
Introduction
Among all organs in the body, the heart has the highest metabolic rate reflecting its extensive workload and energy demand1. Mitochondria generate the majority of energy needed for the activities of the heart2 and are thus crucial organelles for the beating heart. The energy is provided by mitochondrial respiration, which involves a series of redox reactions in the four mitochondrial inner membrane (MIM) electron transport complexes (ETC), followed by subsequent generation of adenosine triphosphate (ATP) by complex V (CV)3. The ETC consists of complex I to IV (CI-CIV) and several associated protein complexes, such as cytochrome c (Cytc), a loosely bound MIM protein that transfers electrons between CIII and CIV. The ETC complexes together with CV constitute the oxidative phosphorylation (OXPHOS) system. The continuous beating heart requires optimal mitochondrial OXPHOS function to sustain its high rate of ATP demand.
Impaired mitochondrial oxygen consumption or respiration, reflecting mitochondrial dysfunction, is involved in a wide range of cardiometabolic disorders4,5,6 and in cardiac aging7,8. Deficiency in mitochondrial CI in the Ndufs4-/- mice causes severe mitochondrial disorders and accelerated pressure overloaded heart failure (HF)9,10. In agreement, a reduced enzymatic activity of CI11 and defects in CI-linked respiration were observed in human HF12,13,14. Similarly, CI-mediated and fatty acid-mediated mitochondrial respiration as well as CI enzymatic activity were compromised in diabetic subsarcolemmal mitochondria15 and atrial myofibers from type 2 diabetic (T2D) patients with diabetic cardiomyopathy16,17. This finding was corroborated by data from animal studies. For instance, a study using New Zealand obese (NZO) mice, characterized by severe obesity, insulin resistance, and a predisposition to T2D, revealed a notable cardiac dysfunction and an impaired mitochondrial respiratory function in cardiac tissue under high-fat diet (HFD) feeding18. Furthermore, the activity of mitochondrial ETC complexes, particularly of CI and CIV, declined with age in rat hearts7. Mice subjected to coronary ischemia reperfusion (IR) were reported to develop cardiac mitochondrial dysfunction6,19. Collectively, the close association between cardiac diseases and compromised mitochondrial respiration underscores the importance of assessing mitochondrial respiratory function of cardiac tissue, which would reveal specific functional deficits and provide therapeutic guidance.
Currently, mitochondrial respiration of cardiac tissue is typically measured using mitochondrial preparations from freshly sampled tissue20. The requirement for fresh tissue presents a severe limitation, since the analysis must be performed shortly after sampling21, which requires a stringent alignment in time and proximity of physician and the specialized analysis lab. This introduces both study limitations and experimental variation due to compromised standardization. In addition, frozen cardiac tissue samples stored in patient biobanks cannot be used, limiting translational applications22. Therefore, a robust method tailored for measuring mitochondrial respiration in frozen cardiac tissue to overcome these limitations is urgently needed.
Frozen samples have lost the coupling between the ETC and the ATP generation capacity due to the disruption of the MIM integrity during the freeze-thaw cycle. Therefore, many efforts have been dedicated to optimize cryopreservation of biopsies to maintain mitochondrial membrane integrity23,24,25,26. However, cryopreservation is a delicate process, making it impractical for sample storage in clinical practice. Evidence from both the spectrophotometric assays of individual ETC complex activities27 as well as the functional analysis of mitochondrial supercomplexs (SCs, e.g., the I-II-III2-IV2 SC)28 indicated that the electron transport activity is maintained in frozen tissue samples despite the uncoupled condition. In respirometry, the uncoupled respiratory state is assessed by the addition of a chemical uncoupler to gain insight in the maximal ETC capacity. A reduction in the maximal respiratory capacity may limit the ability to cope with stressors, resulting in mitochondrial dysfunction29. The situation in frozen samples represents the condition in which uncoupled maximal respiration is assessed. By directly supplying ETC substrates as well as loosely attached ETC components, such as Cytc, it would be possible to measure the respiratory capacity that is comparable to uncoupled maximal respiration in conventional respirometry using fresh tissue. Indeed, two methods were recently explored to measure mitochondrial CI respiratory capacity in frozen patient muscle biopsies using the Oroboros O2k30 and in various tissue samples using the Seahorse XF Analyzer22. However, a robust method using high-resolution respirometry to assess cardiac mitochondrial respiratory capacities in frozen tissue, capable of detecting respiratory activities of multiple mitochondrial complexes within a single round of measurement, is not yet available.
Compared to the Seahorse XF Analyzer, the multiple additions possible in the Oroboros O2k allow for detailed measurement of mitochondrial respiration mediated by various mitochondrial complexes within a single round. Therefore, we aimed to develop a method, using the Oroboros O2k, to measure mitochondrial respiratory capacities in frozen cardiac tissue by Tailored Substrate-Inhibitor Titration of mitochondrial complexes (named TSIT). Frozen cardiac tissue samples from various physiological and pathophysiological mouse models and human donors with potential mitochondrial dysfunction were used to further test the sensitivity and robustness of the application of TSIT. New insights into mitochondrial function of disease models using TSIT were investigated, and SC analysis to explain feasibility of TSIT in frozen samples was performed.
Results
High-resolution respirometry of fresh cardiac tissue reveals significant differences in mitochondrial respiration between atria and ventricles in mice
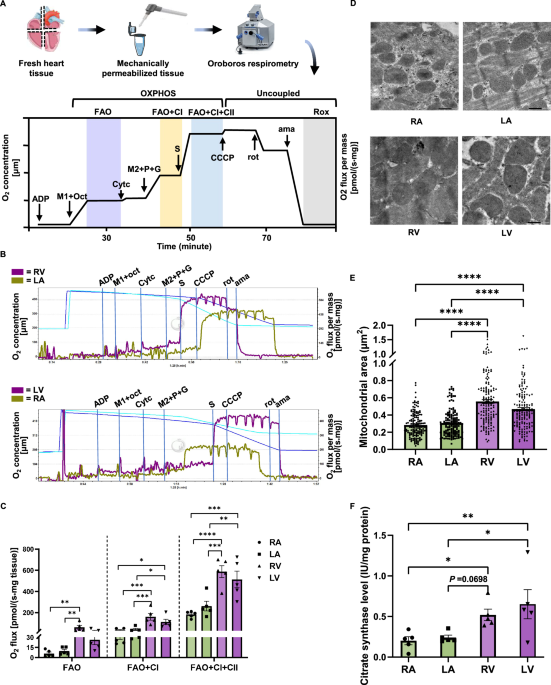
Mitochondrial respiration of fresh mouse cardiac tissues from both atria and ventricles was measured using high-resolution respirometry according to the SUIT-002 protocol (Oroboros O2k) (Fig. 1A). We found that the FAO + CI-linked, as well as FAO + CI + CII-linked mitochondrial OXPHOS respiration were significantly higher for ventricles (RV and LV) than for atria (RA and LA) (Fig. 1B, C). No significant difference between the atria (RA vs LA) or between the ventricles (RV vs LV) was found. Negligible changes in respiration upon addition of Cytc indicated the intactness of mitochondrial outer membrane. No difference was observed between mitochondrial respiration in the OXPHOS state and the uncoupled maximal ETS state (Figure. S2), which was in line with previous study that the ETS state was found to be similar to the value of ADP-stimulated respiration (OXPHOS states) in mouse tissue20. Furthermore, we observed a larger mitochondrial size (Fig. 1D, E) and mass (Fig. 1F) in ventricular tissue than those in atrial tissue, which might explain the differences in mitochondrial respiration between atria and ventricles. Taken together, these data indicate evident differences in mitochondrial respiration and morphology between atrial and ventricular tissues in mice.

