Main
The broad range of applications that employ bacteria and viruses for therapy mirrors the diversity of microbes themselves. Distinct bacteria target different tissues, microbiomes and even intra- versus extracellular spaces. Ranging from skin-colonizing Staphylococcus epidermidis to lung-homing Mycoplasma pneumoniae, to the intracellularly growing Listeria monocytogenes and Salmonella typhimurium, different bacteria are each equipped with capabilities uniquely exploitable for synthetic engineering1,2,3,4,5. Analogously, the array of viral families under investigation for therapy is similarly broad, with applications exploiting the different genomic structures, natural tropisms and life cycles of each. Examples include small DNA viruses such as adeno-associated virus for non-immunogenic tissue targeting, minus-strand RNA viruses such as rabies virus for retrograde neuronal tracing and plus-strand RNA viruses such as the poliovirus-derivative ‘PVSRIPO’ for oncolytic purposes6,7,8,9,10. Thus, microbes of broadly distinct cellular proclivities have each found utility for a specific application niche.
Bacteria and viruses are generally considered separately in approaches to therapeutic delivery. Yet, during natural co-infection, some viruses and bacteria directly bind to one another, leading to enhanced fitness11,12,13. Similarly, applications in synthetic biology are beginning to engineer coordination between multiple interacting entities with unique properties that can together produce a consortium achieving a collective objective14,15,16,17,18,19,20,21. In this work, we consider synthetic approaches for viral and bacterial cooperation to overcome key challenges in applying microbially delivered therapies to combat cancer. While oncolytic viruses have shown efficacy in clinical trials, pre-existing humoral immunity can limit the ability of systemically delivered viral particles to reach their target tumours22. Meanwhile, engineered bacteria therapies can deliver genetic payloads to tumour cells, but typically remain trapped locally within the tumour core and are limited in their ability to influence peripheral or distant tumour cells. Indeed, clinical trials with strains such as attenuated Salmonella VNP200009 have yet to demonstrate significant clinical efficacy and show dose-related toxicity23,24.
Here we address these limitations by engineering a platform called CAPPSID (Coordinated Activity of Prokaryote and Picornavirus for Safe Intracellular Delivery). This system consists of a synthetic bacteria–virus partnership to deliver an oncolytic picornavirus into tumours via systemic delivery, where the bacteria can cloak the virus from circulating antiviral antibodies. Then, once inside the tumour, the bacteria can launch the virus and spread. Specifically, CAPPSID relies on an engineered S. typhimurium to act as a dynamic and synthetic ‘capsid’ to transcribe and deliver viral RNA inside cancer cells, launching a virus that can directly lyse surrounding cells. We then additionally engineer the virus to require a bacterially donated accessory enzyme necessary for viral maturation and subsequent spread, thereby constraining viral replication to one additional infection cycle beyond the originating, bacteria-containing cell (Fig. 1). Together, this bacteria–virus cooperation enables tumour inhibition and yields prolonged engineered viral replication. Being presumably the first example of direct engineered cooperativity between bacteria and oncolytic viruses, this work demonstrates bespoke interacting communities of programmable medicines.

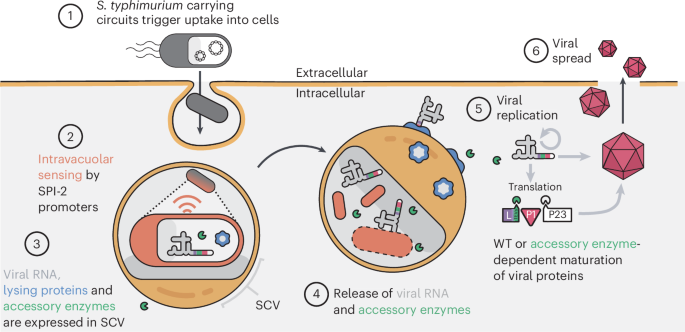
(1) S. typhimurium carrying synthetic circuits enter mammalian cells via natural effectors encoded on Salmonella pathogenicity island 1 (SPI-1). (2) Internalized S. typhimurium within a Salmonella containing vacuole (SCV) sense the intravacuolar space and trigger activation of SPI-2 promoters. (3) Engineered SPI-2 promoters are then used to drive the production of viral RNAs (poliovirus replicon, Senecavirus A (SVA) or engineered SVA), lysing proteins hemolysin E (HlyE) and E from phage φX174, and accessory enzyme. (4) Upon successful bacterial and vacuolar lysis, viral RNAs and accessory enzyme are released into the host cytoplasm. (5) Wild-type (WT) viral RNAs are translated in the cytoplasm and viral replication is initiated. The maturation of viral particles may be engineered to require the accessory enzyme for complete maturation. (6) Infectious particles are released into the extracellular space to infect neighbouring cells. Since S. typhimurium bacteria act as a viral ‘capsid’, we have named the platform Coordinated Activity of Prokaryote and Picornavirus for Safe Intracellular Delivery (CAPPSID).
Results
Engineered S. typhimurium autonomously launches viral RNA
To establish CAPPSID as a bacterial platform capable of delivering viral RNAs into cells, we focused on S. typhimurium, a naturally facultative intracellular bacterium. S. typhimurium achieves invasion into host cells via macropinocytosis and survives within the Salmonella containing vacuole (SCV) by expressing a battery of genes encoded on Salmonella pathogenicity islands 1 and 2 (SPI-1 and SPI-2), respectively25. By using Salmonella to release viral RNA, the direct translation of its viral gene products bypasses the need for nuclear translocation of plasmid-encoded therapeutics and their subsequent expression before induction of apoptosis or pyroptosis by Salmonella26,27. To efficiently deliver long nucleic acids by bacteria, associated challenges include sufficient RNA production, robust RNA integrity, RNA escape into the host cytoplasm and the need for the host to survive through protein translation.
To control RNA release, we employ natural spatial cues transduced by Salmonella to sense the intracellular phase of their life cycle, using the SPI-2 promoters of the genes sseA and sseJ to trigger activation28,29,30 (Fig. 2a). When these promoters are fused to mCherry, we observed rapidly increasing expression of both promoters only after entering mammalian cells, indicating that they are tightly regulated and suitable for intracellular cargo expression, consistent with previous reports4,28,31,32,33,34,35,36,37 (Fig. 2b,c and Supplementary Fig. 1a).

a, Intracellularly, SPI-2 promoter PsseA drives mCherry and T7 polymerase, while PsseJ drives the lysis proteins. Therefore, intravacuolar S. typhimurium lyse themselves and the SCV, releasing mCherry and T7-driven poliovirus-replicon RNA into the cytoplasm where replication and translation produce reporter GFP. b, Micrographs of HeLa cells inoculated with S. typhimurium at MOI 50 carrying constitutive Ptac-GFP and intracellularly activated PsseA-mCherry plasmids. Top: 0 h.p.i. Bottom: 12 h.p.i. Scale bar, 50 μm. c, Quantification of PsseA activation shown as the mean fluorescence intensities (MFI) of mCherry divided by GFP, where each dot represents a single HeLa cell. At each timepoint, the average over all cells is plotted as a red line. The initial value was taken at 1 h.p.i. d, Top left: circuit diagram of proteins produced by PsseA activation and T7-driven poliovirus replicon. Bottom left: schematic of smFISH probes binding to the 3′ end of the viral RNA transcribed by bacteria. Right: micrograph showing DAPI staining of both mammalian and bacterial DNA (blue), PsseA-mCherry fluorescence from S. typhimurium inside SCVs (orange) and fluorescent signal from probes specific to viral RNA (red). Scale bar, 20 μm. e, Top: DIC and mCherry signals of HeLa cells with S. typhimurium at MOI 50 carrying a PsseA-mCherry plasmid. Bottom: HeLa cells with S. typhimurium at MOI 50 carrying mCherry reporter and lysing proteins, showing mCherry signal diffu sing through the host cytoplasm. Scale bar, 50 μm. f, Fraction of GFP-positive cells following inoculation in HCT116, H446, HeLa, 4T1 and B16 cells. GFP is expressed from poliovirus replicon (first three rows) or via plasmid with GFP driven by a mammalian pEF promoter, from strains that either lyse with HlyE and E proteins, the E protein alone, or do not lyse. g, Bacteria with lysing circuit and virus-encoding plasmid were used to inoculate HeLa cells. DAPI indicates nuclear staining. GFP fluorescence is derived from the poliovirus-replicon reporter. Red fluorescence signal is an anti-dsRNA antibody (J2) indicating active replication of viral RNA. Scale bar, 50 μm.
