terminus. We selected the anti-RNF43 nanobody with Gln at N-terminus (N-Gln) and Δ15 Pd2,6ST, one recombinant truncated enzyme with α2,6-trans-sialidase and α2,6-sialyltransferase activity, with Cys at N-terminus (N-Cys)40. According to the SDS-PAGE, though the release of Gln-NbRNF43 needs at least 20 h’s incubation, the release of Cys-Δ15 Pd2,6ST was almost achieved within 8 h. Both target proteins were efficiently obtained following incubation with InsP6, demonstrating the broad applicability and practicality of the developed approach (Fig. 5d, e). We also fused the engineered CPD to the N-terminus of the EGFR-targeting nanobody (N-Ala), Sortase A enzyme (N-Gln), and fluorescent protein TurboGFP (N-Glu), respectively. The SDS-PAGE and LC-MS results indicated that the efficient hydrolysis at L234 in the fusions successfully released these POIs with native N-terminus in the presence of InsP6 (Fig. 5f–h). We also surprisingly found that, different from the model protein NbHer2, the tested protein TurboGFP with acidic N-terminal amino acid could be achieved in high purity, either Asp or Glu (Fig. 5h and Supplementary Fig. 28), indicating that D/E is also compatible with this system with suitable POI. In conclusion, the successful application across various POIs highlights the promising and potential of our strategy.
Mechanism study of the cleavage process of the engineered nCPD
To investigate the potential cleavage mechanism of engineered CPD, we site-mutated the catalytic residue Cys144 into Ala to eliminate the hydrolysis activity of nCPD33, and generated nCPDC144A-Gly-NbHer2. As illustrated in Fig. 6a, the nCPDC144A-Gly-NbHer2 kept intact in the presence of InsP6 (lane 1 vs. lane 3), indicating that C144A eliminates the hydrolysis activity of nCPD successfully. However, the inactive nCPDC144A-Gly-NbHer2 could be cleaved when co-incubated with the same amount of active nCPD-Gly-NbHer2 in the presence of InsP6, yielding the fractured nCPDC144A (23952 Da) and nCPD (23984 Da) (Fig. 6b, c). These results indicated the in trans processing of the engineered nCPD. The SDS-PAGE showed nearly complete consumption of nCPD-Gly-NbHer2 in 60 min, while the mixed group of nCPDC144A-Gly-NbHer2 and nCPD-Gly-NbHer2 contained the full-length fusion proteins (Fig. 6a, lane 13 vs. 14). The slower cleavage rate of the mixed group may result from the lower concentration, half of the nCPD-Gly-NbHer2 group, of active nCPD in the mixture. Deep analysis of the LC-MS profiles of the mixed group revealed that the ratio of released nCPDC144A and nCPD is 23.34: 100 at 5 min time point (Fig. 6b, zoom in part), while 58.82: 100 at 30 min time point (Fig. 6c, zoom in part), indicating that the cleavage of nCPD-Gly-NbHer2 is preferred and the percentage of in trans cleavage product increased as time went. Taken together, these results indicated that, though in trans processing exists, the cleavage process is inclined to an auto-processing over in trans processing.

SDS-PAGE (a) and LC-MS (b, c) analysis of nCPDC144A-G-NbHer2 and nCPD-G-NbHer2 co-incubated with 1 mM of InsP6 in 50 mM Tris-HCl at 37 °C. b 5 min; c 30 min. d The predicted structure of nCPD-(G4S)3-VDALA. e The overlapped structure of nCPD-(G4S)3-VDALA and reported CPD (PDB ID: 3EEB). f The predicted structure of nCPD-(G4S)3-VDALA-NbHer2-His6.
We also turn to AlphaFold2 for the prediction of conformational structures of the engineered CPD or the fusion proteins. We first predicted structure of ‘nCPD-(G4S)3-VDALA’ from Fusion 9, and found that the flexible GS linker facilitates the accessibility of L234 to the catalytic center, in which the catalytic site is labeled in red (Fig.6d). In contrast, in the reported structure of native, processed CPD without GS linker (cyan, PDB ID: 3EEB)33, the N-terminus is more accessible to the catalytic residue (red) than C-terminus (Fig.6e). In addition, the highly overlap of the engineered and reported CPD structures revealed a high score of prediction. Further, we predicted the conformational structure of Fusion 9, which clearly illustrated that the cleavage sequence ‘VDALA’ (green) is close to the catalytic residue (red) of nCPD (Fig.6f), indicating the high possibility of auto-processing approach of the fusion proteins with the engineered nCPD.