A Schematic representation of the sample preparation and respirometry protocol. Fresh dissected cardiac tissues from the right atrium (RA), left atrium (LA), right ventricle (RV), left ventricle (LV) of 6-month-old mice were weighed, homogenized, and immediately measured using high-resolution respirometry according to the SUIT-002 protocol as illustrated. Fatty acid oxidation-boosted respiration (FAO, purple) was determined at 7.5 mM ADP, 0.3 mM malate (M1), and 0.5 mM octanoylcarnitine (oct). 10 µM cytochrome c (Cytc) was then added to test mitochondrial outer membrane integrity. FAO+complex I (CI)-linked respiration (FAO + CI, yellow) was determined at an additional 2 mM malate (M2), 5 mM pyruvate (P), and 10 mM glutamate (G). FAO + CI+complex II (CII)-linked respiration (FAO + CI + CII, blue) was determined at an additional 10 mM succinate (S). FAO + CI + CII-linked uncoupled maximal respiration was determined at an additional titration of 1 μl of 0.1 μM carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (CCCP). Residual oxygen consumption (Rox, gray) was determined at an additional 2 µM rotenone (rot) and 5 μM antimycin A (ama). Icons are provided by Servier Medical Art (https://smart.servier.com/), licensed under CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/). B Representative traces of fresh RA, LA, RV, and LV tissues from BL6JRcc(BL6J)-Nnt+/Wuhap mice. Purple traces indicate the ventricular O2 flux, and olive-green traces indicate the atrial O2 flux. C Quantification of FAO-, FAO + CI-, and FAO + CI + CII-linked mitochondrial respiration in fresh atrial and ventricular tissues (n = 5). D Representative images of mitochondrial morphology in RA, LA, RV, and LV tissues, scale bar = 500 nm. E Quantification of mitochondrial size in atrial and ventricular tissues (n = 150 mitochondria from three biological replicates). F Citrate synthase levels, an indicator of mitochondrial mass, in fresh RA, LA, RV, and LV tissues (n = 5). Data are expressed as mean ± standard error of the mean. One-way analysis of variance (ANOVA) followed with a post-hoc Fisher’s LSD test was used. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Establishment and optimization of TSIT using mouse frozen cardiac tissue
Previously, either frozen tissue homogenate without centrifugation30 or with low-speed centrifugation22 was used in mitochondrial respirometry. To test the centrifugation effects in TSIT, we compared the mitochondrial respiratory capacities of frozen LV tissue homogenates with and without centrifugation at 1000 × g for 10 min using the same amount of tissue homogenate. We found that centrifugation significantly reduced the mitochondrial respiratory capacities (Fig. S3A, B), indicating loss of mitochondria due to centrifugation prior to the measurement. In addition, after correcting the O2 flux to the protein concentration, a larger standard deviation in mitochondrial respiratory capacities was found in centrifuged samples compared with the uncentrifuged samples, although the significant differences between centrifuged and uncentrifuged samples were less abundant (Fig. S3C). Therefore, whole tissue homogenate without centrifugation was applied in TSIT.
The conventional SUIT protocol using fresh tissue showed a smaller mitochondrial respiration through the FAO pathway compared to CI- and CII-linked respiration (Fig. 1C). We explored whether FAO-linked respiratory capacity could be assessed in frozen LV tissue. However, this was not feasible when using theoretically relevant FAO substrates and components, specifically 40 µM palmitoyl-CoA, 40 µM FAD+, and 1.2 mM CoASH (Fig. S4). In contrast, a substantial O2 flux after the subsequent addition of acetyl-CoA (CI-linked respiratory capacity) and succinate (CII-linked respiratory capacity) indicated that the acetyl-CoA-mediated TCA cycle and the mitochondrial electron transport capacity from, respectively, CI and CII to CIII and CIV were preserved in the frozen tissue (Fig. S4). Therefore, we further focused on the CI- and CII-linked respiratory capacities.
Since the acetyl-CoA-mediated TCA cycle generated a limited amount of the CI substrate NADH, we then directly titrated NADH to measure the maximum potential of CI-linked respiratory capacity in frozen tissue. The results showed that the addition of NADH resulted in a flux that was roughly 4 times that of the CI-linked respiratory capacity meditated by acetyl-CoA in frozen moue LV tissue (Fig. 2). Nevertheless, CI-linked respiratory capacity with addition of acetyl-CoA and the necessary substrates for TCA cycle better mirrors physiological conditions wherein the TCA cycle and the ETC are functionally coupled for NADH production, and hence, we decided to measure the acetyl-CoA-mediated CI-linked respiratory capacity in the following tests of mitochondrial function in various frozen cardiac samples using TSIT.

A Representative trace of frozen left ventricular (LV) tissue. Basal respiratory capacity was determined at 2 mM malate (M), 100 µM NAD+, and 10 µM cytochrome c (Cytc). CI-linked respiratory capacity (purple) was determined at titration of 15 µl of 20 mM acetyl-CoA to mimic the physiological condition. Maximum CI-linked respiratory capacity (CImax, olive-green) was determined with stepwise titration of 20 μl of 10 mM NADH. The initial NADH titration caused increase in O₂ flux that was not maintained, likely due to consumption by enzymes such as malate dehydrogenase and nicotinamide nucleotide transhydrogenase. Continued titration led to a steady-state O₂ flux, which was used for quantification. Complex II-linked respiratory capacity was determined at 1 µM rotenone (rot) and titration of 20 µl of 1 M succinate (S). Mitochondrial respiratory capacity was calculated as O2 flux subtracted from residual oxygen consumption, which was determined at an additional 5 mM malonate (mna) and 5 µM antimycin A (ama). B Quantified O2 flux showed that the CImax (olive-green) was roughly 4 times that of the CI-linked respiratory capacity (purple) mediated by acetyl-CoA in frozen LV tissue (n = 3). Data are expressed as mean ± standard error of the mean. A two tailed unpaired Student t-test was used. **P < 0.01. **P < 0.01.