Because prokaryotes do not incorporate 5′m7G caps required for typical mammalian translation38, we chose to deliver viral RNAs that rely instead on cap-independent translation using the internal ribosome entry site (IRES) from Picornaviridae39. To evaluate how such a platform might function across a range of cell lines, we first utilize a poliovirus replicon with GFP in place of its structural proteins as our viral RNA reporter, which has been shown to be replication competent in numerous cell types40. This replicon, with a fully active viral polymerase, serves to self-amplify the viral RNA but is unable to spread. Notably, the robust GFP production we observed upon direct RNA transfection was reduced by 50- to 1,000-fold when the viral polymerase was mutationally inactivated, supporting the use of self-amplifying RNA for increased cargo expression (Supplementary Fig. 1b). To couple transcription of this viral RNA production to intracellular sensing, the PsseA promoter of S. typhimurium drives expression of the highly processive T7-RNA polymerase, which in turn transcribes the viral RNA from its complementary DNA (cDNA) genome encoded on a plasmid. When this circuit is transformed into S. typhimurium LH1301 (ΔaroA, ΔphoPQ) and used to invade HeLa cells, these bacteria produce the full-length viral RNA, as measured by single-molecule fluorescence in situ hybridization (smFISH) using probes against the 3′ end of the 5.5 kb poliovirus replicon (Fig. 2d).
Once transcribed, the viral genome must escape the bacterium and translocate through the SCV into the cytoplasm of the mammalian host to begin replication. To optimize efficiency of this translocation, we added to our circuit two distinct lytic proteins: (1) lysis protein E from phage φX174 that disrupts bacterial membranes4,41,42,43, allowing the viral RNA to exit the lysed bacterium, and (2) hemolysin E (HlyE) which forms pores in the SCV, allowing the viral RNA to enter the host cytosol44. Genes encoding these proteins are expressed under the control of intracellular sensing promoter PsseJ and complemented by a deletion of the sifA gene, which further disrupts SCV integrity4,45. When S. typhimurium carries this circuit into HeLa cells, mCherry appears to diffuse out of the SCV, filling the cytoplasm of the host cell, while in the absence of these lytic proteins, mCherry remains punctate, indicating restricted localization within vacuoles (Fig. 2e).
Finally, we coupled the viral transcription and lysis circuits together in S. typhimurium to evaluate whether the poliovirus replicon encoding GFP could be delivered into a range of cell types and launch replication. We observed intense GFP signals indicative of successful viral delivery and replication in both mouse and human cell lines including 4T1, B16, HCT116, HeLa, MC38 and H446 cells, with varying efficiencies (Fig. 2f and Supplementary Fig. 1c). Furthermore, lysis via E and HlyE enabled dramatically more efficient delivery than in the absence of lytic proteins, or when S. typhimurium delivered a plasmid encoding a mammalian-promoter-driven GFP, across most cell lines (Fig. 2f and Supplementary Fig. 1d,e). Finally, to confirm that this was not simply passive translation of the incoming genomic viral RNA, we stained cells with an antibody against long double-stranded RNA (dsRNA), a product of active viral replication, and observed positive signals in cells that were also GFP positive (Fig. 2g). Together, these data show that CAPPSID is capable of successfully delivering actively replicating viral RNA.
CAPPSID delivery of full-length virus clears SCLC tumours
We next tested the ability of this system to deliver a therapeutically relevant full-length oncolytic virus, Senecavirus A (SVA), known to efficiently infect neuroendocrine-like cells, including H446 small-cell lung cancer (SCLC)46,47,48,49. Because cells infected with S. typhimurium frequently die via induction of apoptosis and pyroptosis26, a spreading virus could infect surrounding S. typhimurium-free cells, thereby augmenting the overall therapeutic effect (Fig. 3a).

a, Schematic of SPI-2-driven full-length viral RNA and lysing proteins. After releasing RNA into host cytoplasm, IRES-mediated translation produces viral proteins necessary for replication and packaging. b, (i) Top: H446 cells inoculated with MOI 25 S. typhimurium carrying SPI-2-driven lysis and reporter plasmid, along with SVA-GFP plasmid. Bottom: time-lapse microscopy at three timepoints of spreading SVA-GFP as launched from bacteria. Scale bar, 500 μm. (ii) Time course of SVA-GFP infection through the 72-h acquisition period, projecting time as a colour where initial events are represented in light blue hues and later events as yellow and red hues. c, (i) Experimental outline of in vivo experiment where nude mice were engrafted with H446 cells on bilateral flanks, and right flanks were IT injected with 2.5 × 106 bacteria when tumours reached ~150 mm3 14 days later. (ii) IVIS images of nude mice injected with NanoLuc substrate IT 2 and 4 days post bacterial injection, demonstrating launch and viral spread. d, (i) Growth kinetics of uninjected left flank tumours after administration of lysing S. typhimurium with WT-SVA (black), lysing S. typhimurium only (blue) and RPMI media control (red) into right flank tumours. Mean relative tumour trajectories plotted with error bars representing s.e.m. of biological replicates (n = 5 tumours per group; ordinary two-way analysis of variance (ANOVA) with Tukey post test). (ii) Growth kinetics of treated right flank tumours (n = 5 tumours per group; ordinary two-way ANOVA with Tukey post test). (iii) Survival curves for mice treated with groups shown in (i) and (ii). Multiple log-rank tests with Bonferroni correction (n = 5 mice per group). e, (i) Left: Experimental timeline: A/J mice were infected with 2.5 × 106 SVA plaque-forming units (p.f.u.s) and then engrafted with N1E-115 cells 7 days later. When tumours were ~150 mm3 (after another ~7 days), mice were injected IV with 2.5 × 106 CAPPSID/NanoLuc-SVA. Right: IVIS images of mice previously infected with SVA receiving a rechallenge at day 14 with either viral particle rechallenge of 2.5 × 106 p.f.u.s (top) or 2.5 × 106 CAPPSID/NanoLuc-SVA (bottom). (ii) Growth trajectories of tumours receiving: CAPPSID/SVA (black) in vaccinated mice; lysing control S. typhimurium (red); vaccinated mice rechallenged with an additional 2.5 × 106 SVA viral particles (blue). (iii) Survival curves for groups in (ii) (log-rank test, n = 5 mice per group).
For this system to function, bacteria and virus must both be able to infect the target population, with the former not directly inhibiting the latter. Using SVA with a GFP reporter (SVA-GFP)50, we measured whether and how viral spread was affected when introduced 1 h after bacterial pre-infection in H446 cells. At 24 h post infection (h.p.i.) there was no measurable reduction in the spread of virus in the presence or absence of bacteria (Supplementary Fig. 2a).