His-tagged nCPD enables one-step output of native target proteins without tags
So far, we have successfully developed the nCPD for expression of target proteins with desired N-terminal amino acid. However, all these tested proteins are fused with a C-terminal His tag for purification. Now, we move forward to construct a nCPD system that could yield a native target protein with desired N-terminus and without tag. To achieve this goal, we inserted the His tag at the N-terminus or C-terminus of the nCPD, and constructed fusion proteins His6/10-nCPD-GS-POI (Fusion 11a-b) and nCPD-His10-GS-POI (Fusion 12), respectively (Fig. 7a). At the first attempt, we fused the His-tagged nCPD with protein Δ15 Pd2,6ST, and the fusion protein was expressed and absorbed to the Ni-NTA resin, which was successively washed by PBS, 10 mM and 25 mM of imidazole. Thereafter, 1 mM of InsP6 in 50 mM Tris-HCl (pH 7.5) was added and the mixture was incubated at room temperature for 12 h and then at 37 °C for 4 h for hydrolysis. According to the SDS-PAGE analysis, an unexpected band with a higher molecular weight than target protein Δ15 Pd2,6ST was found in the fusion proteins with N-terminal His-tagged nCPD, either Fusion 11a (Fig. 7b) or 11b (Fig. 7c). However, the Fusion 12 with nCPD-His10 showed acceptable purity of the target protein Δ15 Pd2,6ST after InsP6-induced cleavage (Fig. 7d, e). Further, we fused the nCPD-His10 with TurboGFP and NbHer2 carrying a GGG at N-terminus, respectively. Both SDS-PAGE and LC-MS analysis indicated the excellent performance of the nCPD-His10 in yielding the tag-free target proteins with desired N-terminal amino acid (Fig. 7f, g).

a The construction of Fusion 11a-b and Fusion 12 with His-tagged nCPD. b SDS-PAGE analysis of one-step output efficiency of His6-nCPD-GS-Δ15 Pd2,6ST; S, supernatant; P, precipitate. c SDS-PAGE analysis of one-step output efficiency of His10-nCPD-GS-Δ15 Pd2,6ST. d SDS-PAGE analysis of one-step output efficiency of nCPD-His10-GS-Δ15 Pd2,6ST. e SDS-PAGE profile of the outputted tag-free Δ15 Pd2,6ST from the cleavage of nCPD-His10-GS-Δ15 Pd2,6ST. f The outputted tag-free TurboGFP. g The outputted tag-free G3-NbHer2. Reaction conditions: 1 mM of InsP6 in 50 mM Tris-HCl, pH7.5 at r.t. or 37 °C.
Conclusion
The development of biological tags significantly enhances the expression of recombination protein either in mammalian cells or in E. coli system. However, the additional tag may challenge the target proteins, prompting the need for effective removal of extra tags post-expression. In this study, we introduced an engineered CPD tag at the N-terminal of the target protein, a well-known self-cleavage tag to promote the protein expression and solubility that can be activated by InsP6, and investigated the potential applications in achieving the target proteins with the desired N-terminal amino acid. After several attempts in engineering the CPD tag, we found that L207 and L214 of CPD are two alternative cleavage sites when CPD is fused to the N-terminus, which tells us the existence of in trans processing of the CPD. We also found that mutating L207 into L207A resulted in the total loss of cleavage activity. According to the reported conformational structure of CPD (PDB ID: 3EEB)33, L207 is in the InsP6-binding pocket and is essential to the activation of CPD for cleavage, which may explain why the inactive cleavage of L207A mutant. To decrease the interference from L207 and keep the cleavage activity of nCPD in the meantime, L207I is supposed to be a feasible alternative. The dual-mutation of the N-fused CPD with L207I/L214A efficiently avoided the undesired cleavage and in-depth studies confirmed the practicality of this strategy in yielding the proteins with the designed N-terminal amino acid. The applications of model protein NbHer2 demonstrated that the protein with N-terminal Pro shows limited cleavage efficiency, and Trp gives undesired cleavage. Additionally, we found that the acidic amino acids of NbHer2 N-terminus, Asp and Glu, besides the designed site, contributed to the alternative cleavage at L4 of NbHer2 as well. However, this phenomenon was not found in another tested POI, TurboGFP, with either Asp or Glu at the N-terminus, suggesting