Furthermore, it is well established that the ETC complexes can form SCs31,32,33. Previous research suggested that TCA cycle enzymes can also form SCs, named metabolon34,35. Moreover, a recent proteomic study showed that knockdown of CS, a key enzyme of the TCA cycle, significantly reduced the expression of mitochondrial ETC enzymes, especially the CI subunits36. Since TSIT demonstrated that mitochondrial respiratory capacity can be assessed in frozen cardiac tissue with acetyl-CoA as substrate for TCA cycle, but not with substrates for FAO (Fig. S3), we hypothesized that the TCA cycle enzymes might couple with the SCs of ETC in the frozen cardiac tissue. To test this hypothesis, we assessed mitochondrial SCs formation in frozen tissue homogenates from mouse LV tissues with blue native gel electrophoresis (BNGE). As expected, BNGE showed that CI-CIV and CI-CIII formed SCs at a high molecular weight of over 1 M kDa with individual CI, CIII, and CIV also being visible (Figs. 3A, and S5, S6). Due to technical limitations of BNGE28, CII, CIII, and CIV individually were observed, while CII-CIV or CII-CIII SCs could not be visualized as expected (Fig. 3B and S5, S6). Intriguingly, we found interactions of CS with CI (Figs. 3C and S5) and of CS with CII (Figs. 3D and S5), indicating the TCA cycle enzymes indeed coupled with ETC complexes, which could explain why measurement of respiratory capacity in frozen tissue homogenate using substrates of the TCA cycle (i.e., acetyl-CoA) could be achieved.

A Representative images of mitochondrial CI-CIV SCs (colored yellow) and individual bands corresponding to CI (colored green) and CIV (colored red). B No CII-CIV SCs were found due to technical limitations of BNGE. Individual bands corresponding to CII (colored green) and CIV (colored red) could be observed. C Representative images of SCs formed with CS and CI (colored yellow) and individual bands corresponding to CI (colored green) and CS (colored red). D Representative images of SCs formed with CII and CS (colored yellow). Individual bands corresponding to CII (colored green) and CS (colored red). Nntwt BL6JRcc(BL6J)-Nnt+/Wuhap mice, CI complex I, CII complex II, CIV complex IV, CS citrate synthase.
The TSIT method reveals significant differences in mitochondrial respiratory capacity between frozen atria and ventricles in mice
To test if the mitochondrial respiration differences between fresh atrial and ventricular tissues could also be observed in the frozen samples using TSIT, mitochondrial respiratory capacities of frozen mouse atrial and ventricular tissues were determined (Fig. 4A). As expected, mitochondrial respiratory capacities could be measured in frozen cardiac tissue with multiple injections of substrates or inhibitors of mitochondrial complexes in a single experimental round. Basal respiratory capacity was determined after the injection of malate, NAD+, and Cytc. Acetyl-CoA was then injected to drive the utilization of malate and NAD+ in the TCA cycle to produce NADH as the substrate for CI, allowing for the measurement of CI-linked mitochondrial respiratory capacity. The third injection with rotenone could fully block the O2 flux, proving that the majority of mitochondrial respiratory capacity upon addition of acetyl-CoA was CI-linked. The fourth series of injections was the titration with succinate to measure the CII-linked mitochondrial respiratory capacity to independently assess CII function, eliminating the confounding effects of CI-linked respiratory capacity present in the SUIT protocol. The last two injections with malonate and antimycin A were to measure Rox for the correction with non-mitochondrial oxygen consumption. Using this method, we observed that acetyl-CoA-mediated mitochondrial CI-linked respiratory capacity and succinate-driven CII-linked respiratory capacity in frozen ventricular tissue were significantly higher than the corresponding respiratory capacity in the frozen atrial tissue (Fig. 4B, C). This was consistent with data obtained using fresh tissue (Fig. 1C). Collectively, these data demonstrate that the optimized TSIT using frozen cardiac tissue homogenates can be performed successfully with comparable reliability and sensitivity to the conventional mitochondrial SUIT respiration measurement using fresh tissue regarding CI- and CII-linked mitochondrial respiration.

A Schematic representation of the sample preparation and the TSIT method for frozen cardiac samples from the right atrium (RA), left atrium (LA), right ventricle (RV), and left ventricle (LV) of 6-month-old mice. Samples were weighed, homogenized, and assessed via high-resolution respirometry using TSIT as illustrated. Basal respiratory capacity (green) was determined at 2 mM malate (M), 100 µM NAD+, and 10 µM cytochrome c (Cytc). Complex I (CI)-linked respiratory capacity (yellow) was determined at an additional 150 µM acetyl-CoA. Complex II (CII)-linked respiratory capacity (blue) was determined at 1 µM rotenone (rot) and titration of 20 µl of 1 M succinate (S). Residual oxygen consumption (Rox, gray) was determined at an additional 5 mM malonate (mna) and 5 µM antimycin A (ama). At least 2 technical replicates were performed. Icons are provided by Servier Medical Art (https://smart.servier.com/), licensed under CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/). B Representative traces of frozen RA, LA, RV, and LV tissues from BL6JRcc(BL6J)-Nnt+/Wuhap mice. Purple traces indicate the ventricular O2 flux, and olive-green traces indicate the atrial O2 flux. C Quantification of basal-, CI- and CII-linked respiratory capacities in frozen atrial and ventricular tissues (n = 6). Data are expressed as mean ± standard error of the mean. One-way analysis of variance (ANOVA) followed with a post-hoc Fisher’s LSD test was used within each mitochondrial respiratory capacity. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
The TSIT method distinguishes differences in mitochondrial respiratory capacity between frozen cardiac tissue of young and old mice
To determine whether TSIT can be applied to distinguish physiologically relevant changes, a well-recognized ageing mouse model was assessed. Using conventional SUIT respirometry protocols, compromised mitochondrial respiration was observed with ageing in mitochondria isolated from fresh mouse LV tissue37, in fresh permeabilized rat ventricular tissue38, and in human skin fibroblasts39. Therefore, we performed TSIT respirometry in young (4-month-old) and old (23-month-old) mice using frozen LV tissue. Our results showed that CII-linked respiratory capacity was significantly decreased with age, while the basal and CI-linked respiratory capacities were also decreased with a trend but not reached statistical significance between young and old mice (Fig. 5A, B). These results illustrated that TSIT could be applied to detect ageing-related mitochondrial dysfunction in the frozen cardiac tissue.