In S. typhimurium containing the lysis circuit, we added SVA-GFP and inoculated H446 cells to look for successful initial delivery events from bacteria and a subsequent capacity for spread throughout the same culture. Here, GFP-positive cells lacking mCherry would indicate spreading viral infection in cells not originally infected by bacteria, whereas double-positive cells contain both bacteria and virus. The first wave of SVA-infected cells was observed at ~8 h following bacteria inoculation, with viral spreading occurring continuously throughout the subsequent 60 h. By 72 h.p.i., effectively all cells were SVA-positive, regardless of initial bacterial infection status (Fig. 3b and Supplementary Video 1). This system demonstrates that even a low fraction of initially infected cells with S. typhimurium could deliver and launch the spread of an oncolytic virus across an entire monolayer of cells.
To determine whether CAPPSID carrying the full-length SVA virus could also achieve this effect in vivo, we used a mouse model engrafted with bilateral hind-flank H446 tumours. Right tumours were injected intratumourally (IT) with lysing S. typhimurium carrying SVA-NanoLuc (a luminescent reporter) and imaged over time for luminescence. At 2 days post infection, right-flank tumours began to show signal. Then, at day 4, the signal was additionally observable in left tumours that had not been injected with bacteria, showing productive viral infection in the right tumours and sufficient titre capable of viral translocation to left-flank tumours (Fig. 3c). In contrast, control bacteria recombinantly expressing their own luminescent reporter luxCDABE (and no viral RNAs) under the control of PsseA, showed no detectable translocation to left tumours over the same time (Supplementary Fig. 2b,c). Tumour volume measurements over more than 40 days showed complete regression of both left and right tumours in the treatment group within 2 weeks, whereas all tumours treated with only buffer or lysing bacteria alone continued to grow until reaching maximum allowable sizes (Fig. 3d and Supplementary Fig. 2d). The striking regression of tumours conferred 100% survival in mice treated with S. typhimurium carrying SVA-NanoLuc, while all control mice treated with RPMI and lysing Salmonella alone succumbed to tumour burden. Mice experienced no decline in weight, and had negligible bacteria in the liver or spleen, despite appreciable loads present in the tumours, suggesting no adverse response to bacterial injections (Supplementary Fig. 2e,f). Furthermore, histology shows viral spread well beyond the local vicinity in the tumour colonized by bacteria (Supplementary Fig. 2g). Taken together, CAPPSID/NanoLuc-SVA can launch an oncolytic viral infection clearing H446 tumours in vivo.
CAPPSID overcomes systemic viral-neutralizing antibodies
Thus far, we have shown successful viral launch using IT-injected bacteria in athymic mice unsusceptible to SVA. Next, we sought to extend the investigation of the platform to a fully immunocompetent model, employing the syngeneic mouse neuroblastoma cell line N1E-115 in A/J mice. After validating in vitro launch in N1E-115 (Supplementary Fig. 2h), we engrafted mice with double hind-flank N1E-115 tumours, and 2 weeks later IT injected CAPPSID/SVA. Following the mice over a course of 1 month, we observed successful attenuation of tumour growth and improved overall survival (Supplementary Fig. 2i).
If CAPPSID can initiate oncolytic infections in tumours when delivered intravenously (IV), we hypothesized that it could also overcome pre-existing immunity against an oncolytic virus. For example, could a bacterially cloaked viral genome escape circulating antiviral antibodies, permitting tumour targeting when systemically delivered? To test this, we first infected mice with wild-type (WT)-SVA particles 7 days before tumour engraftment and confirmed the presence of resulting circulating neutralizing antibodies against the virus (Supplementary Fig. 2j). Then, after sufficient tumour growth, we IV treated the mice with CAPPSID/SVA after pre-dosing with innate immune antagonists, and tracked the effects on tumour progression and luminescence over time. We observed that previously vaccinated mice were completely refractory to viral particle rechallenge, resulting in no detectable viral luminescence within the tumour and no survival benefit compared with mock treatment (Fig. 3e). In contrast, previously SVA-infected mice that received bacteria in the form of CAPPSID/SVA showed clear luminescence within the tumour as well as improved survival (Fig. 3e and Supplementary Fig. 2j,k). Assaying animal health and concurrent biodistribution of these bacteria when delivered IV showed substantial enrichment within the tumours, with little to no bacteria detectable in either the spleen or the liver (Supplementary Fig. 2l) and no meaningful decrease in mouse weight. Together these data demonstrate the ability of CAPPSID to safely attenuate tumour growth in a fully immunocompetent model when delivered either IT or systemically, even in the presence of neutralizing antibodies.
Engineered cooperation enables control over viral spread
While potentially promising, some viruses considered pre-clinically as oncolytics can spread systemically and cause undesirable side effects51. In contrast, non-spreading viral RNAs generated through the deletion of structural proteins to yield ‘self-amplifying RNA’ have recently gained interest as a therapeutic medium52 but still show limited persistence due to an inability to re-infect cells. Methods to improve targeting and safety profiles of oncolytic viruses are achieved either at the cell-surface level, where the receptor-binding domain is altered to recognize a target cell, or intracellularly, where replication of the virus is modulated positively or negatively by cell-type specific cytoplasmic or nuclear determinants53. Here we wondered whether we could exploit our CAPPSID platform to control and enhance the virus using the bacteria which are naturally restricted to the immunoprivileged tumour core.
In the life cycle of picornaviruses, all proteins are first translated as a single large open reading frame, termed a polyprotein, that is then cleaved into constituents by virally encoded proteases. These cleaved proteins act as mature replicase proteins and viral structural proteins required for continued spreading infection54. We hypothesized that shifting a cleavage event within the structural proteins to an orthogonal protease expressed by the same bacteria would enable complete viral particles to be produced, leading to a wave of infection beyond the bacterially infected cells, further enhancing the spatial and temporal reach. In those cells subsequently infected by virus and not bacteria, the viral replication could cause cytopathic effects, but spread no further (Fig. 4a). Due to its potential for recombinant expression and thorough characterization, we chose tobacco etch virus protease (TEVp) as the orthogonal protease55. Furthermore, TEVp has the flexibility to recognize nearly all residues at the final position of the cognate TEV cleavage site (TEVs) (ENLYFQ^G) where cleavage occurs between the last two amino acids55. This allows for the ability to retain the native N-terminal residue of the downstream protein following successful cleavage.