to us that various POIs may exhibit different cleavage behavior. Further mutation of Cys144, a key catalytic residue, into Ala deprived the hydrolytic activity of nCPD. The fusion proteins with nCPDC144A, however, could be cleaved by incubating with the one with active nCPD, indicating the existence of in trans processing. Meanwhile, we surprisingly found that the native nCPD, rather than nCPDC144A, is the predominant component of the released nCPD at the beginning, and the percentage of nCPDC144A increases as time extends, which demonstrates the possibility of the auto-processing of nCPD from fusion proteins with a proper GS linker. These results were further confirmed by conformation prediction of the engineered nCPD by AlphaFold2, which suggests that the cleavage site ‘ALA’ coincidentally locates in the catalytic center of the engineered nCPD, maybe a result resulted from the flexible GS linker. In the last part, we moved the His tag from the POI’s C-terminus to the nCPD fragment, either at the N-terminus or C-terminus of nCPD, and successfully developed a one-step approach for the purification of a POI with the desired amino acid and without any tag. Overall, we suppose that this approach could customize diverse N-terminal amino acids and introduce functional amino acid, which advances the research on N-terminal modification and functionalization.
Materials and methods
Materials
All designed primers and DH5α cells were ordered from Tsingke Biotech Co., Ltd (Beijing, China). The Pfu DNA polymerase and other general PCR-related reagents were from TransGen Biotech Co., Ltd. (Beijing, China). The Dpn I enzyme, Seamless Cloning kit, DNA Gel extraction kit and Ni-NTA Sepharose 6FF (His-Tag) were bought from Sangon Biotech Co., Ltd. (Shanghai, China). DNA extraction kit was obtained from Vazyme Biotech Co., Ltd. (Nanjing, China). Protein Marker and BL21 cells were from Yeasen Biotechnology Co., Ltd. (Shanghai, China). InsP6 was obtained from Shanghai Haohong Scientific Co., Ltd (Shanghai, China). Amicon centrifugal ultrafiltration devices were obtained from Merck-Millipore (Germany). The pET22b-CPDSalI vector was from Addgene16 (Addgene plasmid # 38251; http://n2t.net/addgene:38251; RRID: Addgene_38251).
High performance liquid chromatography and high-resolution mass spectroscopy (HPLC-HRMS)
The mass spectra of proteins were recorded under the extended mass range mode (high 20,000 m/z, 1 GHz) and the data were collected in the mass range of 500–2500. Key source parameters: Cone gas 50 L/h; desolvation gas 800 L/h; source temperature of 120 °C; desolvation temperature of 600 °C; capillary voltage of 3000 V; collision cell RF offset of 600 volts; collision cell RF gain of 0.
Plasmid construction
To construct CPD-NbHer2-His6, two independent PCR assays were conducted to acquire pET26b-Her2 nanobody-His6 with homologous arm and the CPD fragment with SalI and XhoI sites was cloned from plasmid contains pET22b-CPDSalI vector16,38. Then assembled two fragments via seamless cloning kit after Gel extraction and subsequently digested by DpnI enzyme. Finally, the recombination plasmid was transfected into DH5α cells and the several single colonies were picked out for DNA sequencing after cultivation at 37 °C (Tsingke Biotech Co., Ltd).
Site-directed mutagenesis PCR were performed to generate specific plasmids using the designed primers. The generated PCR product was respectively digested and transfected into DH5α cells. Then picked the single colonies out for amplification at the next day and sent for sequencing.
General procedure for protein expression and purification
The correct plasmids identified via DNA sequencing were transfected into BL21 cells. After incubation overnight at 37 °C, the single colonies were inoculated into 5 mL of LB medium with 100 μg/mL ampicillin, and were cultured at 37 °C with 200–220 r/min. Next day, the cloudy bacterial fluid was transferred into appropriate volume of LB medium, and was continuously cultivated until the OD600 was about 0.8. Then, 1 mM IPTG and 10% glycerol were added to induce protein expression. The medium was shaken at lower speed overnight at 18 °C. Thereafter, the cells were harvested by centrifugation and kept at −80 °C.