A Representative traces of frozen left ventricular (LV) tissues from young (4-month-old, purple) and old (23-month-old, olive-green) mice. B Quantification of basal-, complex I (CI)-, and complex II (CII)-linked respiratory capacities in the frozen LV tissues of young (purple) and old (olive-green) mice (n = 6). Data are expressed as mean ± standard error of the mean. A two tailed unpaired Student t-test was used within each mitochondrial respiratory capacity. *P < 0.05. M+Cytc+NAD+ malate+cytochrome c+NAD+, rot rotenone, S succinate, mna malonate, ama antimycin A.
The TSIT method differentiates the impairment of mitochondrial respiratory capacity in frozen cardiac tissue of pathophysiological disease models
Mitochondrial dysfunction has been associated with a broad range of human diseases. We aimed to apply our TSIT method to measure mitochondrial respiratory (dys)function in frozen cardiac tissue samples from multiple pathophysiological disease models.
To firstly validate TSIT in a genetically modified mouse model with impaired mitochondrial function, we assessed respiratory capacities in frozen cardiac tissue from a CI-deficient (Ndufs4−/−) mouse model. Reduced activity of mitochondrial CI is expected to significantly impact CI-linked respiration in the Ndufs4−/− mouse heart. Indeed, a significantly decreased basal and CI-linked respiratory capacities was observed in frozen LV tissue from Ndufs4−/− mice compared with the WT control, while CII-linked respiratory capacity remained unchanged (Fig. 6A, B), proving the validity and sensitivity of TSIT.

A Representative traces of frozen LV tissues from Ndufs4 knockout (Ndufs4−/−; olive-green) and wildtype (WT; purple) mice. B Quantification of basal-, complex I (CI)-, and complex II (CII)-linked respiratory capacities in frozen LV tissues in WT and Ndufs4−/− mice (n = 6). A two tailed unpaired Student t-test was used within each mitochondrial respiratory capacity. C Representative traces of frozen left ventricular (LV) tissues from mice subjected to ischemia reperfusion injury (IR; purple) and sham control (olive-green). D Quantification of basal-, CI-, and CII-linked respiratory capacities in frozen LV tissues from the IR and the sham control (n = 6). A two tailed unpaired Student t-test was used within each mitochondrial respiratory capacity. E Representative traces of frozen LV tissues from C57BL/6J mice fed with a standard chow diet (BL/6J control; purple), and NZO mice fed with a standard chow diet (NZO + SD; olive-green) or a high fat diet (NZO + HFD; light blue). F Quantification of basal-, CI-, and CII-linked respiratory capacities in frozen LV tissues from BL/6J, NZO + SD, and NZO + HFD (n = 6). One-way analysis of variance (ANOVA) followed with a post-hoc Fisher’s LSD test was used within each mitochondrial respiratory capacity. Data are expressed as mean ± standard error of the mean. *P < 0.05, **P < 0.01, ***P < 0.001. M+Cytc+NAD+ malate+cytochrome c+NAD+, rot rotenone, S succinate, mna malonate, ama antimycin A.
Then, a mouse cardiac IR model was employed since impaired mitochondrial metabolism, including oxidative stress, mitochondrial calcium overload, and impaired ETC and OXPHOS, were reported during cardiac IR injury40,41. Prolonged ischemia was shown to induce a significant reduction in CI and CII-linked oxygen consumption in rat subsarcolemmal mitochondria isolated from fresh cardiac tissue42. In agreement, our TSIT method was able to detect a significant impaired CI- and CII-linked respiratory capacities in frozen LV tissue from IR mice compared with the sham control (Fig. 6C, D).
Previous research has shown impaired CII-linked respiration in heart tissue from a diabetic FVB/N mouse model, which was induced by the combination of low-dose streptozotocin injection and HFD2. Similarly, the overall maximum mitochondrial respiration was found to be decreased in fresh LV tissue from HFD-treated NZO mice, a well-established model for obesity and T2D18. However, the specific CI- or CII-linked cardiac mitochondrial function of NZO mice has not been comprehensively investigated. To address this gap, we used TSIT to assess mitochondrial respiratory capacities in frozen LV tissue of NZO mice, with or without HFD exposure. TSIT detected a significant reduction in both CI- and CII-linked respiratory capacities in the NZO + HFD group compared with the BL/6J control (Fig. 6E, F). Moreover, the CII-linked respiratory capacity in the NZO + HFD group was significantly lower than that in the NZO + SD group (Fig. 6E, F). This finding newly identifies respiratory impairments of specific mitochondrial complexes in the NZO mouse model of obesity and T2D, proving novel insights and potential therapeutic targets.
We further tested the association of the metabolon component CS with ETC SCs component CI in these three pathophysiological disease models. CI-containing SCs were impaired in the LV tissue of Ndufs4−/− mice (Fig. S7A, B), IR mice (Fig. S7C, D), as well as in NZO + SD and NZO + HFD mice (Fig. S7E, F). CS-containing SCs were also impaired in the Ndufs4−/−, IR, and NZO + HFD groups compared with their corresponding control groups, whereas the interactions of CS with CI were not significantly influenced (Fig. S7).
Taken together, these observations strongly support the conclusion that TSIT is validate, specific, and capable of detecting mitochondrial respiratory differences across various pathophysiological conditions using frozen cardiac tissue.
The TSIT method detects mitochondrial damage in human frozen cardiac tissue
To promote the translational application of TSIT in clinical practice, we measured mitochondrial respiratory capacities using frozen heart tissue homogenates from human donors. Five LV samples with cardiac pathology (CP) and four LV samples with no/low cardiac pathology (NCP) were used (Table S1). We could thus measure mitochondrial respiratory capacities in frozen human tissue using the same TSIT method. The TSIT was able to detect a significantly impaired CII-linked respiratory capacity in frozen LV tissue from the CP compared with the NCP, while no significant difference in basal- and CI-linked respiratory capacities between the two groups was observed (Fig. 7A, B).