a, Schematic of potential orthogonal cleavage sites flanking the structural viral proteins to reprogramme for exogenous TEV protease recognition (white-to-green graded arrowheads). b, H446 cells stably expressing TEV protease (bottom row) and WT H446 cells (top row) were subsequently transfected with WT-SVA or SVA containing TEV cleavage sites in place of native recognition sequences between structural proteins. Images taken at 24 h post transfection. Scale bar, 500 μm. c, Illustration of TEV-mediated cleavage between l-protein and VP4. TEV site is cleaved by S. typhimurium-provided TEV protease, while all other cleavage sites are naturally cleaved by the SVA-protease 3Cpro. d, Fraction of GFP-positive cells after transfection of WT-SVA-GFP (grey), TEVs-SVA-GFP into H446 cells not expressing protease (dashed blue) and TEVs-SVA-GFP into H446 cells expressing TEVp (solid blue) at 12, 24 and 48 h. Data are mean ± s.d. across four biological replicates for each timepoint, with a total of 17,158, 8,107 and 9,309 cells, respectively, in the +TEVp condition, and 6,743, 9,107 and 4,040 cells, respectively, in the −TEVp condition. e, H446 cells inoculated with S. typhimurium carrying TEVs-SVA-GFP with (right) and without (left) TEVp at MOI 50, followed by time-lapse imaging. Temporal information is shown as a projection into colour space (gradient from blue to red) over 72 h. f, Same as in d but with mutationally resistant TEV site (rTEVs, ENLYCQ^G). Data are mean ± s.d. across four biological replicates for each timepoint, with a total of 12,672, 6,434 and 11,617 cells, respectively, in the +TEVp condition, and 3,285, 8,339 and 5,107 cells, respectively, in the −TEVp condition. g, Normalized viral titre from bacterially delivered SVA variants with or without protease. Hind-flank H446 tumours IT injected with lysing S. typhimurium carrying WT-SVA-NanoLuc without TEV protease, TEVs-SVA-NanoLuc with and without TEV protease, and rTEVs-SVA-NanoLuc with TEV protease were collected at 18 h.p.i. Then, naive H446 cells were inoculated with clarified freeze thawed tumour homogenate. Each point represents one tumour. Data were normalized to the mean of the group receiving bacterially delivered WT-SVA. Two-way ANOVA evaluation determined no significant difference (P = 0.46) in means of the groups receiving WT-SVA-NanoLuc, TEVs-SVA-NanoLuc with TEV protease and rTEVs-SVA-NanoLuc with TEV protease. h, Luminescent signals from nude mice with bilateral hind-flank tumours injected with S. typhimurium delivering rTEVs-SVA-NanoLuc with (black) and without (red) TEV protease. Each point on the graph illustrates the mean ± s.d. of the luminescent signal of n = 10 tumours at each timepoint, with significant difference between groups (Wilcoxon signed-rank test, two-tailed). LOD, limit of detection.
Investigating which natural cleavage sites might be amenable to TEVs substitution, we replaced all four cleavage sites flanking the structural proteins, abrogating the natural cleavage sequence in the process. We engineered each of these four potential variants and transfected them either into WT H446 cells, or H446s constitutively expressing TEVp, and looked for conditional spreading (Fig. 4b,c). When the TEVs was placed between the non-structural leader protein and the first structural protein, VP4, the spreading of this variant became entirely dependent on TEVp and was capable of infecting surrounding cells at a rate equivalent to WT virus (Fig. 4d). Replacing the other natural cleavage sites with TEVs, however, resulted in TEV-independent spread or abrogation of spread entirely.
To couple the spread of this variant to co-infecting bacteria, we engineered lysing S. typhimurium to also express TEVp under the control of a second PsseA promoter. In addition, we incorporated a series of mutations in TEVp previously shown to improve the solubility of the protease56,57 (Supplementary Fig. 3a). When the bacteria delivered a TEVp-dependent virus without bacterially produced TEVp, the virus launches but then fails to spread, as expected (Fig. 4e, left). However, when the S. typhimurium simultaneously delivered both the TEVp-dependent virus as well as TEVp, localized foci of spreading infection appeared (Fig. 4e, right).
We next proceeded to evaluate this platform in vivo, first aiming to characterize the stability of the engineered genome, owing to the high error rate of RNA-dependent RNA polymerases and potential to mutate away from TEVp dependence58. Tumours were injected IT with S. typhimurium delivering wild-type SVA-NanoLuc and compared to TEV-dependent virus (TEVs-SVA-NanoLuc) with co-delivered protease. Over the course of 1 week, the luminescence of the group receiving bacterially delivered WT virus continued to increase rapidly. In contrast, the signal from tumours injected with bacterially delivered TEV-dependent SVA along with protease remained lower than WT through day 8, but then began to increase (Supplementary Fig. 3b, top). Sequencing of viral RNA extracted from tumours that received the TEVs-SVA-NanoLuc revealed that 3 out of 5 had a single-nucleotide polymorphism (SNP) in the TEVs in >95% of total reads, yielding two different ways of producing an identical phenylalanine-to-leucine (F→L) substitution (Supplementary Fig. 3b, bottom). When this mutation was cloned into the viral genome and transfected directly into H446 with or without TEVp, we observed that this mutation was indeed sufficient to achieve TEVp-independent spreading (Supplementary Fig. 3c). Examination of the natural cleavage site at this position revealed that a leucine recapitulates the amino acid normally present immediately upstream of the native scissile Q^G site.
To prevent this ‘escape’ mutation from occurring, an optimal TEVs sequence would be one where the codon for phenylalanine requires more than one SNP to revert into a leucine. While no codon like this for phenylalanine exists, previous interrogation of TEVs revealed that a cysteine substitution at the phenylalanine site maintained TEVp-mediated cleavage while also being two SNPs away from mutating to a leucine59. Indeed, an SVA variant with the modified TEVs sequence of ENLYCQ^G only spread conditionally in the presence of TEVp, although at slightly reduced efficiency compared with the WT TEVs (Fig. 4f and Supplementary Fig. 3c). Thus, we were able to construct a mutationally resistant variant of TEVp-dependent SVA (denoted rTEVs-SVA).
Mice carrying double hind-flank H446 tumours were injected IT with CAPPSID rTEVs-SVA with and without TEVp expression, or with WT-SVA alone. Then, 24 h following injection of bacteria, tumours were collected, homogenized and assayed for luminescence ex vivo as a readout for replication originating from bacterial launch, as well as for viral titre measurements as an indication of successful packaging of the virus. The initial luminescence as measured ex vivo was statistically indistinguishable between groups, showing equivalent initial delivery of WT virus from bacteria compared to TEVp-dependent virus (Supplementary Fig. 3d). Similarly, the number of viral particles produced by cells infected with TEVp-dependent virus was also statistically the same as WT virus delivery at launch (Fig. 4g). In contrast, no infectious particles were recovered when the TEVp-dependent virus was delivered in the absence of TEVp, as expected (Fig. 4g). Furthermore, when naive cells in vitro were infected with tumour-collected WT virus, spreading was observed, while tumours containing TEVp-dependent virus showed initial replication and no further spread (Supplementary Fig. 3e). Together, these data suggest that the initial launch and production of infectious viral particles are equally efficient between both WT and TEV-dependent viruses, and that engineered virus launched from bacteria was indeed TEVp dependent. Finally, when a cohort of mice injected with lysing S. typhimurium delivering TEVp-dependent virus with and without protease was measured longitudinally, we observed that luminescence from this mutation-resistant variant continued to remain present for 2 weeks following a single injection, while virus delivered without protease showed a complete loss of signal over the same period. Over this time course, no increasing luminescent signals were observed, suggesting that reversion to TEVp independence did not occur (Fig. 4h and Supplementary Fig. 3f).
Discussion
This work explored synthetic strategies to design distinct levels of cooperation between two clinically relevant microbes, S. typhimurium and SVA. By utilizing bacteria as a dynamic and engineerable ‘synthetic capsid’, we delivered replicons and full-length viral RNAs into the host cytoplasm. Using CAPPSID, we entirely clear subcutaneous SCLC tumours and overcome systemic neutralizing antibodies when injected IV in fully immunocompetent mice. In addition, the bacterial launch of replicons into a range of mouse and human cell lines demonstrates the ability to deliver non-spreading self-amplifying viral RNAs beyond their natural tropism. Noting that the bacteria can simultaneously deliver proteins and nucleic acids, we devised interactions by engineering a virus whose protein maturation depends on a bacterially provided protease when substituting a synthetic cleavage site. Together, we developed a multilayered engineering approach for coordinating a two-microbe system for oncolytic applications. The platform was further able to show how coordination can dramatically improve the persistence of the virus compared with a replicon when supplemented with an engineered requirement for an accessory enzyme for viral spreading. While SVA does not cause mouse toxicity, the development of new strategies to achieve targeted replication and overcoming systemic neutralization have remained central challenges in the advancement of novel virotherapies. Through these efforts, our CAPPSID system demonstrates the ability of viruses to extend the tumouricidal effect of bacteria.