For protein purification, the harvested cells were resuspended in 1 × PBS and were sonicated. The supernatant was collected after centrifugation and was further incubated with 1–2 mL of Ni-NTA resin for 2 h at 4 °C. Then, the mixture was successively eluted with 10 mM, 25 mM, 50 mM, 100 mM and 250 mM imidazole. Next, the SDS-PAGE analysis was conducted to identify which elution contains target protein. The fractions containing product were collected, ultrafiltrated and concentrated via 10 kDa or 30 kDa MWCO Amicon devices. After exchanged the imidazole to PBS buffer, the obtained protein was stored at −20 °C.
General procedure for nCPD tag removal of various engineered nCPD-POI
The fusion protein (2 mg/mL) was incubated with InsP6 (1 mM) in a Tris-HCl buffer (50 mM, pH 7.5) at 37 °C. An aliquot of 2.0 μg was taken out for SDS-PAGE at the defined intervals. And 4.0 μg of protein was used for LC-MS determination. Note: in the hydrolysis of Fusion 1 and Fusion 2, 1 × PBS buffer was used for the cleavage.
General procedure to purify Xaa-POI
The fusion protein (2 mg/mL) was incubated with InsP6 (1 mM) in a Tris-HCl buffer (50 mM, pH 7.5) at 37 °C. Once the SDS-PAGE and LC-MS analysis indicated the completion of the cleavage reaction, the Ni-NTA resin was added into the solution and incubated at 4 °C for 2 h. Then, 10 mM and 25 mM imidazole were successively used to wash the cleaved nCPD fragment. Next, higher concentration imidazole solutions were added to elute the Xaa-POI. The fractions containing product were collected, ultrafiltrated and concentrated by MWCO Amicon devices.
nCPD removal of the nCPD-NbRNF43, nCPD-Δ15 Pd2,6ST, nCPD-NbEGFR, nCPD-Sortase A and nCPD-TurboGFP to generate NbRNF43, Δ15 Pd2,6ST, NbEGFR, Sortase A and TurboGFP with desired N-terminus
The recombinant nCPD-POIs were constructed and expressed according to the above-mentioned protocols. Then, the fusion proteins (40 μg) were respectively incubated with InsP6 (1 mM) at 37 °C in 20 μL of 50 mM Tris-HCl, pH 7.5. All reactions were monitored by SDS-PAGE and LC-MS analysis at the specified intervals to detect the product.
General procedure for one-step output of native target proteins without tags
His6/10-nCPD-GS-POI and nCPD-His10-GS-POI were respectively constructed via the above-mentioned protocols, and all expressed by E. coli expression system. The expressed cells were collected and sonicated, then the supernatant were separated by centrifugation. The Ni-NTA resin was mixed with the supernatant and co-incubated at 4 °C more than 2 h. Next, the Ni-NTA-supernatant mixture were loaded in the empty affinity chromatography column, the flow through solution collected and re-loaded in the column. Then, the PBS buffer, and imidazole at a low concentration like 10 mM, 25 mM or 50 mM was successively added to wash the non-specific protein attached to resin. Subsequently, 50 mM Tris-HCl buffer at pH 7.5 was added in the column to replace the imidazole, and the buffer contains 1 mM InsP6 was supplemented to induce the cleavage. After induced at room temperature for 12 h and incubated at 37 °C for another 4 h, collected the flow through solution. The PBS buffer was added to wash the tag-free POIs in the column. Finally, the imidazole at high concentration was used to elute the resin.
Structure prediction
The 3D structures of proteins were predicted by Swiss-Model (https://swissmodel.expasy.org), or ColabFold41 (https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb) based on AlphaFold2.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data supporting the findings of this study are available within the main text and the Supplementary Information. The relevant sequences and LC-MS profiles are provided in the Supplementary Information file. The uncropped gel images are available in the Supplementary Information Fig. 29.
References
-
Assenberg, R., Wan, P. T., Geisse, S. & Mayr, L. M. Advances in recombinant protein expression for use in pharmaceutical research. Curr. Opin. Struct. Biol. 23, 393–402 (2013).