A Representative traces of frozen left ventricular (LV) tissue from human donors with no/low cardiac pathology (NCP, purple) and with cardiac pathology (CP, olive-green). B Quantification of basal-, complex I (CI)-, and complex II (CII)-linked respiratory capacities in frozen LV tissues from NCP (n = 4) and CP (n = 5) groups. A two tailed unpaired Student t-test was used within each mitochondrial respiratory capacity. C Representative Western blotting plots of individual mitochondrial CI to CV in frozen LV tissue from NCP and CP groups. VDAC1 protein served as the loading control. D Quantification of relative complex protein level of mitochondrial CI to CV to the loading control VDAC1. A two tailed unpaired Student t-test was used within each mitochondrial complex protein level. E Pearson correlation analysis between CII-linked respiratory capacity and relative CII protein level to VDAC1 in frozen LV tissue from NCP and CP groups. Data are expressed as mean ± standard error of the mean. *P < 0.05, **P < 0.01. M+Cytc+NAD+ malate+cytochrome c+NAD+, rot rotenone, S succinate, mna malonate, ama antimycin A.
For further confirmation, Western blotting of mitochondrial complexes was performed. A significant decrease in CII protein level in the CP group compared with the NCP group was observed, with no difference between the two groups in the other mitochondrial complexes (Fig. 7C, D). Correlation analysis showed that CII-linked respiratory capacity measured by TSIT was significantly correlated with relative CII protein expression (Fig. 7E). This finding strengthens that TSIT is a reliable and sensitive method for detecting impairments in specific mitochondrial complex-mediated respiratory capacity in human samples with cardiac pathology.
Discussion
To the best of our knowledge, this study is the first to develop and apply a high-resolution respirometry method to assess mitochondrial function across multiple mitochondrial complexes in frozen cardiac tissue using tailored substrate-inhibitor titration (TSIT). TSIT measures oxygen consumption following electron delivery by the TCA cycle as well as by direct CI and CII substrates. The small amount of frozen sample material, a straightforward preparation of tissue homogenates, and multiple titrations with reagents for mitochondrial ETC complexes allow to measure detailed mitochondrial respiratory capacities in frozen cardiac tissue of mice and humans. Using cardiac tissues from the same batch of mice, TSIT identified differences in mitochondrial respiratory capacities between frozen atrial and ventricular tissues, consistent with those identified using the conventional SUIT protocol with fresh tissue. Moreover, by testing in both physiological and pathophysiological models, TSIT effectively distinguished primary differences or impairment in mitochondrial respiratory function of diseased frozen mouse and human cardiac tissue samples compared with the controls. Therefore, evaluating mitochondrial function in frozen cardiac tissue using the TSIT method could significantly contribute to the laboratory assessment of patients with cardiac diseases.
This study newly compared the mitochondrial differences between atrial and ventricular tissue samples in mice using both the conventional SUIT and the newly developed TSIT protocols. The differences observed in mitochondrial respiratory capacities between frozen atrial and ventricular tissue were in line with those of Lemieux et al. using fresh tissue from failing human hearts13. They found that respiration was higher in the ventricles, corresponding to the higher specific CS activity in ventricular compared to atrial tissue. In addition, they also showed that mitochondrial respiration per tissue mass was identical in the right and left atrial tissue13, which is similar with what we found in mice. These concordant results from human and mouse cardiac tissue indicate that the TSIT method can reliably reveal physiologically relevant differences in mitochondrial function.
Mitochondrial functional evaluation is critical for characterizing the pathophysiological differences and metabolic abnormalities in heart diseases. Our results demonstrate that TSIT can effectively identify mitochondrial ETC defects in frozen cardiac tissue from various mouse (patho)physiological models of aging, CI deficiency, IR injury, obesity, and T2D, all of which are known or have potential for causing cardiac mitochondrial dysfunction43,44,45,46,47,48,49. TSIT detected the specific impairment of CI-, but not CII-linked, respiratory capacity in CI deficiency mouse model, highlighting its validity and specificity. Notably, beyond the established knowledge of impaired cardiac mitochondrial function in diabetic mouse models2,18,50, specific complex-linked respiration in the NZO mouse model of obesity and T2D has not been investigated. TSIT newly diagnosed both CI- and CII-linked respiratory dysfunction in the NZO mice that had not been previously clarified using conventional protocols with fresh tissue. Furthermore, TSIT also detected an impaired CII-linked respiratory capacity in the frozen human LV tissue with cardiac pathology compared with the control. This finding was further supported by the changes in protein expression of individual mitochondrial CII subunit. These results suggest that TSIT is a sensitive and reliable method for diagnosis of mitochondrial dysfunction in frozen stored samples in clinical practice. In summary, the successful application of TSIT in the evaluation of mitochondrial respiratory function under both physiological and pathophysiological conditions underscores the sensitivity and robustness of the method. TSIT provides new insight into mitochondrial function of disease models, enabling more detailed and comprehensive research findings.
The successful assessment of CI- and CII-linked mitochondrial respiratory capacities using TSIT indicated that the ETC SCs, specifically SCs formed by CI and CII to CIV, maintained their associations for electron transport and oxygen consumption. Based on the ‘plasticity model’ of mitochondrial SCs theory51, the mitochondrial complexes can be functional either individually or as part of SCs detected by BNGE. Most CI appeared to form SCs with other complexes, including CIII and CIV, or CII, CIII, and CIV31,32. We indeed observed SCs containing CI and CIV formed in frozen mouse cardiac tissue, possibly enhancing efficiency of electron transport from CI to CIV, where oxygen is consumed. Notably, due to the technical limitation of BNGE, no CII-CIV SCs could be detected, although it has been recently proven that CII forms SCs with CI and CIV28,51,52. Inspired by our observation that acetyl-CoA could drive CI-linked mitochondrial respiratory capacity, we analyzed whether SCs between CS and ETC complexes existed in the frozen cardiac tissue. This revealed physical interactions between the important TCA cycle enzyme CS with CI, as well as CII. To our best knowledge, this is the first study to show that the TCA enzymes form SCs with the ETC complexes, explaining why respiratory analysis in frozen tissue is possible. Taken together, in frozen cardiac tissue, not only the mitochondrial ETC-formed SCs, but also the TCA metabolon is preserved and coupled to the ETC SCs.