Delivery of nucleic acids by bacteria, or bactofection, has been previously applied, for example, by Agrobacterium for CRISPR/CAS9 gene editing in wheat60, and L. monocytogenes, Escherichia coli and S. typhimurium for small interfering RNAs, short open reading frame (ORF)-containing RNAs and plasmids27,34,61,62,63,64,65,66,67,68,69. Leveraging the flexible tools available for genetic engineering in S. typhimurium, we were able to deliver large viral RNAs across a broad range of cell types compared with those previously reported via engineered S. typhimurium64. Building on previous work for intracellular delivery, the active replication of viral RNA causes a cytopathic effect in its initial host cell while also enabling spread to surrounding cells uninfected by bacteria, thereby enhancing therapeutic range. While SVA does not natively infect nude mice, we additionally show efficacy in a systemically delivered, fully immunocompetent model susceptible to SVA46. This consortium of cooperative microbes likely elicits these results through a range of mechanisms, including direct cytopathic effect, innate immune activation via microbial pathogen- and damage-associated molecular patterns, as well as neoantigen cross-presentation in the context of an intact adaptive immune system.
Our efforts to insert an orthogonal cleavage site into the virus highlight the importance of addressing the mutability of RNA viruses. RNA-dependent RNA polymerases incorporate an incorrect base at a rate of roughly 1 in 10,000 (refs. 70,71,72,73). Here we attempted to mitigate mutational escape by first identifying the most common escape mechanism in vivo and limiting the likelihood of such reversion by requiring two independent mutations to simultaneously occur, thereby geometrically reducing the escape probability. However, alternative types of mutation, such as wholesale deletions of our orthogonal sequence may also occur, although these were not observed here. Insertion of additional TEV sites, or even additional protease/cleavage site pairs, could further increase the robustness of this system and enable construction of logic-gated viral replication and spread.
By developing a bacterially delivered platform for viral RNA, we show successful launch of a viral infection capable of eradicating tumours, the ability to cloak and deliver viral genomes into tumours in mice with humoral immunity, and that viral spreading controlled in trans by a bacterially donated protease can enhance persistence compared with a replicon alone. Together, this engineered microbial consortium produces a potent therapy that overcomes the limitations of singular approaches.
Methods
Bacterial cell growth
All bacteria were grown in LB Lennox broth. For LH1301, cultures were supplemented with ampicillin (100 μg ml−1) and spectinomycin (200 μg ml−1) for viral-encoding plasmids and lysis circuit plasmids, respectively. Plasmids were cloned into NEB10β and maintained at 100 μg ml−1 ampicillin or 100 μg ml−1 spectinomycin. To prepare strains of LH1301 containing these plasmids, the cells were electroporated, recovered and plated on LB agar. All liquid cultures were grown overnight in a 30 °C incubator with shaking. For all in vitro and in vivo experiments, cultures were grown overnight at 30 °C to stationary phase, and then diluted 1:100 the next morning and grown for ~3 h at 37 °C until reaching an optical density at 600 nm (OD600) of 0.5.
Plasmid construction
Plasmids were constructed using HiFi Assembly (NEB NEBuilder HiFi DNA Assembly Master Mix) and transformed into NEB10β. SVA-containing vectors were cloned into the same p15A/amp backbone flanked by a 5′ T7 promoter, 3′ poly(A) tail, HDV ribozyme and T7 terminator. All lysis circuits were constructed by synthesizing gBlocks from IDT and cloned into plasmids with SC101 origins of replication. To validate constructs, whole plasmid sequencing was performed by Plasmidsaurus.
Bacterial genome engineering
The sifA gene was deleted using the λ-Red recombination system. Linear DNA containing CmR flanked by FRT sites were PCR amplified using pkD3 plasmid and electroporated into LH1301 carrying pKD46 plasmid. Chromosomal deletions were verified by PCR and Sanger sequencing.
Mammalian cell maintenance
HeLa (CRL-1958), 4T1 (CRL-2539), B16 (CRL-6475), HCT116 (CRL-247), N1E-115 (CRL-2263) and H446 (HTB-171) were acquired from ATCC, and MC38 from Kerafast (ENH204-FP). All cell lines were cultured in RPMI medium supplemented with 10% fetal bovine serum (FBS, Gibco) and 1× MEM non-essential amino acids (NEAA) in a 37 °C tissue culture incubator with 5% CO2. To stably and constitutively express TEVp in H446, cells were selected and regularly grown with 0.25 μg ml−1 puromycin after transfection with PB-PEF-Puro-F2A-TEVp and piggyBac supertransposase.
S. typhimurium in vitro invasion assay
Cultures of S. typhimurium were grown to stationary phase overnight and diluted 1:100 the following morning into LB Miller medium supplemented with antibiotics. Cultures were grown at 37 °C to OD600 of 0.4–0.6, when they were spun down and resuspended in 1 ml RPMI + 1% FBS. Bacteria were added at a multiplicity of infection (MOI) of 50 into 24-well plates of mammalian cells split 24 h earlier and spun down in the plate at 200 × g for 5 min. Plates were then incubated at 37 °C and 5% CO2 for 30 min. Then, the wells were thoroughly washed with RPMI to remove bacteria, incubated for another 30 min at 100 μg ml−1 gentamicin in RPMI + 10% FBS to kill residual extracellular bacteria and then replaced with 25 μg ml−1 gentamicin in RPMI + 10% FBS for the duration of the experiment. To stain nuclei, NucBlue (Invitrogen, R37605) was added at 10–20 μl ml−1 of media.
Viral transfection and production
To produce virus, H446 cells were transfected using Lipofectamine MessengerMax at 1 μl reagent per 500 ng RNA per well in a 24-well plate, and scaled up as needed. At 48 h post transfection, cells were collected, freeze thawed three times to disrupt membranes and release viral particles, and clarified by centrifugation at 16,000 × g for 10 min. Virus was titred on H446 by using serial dilutions and identifying the concentration of virus infecting roughly 50% of cells in a well, plated at 100,000 cm−2 24 h earlier, by imaging luminescence or GFP reporter signal, defined as MOI 1.
Animal tumour models
All animal experiments were approved by the Institutional Animal Care and Use Committee (Columbia University, protocol AC-AABQ5551). Female nude mice aged 6–8 weeks from Charles River were grafted with bilateral subcutaneous hind-flank tumours of 5 × 106 H446 cells in 50% reduced growth factor Matrigel (Corning). Tumours were grown until reaching ~150 mm3 over ~2.5 weeks. Alternatively, female A/J mice aged 6–8 weeks from Jackson Labs were grafted with subcutaneous hind-flank tumours of 5 × 105 N1E-115 cells in 50% reduced growth factor Matrigel (Corning), and grown for 10 days until reaching ~150 mm3. Then, 2.5 × 106 bacteria in 25 μl RPMI (without phenol red) were injected IT, or IV in 100 μl. For IV delivery of bacteria in A/J mice, subjects also received 250 μg each of IL1R-, TNFα- and IFNAR1-antagonizing antibodies 2 h before bacterial injection (BioXCell, BE0256, BE0058, BE0241). For in vivo imaging system (IVIS) imaging, mice were subtumourally injected with 25 μl Nano-Glo Fluorofurimazine In Vivo Substrate (Promega N4100) at 8.8 nM. Tumour volume was quantified using digital calipers to measure the length and width of each tumour (V = 0.5 × L × W2). The protocol requires animals be euthanized either when tumours reach 2 cm in diameter or upon veterinary staff recommendation. Survival curves were generated on the basis of tumour burden, as all mice survived engraftment and were not directed for euthanasia before reaching tumour burden limit. Mice were randomized into various groups in a blinded manner.