-
Tsuda, M. & Nonaka, K. Recent progress on heterologous protein production in methylotrophic yeast systems. World J. Microbiol. Biotechnol. 40, 200–212 (2024).
-
Fu, Y. et al. Improvement strategies for transient gene expression in mammalian cells. Appl. Microbiol. Biotechnol. 108, 480–490 (2024).
-
McDonald, B. & Schmidt, M. H. H. Structure, function, and recombinant production of EGFL7. Biol. Chem. 405, 691–700 (2024).
-
Saroha, P., Patil, R. S. & Rathore, A. S. Recent advancements in soluble expression of recombinant antibody fragments in microbial host systems. Prep. Biochem. Biotechnol. 55, 131–140 (2025).
-
Jiang, R. et al. Strategies to overcome the challenges of low or no expression of heterologous proteins in Escherichia coli. Biotechnol. Adv. 75, 108417–108435 (2024).
-
Rashid, M. H. Full-length recombinant antibodies from Escherichia coli: production, characterization, effector function (Fc) engineering, and clinical evaluation. MAbs 14, 2111748–2111767 (2022).
-
Popova, L. G., Khramov, D. E., Nedelyaeva, O. I. & Volkov, V. S. Yeast heterologous expression systems for the study of plant membrane proteins. Int. J. Mol. Sci. 24, 10768–10796 (2023).
-
Zhao, M., Ma, J., Zhang, L. & Qi, H. Engineering strategies for enhanced heterologous protein production by Saccharomyces cerevisiae. Microb. Cell Fact. 23, 32–47 (2024).
-
Rosano, G. L. & Ceccarelli, E. A. Recombinant protein expression in Escherichia coli: advances and challenges. Front. Microbiol. 5, 172 (2014).
-
Kaur, J., Kumar, A. & Kaur, J. Strategies for optimization of heterologous protein expression in E. coli: roadblocks and reinforcements. Int. J. Biol. Macromol. 106, 803–822 (2018).
-
Raran-Kurussi, S. & Waugh, D. S. A dual protease approach for expression and affinity purification of recombinant proteins. Anal. Biochem. 504, 30–37 (2016).
-
Byun, K. T. et al. Development of an anti-HER2 single-chain variable antibody fragment construct for high-yield soluble expression in Escherichia coli and one-step chromatographic purification. Biomolecules. 13, 1508–1919 (2023).
-
Wang, Y. et al. The functional characteristics and soluble expression of saffron CsCCD2. Int. J. Mol. Sci. 24, 15090–15101 (2023).
-
Zhou, L. et al. Expression of melittin in fusion with GST in Escherichia coli and its purification as a pure peptide with good bacteriostatic efficacy. ACS Omega 5, 9251–9258 (2020).
-
Shen, A. et al. Simplified, enhanced protein purification using an inducible, autoprocessing enzyme tag. PLoS ONE 4, e8119–e8129 (2009).
-
Smyth, D. R., Mrozkiewicz, M. K., McGrath, W. J., Listwan, P. & Kobe, B. Crystal structures of fusion proteins with large-affinity tags. Protein Sci. 12, 1313–1322 (2003).
-
Zhao, X., Li, G. & Liang, S. Several affinity tags commonly used in chromatographic purification. J. Anal. Methods Chem. 2013, 581093 (2013).
-
Booth, W. T. et al. Impact of an N-terminal polyhistidine tag on protein thermal stability. ACS Omega 3, 760–768 (2018).
-
Yadav, D. K., Yadav, N., Yadav, S., Haque, S. & Tuteja, N. An insight into fusion technology aiding efficient recombinant protein production for functional proteomics. Arch. Biochem. Biophys. 612, 57–77 (2016).
-
Hwang, P. M., Pan, J. S. & Sykes, B. D. Targeted expression, purification, and cleavage of fusion proteins from inclusion bodies in Escherichia coli. FEBS Lett. 588, 247–252 (2014).
-
Srzentić, K. et al. Chemical-mediated digestion: an alternative realm for middle-down proteomics? J. Proteome Res. 17, 2005–2016 (2018).
-
Lu, W. et al. Split intein facilitated tag affinity purification for recombinant proteins with controllable tag removal by inducible auto-cleavage. J. Chromatogr. A 1218, 2553–2560 (2011).