Although TSIT is capable of recapitulating the measurement of mitochondrial respiratory capacities, as is the traditional SUIT protocol with fresh tissue, the mitochondrial respiratory capacities measured by TSIT should be regarded as the maximal uncoupled respiration, but not OXPHOS-related respiration in the context of conventional respirometry theory using fresh tissue20. This is because the MIM disruption in the freeze-thaw process leads to dissipation of mitochondrial membrane potential and thus to full uncoupling, allowing respiration with a maximal flow within the ETC system. In addition, different reagents were required in TSIT compared with the SUIT protocol. For frozen homogenates of human muscle biopsies, an absence of oxygen consumption was seen in response to pyruvate and malate, routinely used reagents in the SUIT protocol designed for fresh tissue22,30. This impairment is probably linked to the inability to convert pyruvate to acetyl-CoA, possibly due to loss of pyruvate dehydrogenase in mitochondrial matrix in frozen tissue. Alternatively, this impairment could be due to the lack of NAD+ as substrate for NADH production. In contrast, in TSIT, acetyl-CoA together with sufficient NAD+, malate, and Cytc were added. Due to the disrupted mitochondrial membranes of frozen tissue, these substrates can feed the TCA cycle to generate NADH. The produced NADH then serves as the direct substrate for CI, allowing for the measurement of CI-linked respiratory capacity. This is confirmed by a specifically reduced CI- but not CII-linked respiratory capacity in the Ndufs4−/− mice and by the fact of complete inhibition of CI-linked respiratory capacity by rotenone in TSIT. The ability of produced NADH to efficiently boost CI-linked mitochondrial respiratory capacity also serves as a proof for SCs formation between TCA cycle enzymes with CI. Furthermore, a hyper-oxygenated condition during respirometry measurement is normally preferred in the SUIT protocol for fresh permeabilized tissue, while a normal oxygen condition was used in TSIT. This is likely because the small amount of frozen tissue homogenate was fully homogeneous, allowing for sufficient oxygen penetration under normal oxygen conditions. Moreover, the respiratory capacity of frozen tissue homogenate remained consistent in hyper-oxygenated and normal conditions (data not presented), thereby eliminating the potential variance due to differences in oxygen level.
Compared with other respirometry protocols using different types of frozen tissues, TSIT showed advantages from various perspectives. Zuccolotto et al., using frozen skeletal muscle tissue, showed a significant correlation between CI-mediated respiratory capacity and enzyme activity of individual CI (r = 0.597)30. However, they did not test the maximum CI capacity driven by NADH as a direct substrate, nor did they include the measurement of CII-linked mitochondrial respiratory capacity, which is particularly crucial in disease models such as obesity or IR injury, as we show here. In TSIT, we are able to sequentially and independently measure CII-linked respiratory capacity by titration of succinate, which donates H+-linked two-electron to FAD bound to the subunit of CII to form FADH2, relaying electrons further via a series of iron-sulfur clusters to ubiquinone (coenzyme Q)53. An evident inhibitory effect of malonate confirmed that the succinate is indeed feeding CII in the frozen cardiac tissue in TSIT54. Another recent respirometry protocol using frozen tissue and the Seahorse XF Analyzer was established by Acin-Perez et al.22. In the process of sample preparation, these authors centrifuged the tissue lysate to remove debris. Nevertheless, our study proved that centrifugation of tissue lysate prior to respirometry resulted in the loss of mitochondrial protein and induced bigger technical variation, therefore, we recommend omitting centrifugation by using whole tissue homogenate, leading to less technical variation and higher sensitivity. Secondly, the Seahorse XF Analyzer allows for only four predetermined injections of (mixed) reagents, limiting options for titration and verification. The TSIT method is more flexible, illustrated by the inclusion of a separate injection of rotenone to confirm measurement of CI-linked respiratory capacity. Lastly, Acin-Perez et al. used NADH to assess CI-linked respiratory capacity, given their hypothesis of an impaired TCA cycle in frozen tissue22. However, their proposition is challenged by our data and the data from Zuccolotto et al.30, showing that the TCA cycle remained functional in frozen cardiac and muscle tissue, respectively, as NADH could be produced by the TCA cycle upon the supply of acetyl-CoA, NAD+, malate, and Cytc. This is also supported by SCs’ formation of ETC SCs with the TCA cycle metabolon. Furthermore, the respiratory capacity assessed by direct titrations of NADH reflects the maximum capacity of CI, beyond its production via the TCA cycle. Therefore, the CI-linked respiratory capacity fueled by the TCA cycle better represents the physiological interplay between metabolon and ETC SCs in NADH production for CI within mitochondria.
Unfortunately, TSIT could not overcome some limitations of using frozen tissue. A limitation of our method is its inability to assess the FAO-linked respiratory capacity, even with the necessary cofactors for FAO. This is probably due to the disassembly, inactivation, or disintegration of FAO-related enzymatic complexes, preventing them from donating electrons to the coenzyme Q or providing acetyl-CoA to the TCA cycle. It is also important to point out that the interference from varying times of freezing cannot be ruled out, although we have no evidence for this based on samples used in this study that were frozen for different periods of time. Similarly, interference from sampling of different atrial or ventricular areas, especially in large human cardiac tissue, cannot be excluded. However, the same limitation exists in conventional respirometry protocols using fresh tissue. These effects need to be studied systematically, in order to provide solid frozen tissue biobanking advise. Furthermore, we showed the potential association of metabolon association with ETC SCs with a single method. However, future studies are warranted to further confirm and detail this conclusion.
In conclusion, the newly developed versatile TSIT using high-resolution respirometry was shown to be a robust and translational method to evaluate mitochondrial respiratory capacities in diverse frozen cardiac tissues, ranging from preclinical animal models to human donors. It effectively distinguished physiological and pathophysiological mitochondrial impairments, uncovering a new diagnosis of mitochondrial dysfunction in cardiac disease models, and advanced mitochondrial function analyses for both laboratory and clinical applications.
Methods
We have complied with all relevant ethical regulations for animal use. All animal procedures required were ethically approved by the Animal Welfare Committee of Wageningen University regarding the mice used for atrial and ventricular tissue comparison (2020.W-0019.008) and the ageing mice (2020.W-0019.007), by the Radboud University Nijmegen regarding the Ndufs4−/− mice (2017.W-0017-007), by the German Institute of Human Nutrition regarding the NZO mice (2347-46-1029), or by Amsterdam Medical Center regarding the IR mice (AVD11800202114603). All ethical regulations relevant to human research participants were followed. Human cardiac samples were collected and characterized at the Amsterdam Medical Center (Table S1). The study conformed to the principles of the Declaration of Helsinki. The institutional ethical review board approved the study (2024.0643), and patients gave written informed consent.
Cardiac tissue collection from mice and human patients
All mice had free access to food and water and were maintained under standard temperature and humidity conditions in the respective animal facilities. To compare the difference in mitochondrial respiration between atrial and ventricular tissues, cardiac tissue samples were collected from 6-month-old BL6JRcc(BL6J)-Nnt+/Wuhap (Nntwt) male mice fed with standard chow, an in-house bred mouse line similar to C57BL/6JRccHsd. The mice were killed by decapitation and dissected immediately. After washing in ice-cold phosphate-buffered saline (PBS), the heart was split into right atrium (RA), left atrium (LA), right ventricle (RV), and left ventricle (LV). The samples were then either immediately snap frozen in liquid nitrogen and stored at −80 °C before the measurement (n = 6), or freshly immersed in the BIOPS buffer55 and used for respiration measurement within 6 h (n = 5).