Biodistribution
After timepoints following bacterial injections indicated in figure legends, mice were euthanized to collect the tumours, spleen and liver. Tissues were weighed and homogenized using a gentleMACS tissue dissociator (Miltenyi Biotec, C-tubes). These homogenates were then tenfold serially diluted and plated on LB agar with chloramphenicol and grown overnight at 37 °C. Colonies were counted and computed as colony-forming units per gram of tissue.
Ex vivo tumour luminescence
Tumours were extracted, weighed and homogenized using the gentleMACS tissue dissociator (Miltenyi Biotec, C-tubes) in 5 ml RPMI + 1% FBS. To measure luminescence, samples were serially diluted tenfold over four orders of magnitude in replicate, and assayed using a plate reader (Tecan Infinite 200 Pro using the i-control software v.3.9.1.0) after adding Nano-Glo In vivo Substrate (Promega).
Ex vivo tumour-associated virus sequencing
Tumours were extracted, weighed and homogenized using a handheld tissue grinder in 2 ml RPMI supplemented with Ambion RNALater (ThermoFisher, AM7020). Total RNA was extracted using the RNEasy kit from Qiagen (74104). Then, cDNA was prepared using Superscript III First Strand Synthesis Supermix kit (ThermoFisher, 18080400) with a poly-dT primer. This output was used as a PCR template with primers against the SVA genome flanking the TEVs site. The linear ~500-bp fragment was read using Nanopore sequencing provided by Plasmidsaurus, with raw reads aligned using Geneious Prime.
Ex vivo tumour viral titring and antibody neutralization
Tumours were extracted, weighed and homogenized using the gentleMACS tissue dissociator (Miltenyi Biotec, C-tubes) in 1 ml RPMI + 1% FBS with antibiotic-antimycotic (Gibco). Homogenate was freeze thawed three times and clarified by centrifugation at 16,000 × g for 10 min. Three tenfold serial dilutions of this homogenized preparation were inoculated on naive H446 cells for 1 h and then replaced with fresh RPMI + 10% FBS. After 12 h, cells were dissociated by pipetting and imaged to count the number of luminescent cells after adding Nano-Glo In vivo Substrat e (Promega, N4100).
For neutralization assays, mice were cheek bled 2 weeks after receiving either 2.5 × 106 viral particles IV or sham. Serum was then isolated by centrifugation and diluted 1:100 with MOI 1 SVA-GFP in 200 μl 1% FBS in RPMI with Pen/Strep. This inoculum was overlaid on H446s plated at 75,000 cm−2 24 h earlier. After 30 min, the inoculum was removed and then time-lapse imaged to observe the number of reporter-positive cells.
Histology
For histology experiments, tumours were excised and fixed in 4% paraformaldehyde, then switched to 70% ethanol, bisected, paraffin-embedded, sectioned at 4 μm and sent for histology services at Histowiz. Sections were stained using anti-lipopolysaccharide (WN1 222-5, Absolute Antibody, Ab00141-23.0) or terminal deoxynucleotidyl transferase dUTP nick end labelling.
Statistics and reproducibility
Statistical tests were performed and corresponding plots created using either GraphPad Prism 10 or Matlab 2023A. The details of the statistical tests are indicated in the respective figure legends. Mice were randomized into different groups before the experiments. Unless otherwise noted, each experiment was reproduced separately at least two times.
ImmunoFISH sample preparation and staining
The ImmunoFISH sample preparation and staining protocol was performed as in ref. 74. Cells were fixed in 4% formaldehyde for 5 min at room temperature (r.t.), washed twice in PBS and placed in 70% ethanol overnight at −20 °C. For staining dsRNA, the next morning, cells were washed twice with PBS; permeabilized in 0.1% Triton-X for 5 min; washed twice in PBS; blocked in 10% normal goat serum (Thermo, 50062Z); washed twice with PBS; incubated with J2 primary antibody (Scicons, 10010200) at 0.5 μg ml−1 in 10% normal goat serum for 2 h; washed twice with PBS; incubated with goat anti-mouse Alexa Fluor 647 secondary antibody (Invitrogen, A-21235) at r.t. for 30 min; washed twice in PBS; incubated with 10% normal goat serum for an additional 10 min at r.t.; and then imaged in PBS.
For smFISH staining, probes were designed to bind the 3′ end of the poliovirus replicon. After fixing and overnight incubation in 70% ethanol, cells were washed twice in PBS; equilibrated in FISH wash buffer containing 2× SSC (Invitrogen, 15557044) and 20% formamide (Ambion, AM9342) for 5 min at r.t.; and hybridized with Stellaris FISH probes labelled with Quasar 670 at 125 nM (Biosearch Technologies; Supplementary Table 1) overnight at 30 °C in hybridization buffer (containing 20% formamide (Ambion, AM9342), 2× SSC, 0.1 g ml−1 dextran sulfate (Fisher Sci, BP1585-Dextran Sulfate), 1 mg ml−1 E. coli tRNA (Roche 10109541001), 2 mM Vanadyl ribonucleoside complex (NEB, S1402S) and 0.1% Tween 20 (VWR, 97062-332) in nuclease-free water). The next morning, the hybridization buffer was removed and cells were washed twice in FISH wash buffer; incubated in FISH wash buffer without probe for 30 min at 30 °C; washed three times with 2× SSC; counterstained with DAPI; and finally imaged in 2× SSC.
Microscopy
Cells were imaged on a Nikon Ti2 with PFS4, a Nikon Motorized Encoded Stage, Lumencor SpectraX Light Engine, custom Semrock filters and a Prime 95B sCMOS camera. Automated acquisition for snapshots and time lapse was programmed in NIS Elements. The scope was equipped with an OKO stage-top incubator with temperature, humidity and CO2 control, enabling long-term imaging. For imaging smFISH, a ×60 objective was used. Otherwise, imaging was performed using a ×10 or ×20 ELWD objective.
Imaging analysis
Images were recorded and processed using NIS-Elements software, Fiji, MATLAB and Python. Time-lapse and smFISH image analysis were performed as in ref. 74.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The main data supporting the results in this study are available within the paper and its Supplementary Information. Sequencing data are available at https://www.ncbi.nlm.nih.gov/bioproject/1282161 (ref. 75).
References
-
Chen, Y. E. et al. Engineered skin bacteria induce antitumor T cell responses against melanoma. Science 380, 203–210 (2023).
-
Mazzolini, R. et al. Engineered live bacteria suppress Pseudomonas aeruginosa infection in mouse lung and dissolve endotracheal-tube biofilms. Nat. Biotechnol. 41, 1089–1098 (2023).
-
Chen, Y. et al. Development of a Listeria monocytogenes-based vaccine against hepatocellular carcinoma. Oncogene 31, 2140–2152 (2012).
-
Raman, V. et al. Intracellular delivery of protein drugs with an autonomously lysing bacterial system reduces tumor growth and metastases. Nat. Commun. 12, 6116 (2021).