-
Chong, S. et al. Utilizing the C-terminal cleavage activity of a protein splicing element to purify recombinant proteins in a single chromatographic step. Nucleic Acids Res. 26, 5109–5115 (1998).
-
Osiro, K. O. et al. Cleaving the way for heterologous peptide production: an overview of cleavage strategies. Methods 234, 36–44 (2025).
-
Santana, S. D., Pina, A. S. & Roque, A. C. Immobilization of enterokinase on magnetic supports for the cleavage of fusion proteins. J. Biotechnol. 161, 378–382 (2012).
-
Ruan, B., Fisher, K. E., Alexander, P. A., Doroshko, V. & Bryan, P. N. Engineering subtilisin into a fluoride-triggered processing protease useful for one-step protein purification. Biochemistry 43, 14539–14546 (2004).
-
Messaabi, A. et al. In vivo thrombin activity in the diatom Phaeodactylum tricornutum: biotechnological insights. Appl. Microbiol. Biotechnol. 108, 481 (2024).
-
Zhang, Y. et al. Rational design of a humanized glucagon-like peptide-1 receptor agonist antibody. Angew. Chem. Int. Ed. Engl. 54, 2126–2130 (2015).
-
Cordingley, M. G., Callahan, P. L., Sardana, V. V., Garsky, V. M. & Colonno, R. J. Substrate requirements of human rhinovirus 3C protease for peptide cleavage in vitro. J. Biol. Chem. 265, 9062–9065 (1990).
-
Andreev, Y. A., Kozlov, S. A., Vassilevski, A. A. & Grishin, E. V. Cyanogen bromide cleavage of proteins in salt and buffer solutions. Anal. Biochem. 407, 144–146 (2010).
-
Li, Y. Self-cleaving fusion tags for recombinant protein production. Biotechnol. Lett. 33, 869–881 (2011).
-
Lupardus, P. J., Shen, A., Bogyo, M. & Garcia, K. C. Small molecule-induced allosteric activation of the Vibrio cholerae RTX cysteine protease domain. Science 322, 265–268 (2008).
-
Prochazkova, K. et al. Structural and molecular mechanism for autoprocessing of MARTX toxin of Vibrio cholerae at multiple sites. J. Biol. Chem. 284, 26557–26568 (2009).
-
Biancucci, M. et al. New ligation independent cloning vectors for expression of recombinant proteins with a self-cleaving CPD/6xHis-tag. BMC Biotechnol. 17, 1 (2017).
-
Tang, F. et al. Selective N-glycan editing on living cell surfaces to probe glycoconjugate function. Nat. Chem. Biol. 16, 766–775 (2020).
-
Zeng, Y. et al. C-terminal modification and functionalization of proteins via a self-cleavage tag triggered by a small molecule. Nat. Commun. 14, 7169 (2023).
-
Giddens, J. P., Lomino, J. V., Amin, M. N. & Wang, L. X. Endo-F3 glycosynthase mutants enable chemoenzymatic synthesis of core-fucosylated triantennary complex type glycopeptides and glycoproteins. J. Biol. Chem. 291, 9356–9370 (2016).
-
Shen, A. et al. Mechanistic and structural insights into the proteolytic activation of Vibrio cholerae MARTX toxin. Nat. Chem. Biol. 5, 469–478 (2009).
-
Cheng, J. et al. Trans-sialidase activity of photobacterium damsela alpha2,6-sialyltransferase and its application in the synthesis of sialosides. Glycobiology 20, 260–268 (2010).
-
Mirdita, M. et al. ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682 (2022).
Acknowledgements
This work was supported by the Natural Science Foundation of China (NSFC, Nos. 22422705, 82325045, 92478204, 22337003, 22277126, and 82204183), and Shanghai Sail Program (No. 22YF1457400). F.T. gratefully acknowledge the support of the SANOFI Scholarship Program.
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Ophelia Bu. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zou, X., Liu, Z., Song, F. et al. Facile expression of proteins with desired N-terminal amino acid via an engineered cysteine protease domain. Commun Biol 8, 1165 (2025). https://doi.org/10.1038/s42003-025-08614-7
-
Received:
-
Accepted:
-
Published:
-
DOI: https://doi.org/10.1038/s42003-025-08614-7