Frozen cardiac tissue samples from one physiological mouse model and three pathophysiological mouse models were utilized to evaluate the sensitivity and applicability of TSIT. In detail, the physiological model of natural aging compared young (4-month-old) and old (23-month-old) mice (Nntwt, n = 6 females each), fed with standard chow diet. The pathophysiological models included: (1) chow fed Ndufs4−/− mice and the control wildtype (WT) mice (n = 6 each; 3 males and 3 females)56; (2) chow fed C57BL/6 N mice subjected to myocardial infarction surgery to mimic IR and sham controls (n = 6 males each)57; and (3) C57BL/6 J mice fed with a standard chow diet (BL/6J control) and the NZO mice fed with a standard chow diet (NZO + SD) and a 35 en% high-fat diet (NZO + HFD), respectively (n = 6 males each)18. LV samples from mouse models above were snap frozen and stored in −80 °C until further analysis.
Frozen human LV free-wall tissue samples from human donors with cardiac pathology (n = 5) or with no/low level cardiac pathology (n = 4) were measured to prove the utility of TSIT in clinical practice. The detailed information about the human donors is listed in Table S1.
Preparation of fresh cardiac tissues with mechanical permeabilization for high-resolution respirometry
Fresh mouse atrial and ventricular tissue samples were kept in ice-cold BIOPS buffer for a maximum of 6 h. For subsequent respirometry analyses, the tissue was quickly dried on filter paper and weighed before being transferred to a 1.5 ml Eppendorf tube with 50 μl of ice-cold BIOPS buffer. The average tissue mass varied between 0.4 and 0.7 mg. Mechanical permeabilization was performed using a hand-held tissue homogenizer (47747-370, VWR) with a disposable pestle (431–0094, VWR) at a speed of 12,000 RPM. To ensure intact mitochondrial membranes in the fresh cardiac tissue, 3 s of mechanical permeabilization for atrial tissue and 1 s for ventricular tissue were used. The procedures were performed on ice throughout the experiment. Fresh tissue preparations without centrifugation were then loaded into the chambers of an Oroboros O2k (series H-0391, Oroboros Instruments) with 2 ml mitochondrial respiration medium MiR05 (6010101, Oroboros Instruments) containing 0.5 mM EGTA, 3 mM MgCl2, 60 mM lactobionic acid, 20 mM taurine, 10 mM KH2PO4, 20 mM HEPES, 110 mM D-sucrose, and 1 g/L bovine serum albumin (BSA; pH 7.1).
Preparation of frozen cardiac tissue homogenates for high-resolution respirometry
Frozen cardiac tissue was quickly thawed in ice-cold BIOPS buffer, dried on filter paper, and weighed before being transferred to a 1.5 ml Eppendorf tube with 50 μl of ice-cold BIOPS buffer. The average tissue mass varied between 0.4 and 0.7 mg for frozen mouse atrial and ventricular tissue, and 1.8–2.2 mg for frozen human ventricular tissue. Tissue was homogenized using the hand-held tissue homogenizer with a disposable pestle. The optimal homogenization time durations for frozen tissue were previously determined to ensure effective tissue disruption while preserving mitochondrial respiratory function. These times, which corresponded to the disappearance of visible tissue debris, were ~5 s for mouse ventricular tissue, 10 s for mouse atrial tissue, and 15 s for human ventricular tissue. The procedures were performed on ice throughout the experiment. Fifty microliters frozen tissue homogenates with or without centrifugation at 1000 × g for 5 min at 4 °C were then loaded into the chambers of an Oroboros O2k with 2 ml mitochondrial respiration medium MiR05.
High-resolution respirometry of fresh cardiac tissue
High-resolution respirometry using mechanically permeabilized fresh cardiac tissue was performed at 37 °C in a hyperoxygenated condition (300–450 μM O2), allowing for a sufficient oxygen concentration gradient into the mechanically permeabilized fresh tissue (https://wiki.oroboros.at/index.php/SUIT-002_O2_pfi_D006). Mitochondrial respiration was assessed by the conventional substrate-uncoupler-inhibitor titration (SUIT) protocol with minor modifications as follows (SUIT-002, https://wiki.oroboros.at/index.php/SUIT-002, Fig. 1A). All concentrations provided are final concentrations, except for carbonyl cyanide m-chlorophenyl hydrazone (CCCP), which was titrated stepwise until a maximal stable flux was achieved, with both the stock concentration and titration volumes specified. Briefly, 7.5 mM adenosine diphosphate (ADP; 5285, Sigma) was added to stimulate the consumption of endogenous fuel substrates without ADP limitation (OXPHOS state). 0.3 mM malate (M1000, Sigma) and 0.5 mM octanoylcarnitine (0605, Tocris) were added to measure OXPHOS respiration through the fatty acid oxidation (FAO) pathway. Ten micrometers Cytc (C7752, Sigma) was then added to test mitochondrial outer membrane integrity. Samples that responded to Cytc addition by a 20% increase in respiration were excluded. Then, 2 mM malate, 5 mM pyruvate (P2256, Sigma), and 10 mM glutamate (G1626, Sigma) were added to measure OXPHOS respiration through FAO and CI (FAO + CI), followed by addition of 10 mM succinate (S2378, Sigma) to measure OXPHOS respiration through FAO, CI, and CII (FAO + CI + CII). Next, titrations with 1 μl of 0.1 μM CCCP (C2759, Sigma) were used to measure the uncoupled maximal electron transport chain capacity (ETS state). In the end, 1 µM rotenone (R8875, Sigma) and 5 μM antimycin A (A8674, Sigma) were added to measure residual oxygen consumption (Rox) or non-mitochondrial respiration. Mitochondrial respiration, calculated as oxygen (O2) flux subtracted from Rox, was normalized to wet tissue weight and expressed as pmol/s*mg tissue.