-
Camacho, E., Mesa-Pereira, B., Medina, C., Flores, A. & Santero, E. Engineering Salmonella as intracellular factory for effective killing of tumour cells. Sci. Rep. 6, 30591 (2016).
-
Mendell, J. R. et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 377, 1713–1722 (2017).
-
Russell, S. et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet 390, 849–860 (2017).
-
Wickersham, I. R., Finke, S., Conzelmann, K. K. & Callaway, E. M. Retrograde neuronal tracing with a deletion-mutant rabies virus. Nat. Methods 4, 47–49 (2007).
-
Desjardins, A. et al. Recurrent glioblastoma treated with recombinant poliovirus. N. Engl. J. Med. 379, 150–161 (2018).
-
Goetz, C., Dobrikova, E., Shveygert, M., Dobrikov, M. & Gromeier, M. Oncolytic poliovirus against malignant glioma. Future Virol. 6, 1045–1058 (2011).
-
Erickson, A. K. et al. Bacteria facilitate enteric virus co-infection of mammalian cells and promote genetic recombination. Cell Host Microbe 23, 77–88.e75 (2017).
-
Robinson, C. M., Jesudhasan, P. R. & Pfeiffer, J. K. Bacterial lipopolysaccharide binding enhances virion stability and promotes environmental fitness of an enteric virus. Cell Host Microbe 15, 36–46 (2014).
-
Kuss, S. K. et al. Intestinal microbiota promote enteric virus replication and systemic pathogenesis. Science 334, 249–252 (2011).
-
Perez, M. et al. A synthetic consortium of 100 gut commensals modulates the composition and function in a colon model of the microbiome of elderly subjects. Gut Microbes 13, 1–19 (2021).
-
El Hage, R., Hernandez-Sanabria, E., Calatayud Arroyo, M., Props, R. & Van de Wiele, T. Propionate-producing consortium restores antibiotic-induced dysbiosis in a dynamic in vitro model of the human intestinal microbial ecosystem. Front. Microbiol. 10, 1206 (2019).
-
Li, L. et al. Hydrogel-encapsulated engineered microbial consortium as a photoautotrophic ‘living material’ for promoting skin wound healing. ACS Appl. Mater. Interfaces 15, 6536–6547 (2023).
-
Aalipour, A. et al. Viral delivery of CAR targets to solid tumors enables effective cell therapy. Mol. Ther. Oncolytics 17, 232–240 (2020).
-
Vincent, R. L. et al. Probiotic-guided CAR-T cells for solid tumor targeting. Science 382, 211–218 (2023).
-
Tanoue, T. et al. A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature 565, 600–605 (2019).
-
Gamboa, L. et al. Sensitizing solid tumors to CAR-mediated cytotoxicity by lipid nanoparticle delivery of synthetic antigens. Nat. Cancer 6, 1073–1087 (2025).
-
Khanduja, S. et al. Intracellular delivery of oncolytic viruses with engineered Salmonella causes viral replication and cell death. iScience 27, 109813 (2024).
-
Roberts, D. M. et al. Hexon-chimaeric adenovirus serotype 5 vectors circumvent pre-existing anti-vector immunity. Nature 441, 239–243 (2006).
-
Gurbatri, C. R., Arpaia, N. & Danino, T. Engineering bacteria as interactive cancer therapies. Science 378, 858–864 (2022).
-
Toso, J. F. et al. Phase I study of the intravenous administration of attenuated Salmonella typhimurium to patients with metastatic melanoma. J. Clin. Oncol. 20, 142–152 (2002).
-
Haraga, A., Ohlson, M. B. & Miller, S. I. Salmonellae interplay with host cells. Nat. Rev. Microbiol. 6, 53–66 (2008).
-
Fink, S. L. & Cookson, B. T. Pyroptosis and host cell death responses during Salmonella infection. Cell. Microbiol. 9, 2562–2570 (2007).
-
Schoen, C. et al. Bacterial delivery of functional messenger RNA to mammalian cells. Cell. Microbiol. 7, 709–724 (2005).
-
Hegazy, W., Xu, X., Metelitsa, L. & Hensel, M. Evaluation of Salmonella enterica type III secretion system effector proteins as carriers for heterologous vaccine antigens. Infect. Immun. 80, 1193–1202 (2012).
-
Xu, X., Husseiny, M. I., Goldwich, A. & Hensel, M. Efficacy of intracellular activated promoters for generation of Salmonella-based vaccines. Infect. Immun. 78, 4828–4838 (2010).
-
Xiong, G. et al. Novel cancer vaccine based on genes of Salmonella pathogenicity island 2. Int. J. Cancer 126, 2622–2634 (2010).
-
Xu, X. & Hensel, M. Systematic analysis of the SsrAB virulon of Salmonella enterica. Infect. Immun. 78, 49–58 (2009).
-
Xu, X. et al. Development of an effective cancer vaccine using attenuated Salmonella and type III secretion system to deliver recombinant tumor-associated antigens. Cancer Res. 74, 6260–6270 (2014).
-
Juárez-Rodríguez, M. D. et al. Live attenuated Salmonella vaccines displaying regulated delayed lysis and delayed antigen synthesis to confer protection against Mycobacterium tuberculosis. Infect. Immun. 80, 815–831 (2012).
-
Manuel, E. R. et al. Enhancement of cancer vaccine therapy by systemic delivery of a tumor-targeting Salmonella-based STAT3 shRNA suppresses the growth of established melanoma tumors. Cancer Res. 71, 4183–4191 (2011).
-
Husseiny, M. I., Wartha, F. & Hensel, M. Recombinant vaccines based on translocated effector proteins of Salmonella pathogenicity island 2. Vaccine 25, 185–193 (2007).
-
Chabloz, A. et al. Salmonella-based platform for efficient delivery of functional binding proteins to the cytosol. Commun. Biol. 3, 342 (2020).
-
Widmaier, D. M. et al. Engineering the Salmonella type III secretion system to export spider silk monomers. Mol. Syst. Biol. 5, 309 (2009).
-
Doamekpor, S. K., Sharma, S., Kiledjian, M. & Tong, L. Recent insights into noncanonical 5’ capping and decapping of RNA. J. Biol. Chem. 298, 102171 (2022).
-
Francisco-Velilla, R., Embarc-Buh, A., Abellan, S. & Martinez-Salas, E. Picornavirus translation strategies. FEBS Open Bio 12, 1125–1141 (2022).
-
Ansardi, D. C., Porter, D. C., Anderson, M. J. & Morrow, C. D. Poliovirus assembly and encapsidation of genomic RNA. Adv. Virus Res. 46, 1–68 (1996).
-
Din, M. O. et al. Synchronized cycles of bacterial lysis for in vivo delivery. Nature 536, 81–85 (2016).
-
Loessner, H. et al. Remote control of tumour-targeted Salmonella enterica serovar Typhimurium by the use of l-arabinose as inducer of bacterial gene expression in vivo. Cell. Microbiol. 9, 1529–1537 (2007).
-
Orta, A. K. et al. The mechanism of the phage-encoded protein antibiotic from ΦX174. Science 381, eadg9091 (2023).
-
Wallace, A. J. et al. E. coli hemolysin E (HlyE, ClyA, SheA): X-ray crystal structure of the toxin and observation of membrane pores by electron microscopy. Cell 100, 265–276 (2000).
-
Beuzón, C. R. et al. Salmonella maintains the integrity of its intracellular vacuole through the action of SifA. EMBO J. 19, 3235–3249 (2000).
-
Reddy, P. S. et al. Seneca Valley virus, a systemically deliverable oncolytic picornavirus, and the treatment of neuroendocrine cancers. J. Natl Cancer Inst. 99, 1623–1633 (2007).
-
Burke, M. Oncolytic Seneca Valley Virus: past perspectives and future directions. Oncolytic Virother. 5, 81–89 (2016).
-
Rudin, C. M. et al. Phase I clinical study of Seneca Valley Virus (SVV-001), a replication-competent picornavirus, in advanced solid tumors with neuroendocrine features. Clin. Cancer Res. 17, 888–895 (2011).
-
Morton, C. L. et al. Initial testing of the replication competent Seneca Valley virus (NTX-010) by the pediatric preclinical testing program. Pediatr. Blood Cancer 55, 295–303 (2010).
-
Poirier, J. T. et al. Characterization of a full-length infectious cDNA clone and a GFP reporter derivative of the oncolytic picornavirus SVV-001. J. Gen. Virol. 93, 2606–2613 (2012).
-
Geisler, A., Hazini, A., Heimann, L., Kurreck, J. & Fechner, H. Coxsackievirus B3—its potential as an oncolytic virus. Viruses 13, 718 (2021).
-
Erasmus, J. H. et al. An alphavirus-derived replicon RNA vaccine induces SARS-CoV-2 neutralizing antibody and T cell responses in mice and nonhuman primates. Sci. Transl. Med. 12, eabc9396 (2020).
-
Miest, T. S. & Cattaneo, R. New viruses for cancer therapy: meeting clinical needs. Nat. Rev. Microbiol. 12, 23–34 (2014).
-
Palmenberg, A. C. Proteolytic processing of picornaviral polyprotein. Annu. Rev. Microbiol. 44, 603–623 (1990).
-
Kapust, R. B., Tözsér, J., Copeland, T. D. & Waugh, D. S. The P1′ specificity of tobacco etch virus protease. Biochem. Biophys. Res. Commun. 294, 949–955 (2002).
-
Cabrita, L. D. et al. Enhancing the stability and solubility of TEV protease using in silico design. Protein Sci. 16, 2360–2367 (2007).
-
van den Berg, S., Löfdahl, P.-A., Härd, T. & Berglund, H. Improved solubility of TEV protease by directed evolution. J. Biotechnol. 121, 291–298 (2006).
-
Aguilar Rangel, M. et al. High-resolution mapping reveals the mechanism and contribution of genome insertions and deletions to RNA virus evolution. Proc. Natl Acad. Sci. USA 120, e2304667120 (2023).
-
Dougherty, W. G., Cary, S. M. & Parks, T. D. Molecular genetic analysis of a plant virus polyprotein cleavage site: a model. Virology 171, 356–364 (1989).
-
Zhang, Z. et al. Development of an Agrobacterium-delivered CRISPR/Cas9 system for wheat genome editing. Plant Biotechnol. J. 17, 1623–1635 (2019).
-
Schaffner, W. Direct transfer of cloned genes from bacteria to mammalian cells. Proc. Natl Acad. Sci. USA 77, 2163–2167 (1980).
-
Narayanan, K. & Warburton, P. E. DNA modification and functional delivery into human cells using Escherichia coli DH10B. Nucleic Acids Res. 31, e51 (2003).
-
Grillot-Courvalin, C., Goussard, S., Huetz, F., Ojcius, D. M. & Courvalin, P. Functional gene transfer from intracellular bacteria to mammalian cells. Nat. Biotechnol. 16, 862–866 (1998).
-
Grillot-Courvalin, C., Goussard, S. & Courvalin, P. Wild-type intracellular bacteria deliver DNA into mammalian cells. Cell. Microbiol. 4, 177–186 (2002).
-
Kong, W. et al. Regulated programmed lysis of recombinant Salmonella in host tissues to release protective antigens and confer biological containment. Proc. Natl Acad. Sci. USA 105, 9361–9366 (2008).
-
Kong, W., Brovold, M., Koeneman, B. A., Clark-Curtiss, J. & Curtiss, R. Turning self-destructing Salmonella into a universal DNA vaccine delivery platform. Proc. Natl Acad. Sci. USA 109, 19414–19419 (2012).
-
Xiang, S., Fruehauf, J. & Li, C. J. Short hairpin RNA-expressing bacteria elicit RNA interference in mammals. Nat. Biotechnol. 24, 697–702 (2006).
-
Guo, H. et al. Targeting tumor gene by shRNA-expressing Salmonella-mediated RNAi. Gene Ther. 18, 95–105 (2011).
-
Weiss, S. & Chakraborty, T. Transfer of eukaryotic expression plasmids to mammalian host cells by bacterial carriers. Curr. Opin. Biotechnol. 12, 467–472 (2001).
-
Acevedo, A., Brodsky, L. & Andino, R. Mutational and fitness landscapes of an RNA virus revealed through population sequencing. Nature 505, 686 (2014).
-
Drake, J. W. Rates of spontaneous mutation among RNA viruses. Proc. Natl Acad. Sci. USA 90, 4171–4175 (1993).
-
Sanjuán, R., Nebot, M. R., Chirico, N., Mansky, L. M. & Belshaw, R. Viral mutation rates. J. Virol. 84, 9733–9748 (2010).
-
Domingo, E., García-Crespo, C., Lobo-Vega, R. & Perales, C. Mutation rates, mutation frequencies, and proofreading-repair activities in RNA virus genetics. Viruses 13, 1882 (2021).
-
Singer, Z. S., Ambrose, P. M., Danino, T. & Rice, C. M. Quantitative measurements of early alphaviral replication dynamics in single cells reveals the basis for superinfection exclusion. Cell Syst. 12, 210–219.e3 (2021).
-
Singer, Z. S. et al. Engineered bacteria launch and control an oncolytic virus. Datasets. NCBI Bioproject https://www.ncbi.nlm.nih.gov/bioproject/1282161 (2025).
Acknowledgements
We thank J. T. Poirier, NYU Langone, for input and the infectious cDNA clone of SVA. T.D. discloses support for the research described in this study from the Department of Defence (BC160541) and the National Institutes of Health (R01EB029750). Z.S.S. discloses support for the research described in this study from the National Institutes of Health (F32CA225145). C.M.R and Z.S.S. disclose support for the research described in this study from the National Institutes of Health (R01AI161444).
Ethics declarations
Competing interests
Z.S.S., J.P. and T.D. have filed provisional patent applications (US Patent application nos. 63/471,863, 63/541,079 and 63/610,152 on 8 June 2023, 28 September 2023 and 14 December 2023) with the US Patent and Trademark Office related to this work and have also filed PCT/US2024/032961 on 7 June 2024 with the World Intellectual Property Organization. The other authors declare no competing interests.
Peer review
Peer review information
Nature Biomedical Engineering thanks John Bell and Martin Fussenegger for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Reporting Summary
Supplementary Video 1
S. typhimurium containing the lysis circuit and SVA-GFP were inoculated on H446 cells to assess initial delivery and subsequent viral spread. Temporal dynamics of GFP-positive cells indicated viral spread beyond bacteria-infected cells, with widespread infection observed by 72 h post inoculation.
Supplementary Data 1
Raw data and sequences used in this study.
Supplementary Table 1
Probe sequences for smFISH against 3Dpol of poliovirus replicon.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Singer, Z.S., Pabón, J., Huang, H. et al. Engineered bacteria launch and control an oncolytic virus. Nat. Biomed. Eng (2025). https://doi.org/10.1038/s41551-025-01476-8
-
Received:
-
Accepted:
-
Published:
-
DOI: https://doi.org/10.1038/s41551-025-01476-8