High-resolution respirometry of frozen cardiac tissue
High-resolution respirometry of frozen cardiac tissue homogenate using TSIT was performed in a normal oxygenated condition (80–180 μM O2) since oxygen easily permeated into the frozen tissue homogenate30. The sequential titration of substates and inhibitors of mitochondrial complexes according to TSIT, given in final concentrations unless otherwise specified for regents that were titrated stepwise, was as follows (Fig. 2A, Table S2): 2 mM malate, 100 µM oxidized nicotinamide adenine dinucleotide (NAD+; N0632, Sigma) and 10 µM Cytc were added to supply key components of the tricarboxylic acid (TCA) cycle and ETC that may have been lost due to MIM damage, in order to determine ‘Basal’ respiratory capacity. Next, key substrates or components for FAO, including 40 µM palmitoyl-CoA (P9716, Sigma), 40 µM flavin adenine dinucleotide (FAD; F6625, Sigma), and 1.2 mM coenzyme A (CoASH, C4282, Sigma), were added to determine FAO-linked respiratory capacity. 150 µM acetyl-CoA (A2181, Sigma) served to boost the TCA cycle to produce NADH from NAD+ for the assessment of the CI-linked respiratory capacity. Then, to surpass the limitation of the CI substrate NADH supplied by the TCA cycle, 20 μl of 10 mM NADH (N8129, Sigma) was directly titrated until maximal steady-state O2 flux was obtained, allowing for determining the maximum CI-linked respiratory capacity. One micrometer rotenone was subsequently added to block CI activity, followed by titration with 20 μl of 1 M succinate (substrate of CII) until maximal O₂ flux was achieved, to assess CII-linked respiratory capacity. An ADP addition test following the titration of acetyl-CoA, NADH, and succinate was performed to confirm the uncoupled OXPHOS in frozen tissue (Fig. S1). Finally, Rox was determined after the sequential addition of 5 mM malonate (inhibitor of CII, M1296, Sigma) and 2.5 µM antimycin A. For each biological sample, at least 2 technical replicates were performed. The order of sample measurements across different groups was randomized to prevent potential systematic errors. Mitochondrial respiratory capacities calculated as O2 flux subtracted from Rox were normalized to tissue wet weight and expressed as pmol/s*mg tissue.
Citrate synthase activity
Citrate synthase (CS) activity, an indicator of mitochondrial mass58, was determined using the CS assay kit according to the manufacturer’s instructions (CS0720, Sigma). Briefly, the corresponding frozen tissue from the same animal used for high-resolution respirometry measurement was weighed and homogenized in lysis buffer (150 μl lysis buffer per mg tissue) with the hand-held tissue homogenizer. The lysis buffer contained 50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% Triton X-100, 1 mM ethylenediaminetetraacetic acid (EDTA), and protease (04693159001, Roche) and phosphatase inhibitor cocktail (04906837001, Roche). After a freeze-thaw cycle (−80 °C freezing for 10 min, 4 °C thawing), the homogenate was centrifuged at 10,000 × g for 5 min at 4 °C to remove cell debris, and 4 μl of supernatant was used to measure CS activity based on the real-time absorbance kinetics at 412 nm in the linear range with an interval of 15 s for 3 min using the plate reader (235197, BioSPX). The CS activity (IU/ml) was corrected by the tissue protein concentration (mg protein/ml) and expressed as IU/mg protein.
Transmission electron microscopy
Atrial and ventricular tissue samples were cut into pieces of about 1 mm3 and fixed in a mixture of 2% glutaraldehyde (16316-10, EMS) and 2% formaldehyde (15700, EMS) in 0.1 M phosphate/citrate buffer overnight at 4 °C. Tissue was then washed with 0.1 M phosphate/ citrate buffer and subsequently postfixed in 1% osmium tetroxide (19130, EMS) for 1 h at room temperature, washed with deionized water, dehydrated in ethanol (30%, 50%, 70%, 80%, 90% and 100% for 5–10 min each), embedded in Spurr resin (14300, EMS) overnight, and polymerized at 70 °C for 8 h. Ultrathin sections (60 nm) were stained with 2% uranyl acetate (22400, EMS) and 3% lead citrate (22410, EMS) for 10 min, and then viewed on copper formvar coated grids under a transmission electron microscope (JEM 1400 Plus Transmission EM). From images of three biological replicates, 150 mitochondria of each group were analyzed. Mitochondrial size and number were analyzed using Fiji software (ImageJ 1.52j).
Separation of mitochondrial supercomplexes by blue-native gel electrophoresis
Frozen mouse cardiac tissue (around 10–15 mg) from 6-month-old Nntwt mice and three pathophysiological mouse models (i.e., Ndufs4−/−, IR and NZO mouse models) was homogenized with the hand-held homogenizer with a disposable pestle in 1 ml ice-cold mitochondrial lysis buffer consisting of 250 mM sucrose, 20 mM Tris-HCl pH 7.4, 1 mM EDTA, and a protease inhibitor cocktail. The tissue homogenate was freed from cell debris and nuclei by centrifugation at 1000 × g for 10 min at 4 °C. The supernatant was then centrifuged at 10,000 × g for 10 min at 4 °C to obtain a mitochondrial enriched pellet, which was resuspended in 100 µl fresh mitochondrial lysis buffer. The protein concentration was measured using DC protein assay kit according to the manufacturer’s instructions (500-0116, Bio-Rad). Then, 60 µg of mitochondria-enriched fraction protein was pelleted at 20,000 × g for 10 min at 4 °C and resuspended in 12 µl solubilization buffer (50 mM NaCl, 50 mM imidazole-HCl, 5 mM 6-aminocaproic acid, 1 mM EDTA, pH 7.0). Thereafter, 540 µg digitonin was added, and incubated on ice for 10 min for further solubilization. Then, the solubilized samples were centrifuged at 20,000 × g for 20 min at 4 °C, and supernatant was mixed with 60 µg Coomassie Brilliant Blue G before loading (27,815, Sigma). NativeMark unstained protein standard (LC0725, Invitrogen) was included. Blue native PAGE analysis was performed using a NativePAGE 3–12% Bis-Tris gel (BN2011BX10, Invitrogen), and electrophoresis was done at 50 V for 30 min and then 150 V for 120 min in an XCell SureLock Mini cell (EI0001, Novex). Protein was transferred to a methanol-activated PVDF membrane (IPFL85R, Sigma) at 30 V overnight at 4 °C (1703930, Bio-Rad). The membrane was then washed with 1x TBST with 2% SDS, pH 7.6 at 60 °C, blocked with Intercept Blocking Buffer (927-60001, LI-COR) for 1.5 h at room temperature, and incubated with primary antibodies anti-CI (A21344, Invitrogen, 1:1000 dilution), anti-succinate dehydrogenase A (SDHA, ab14715, Abcam, 1:1000 dilution), anti-CIII (14742-1-AP, Proteintech, 1:1000 dilution), anti-CIV (ab16056, Abcam, 1:1000 dilution), or anti-CS (14309S, Cell signaling, 1:1000 dilution) overnight at 4 °C. The membrane was then incubated with secondary antibodies IRDye 800CW Donkey anti-mouse (926-32212, LI-COR, 1:5000 dilution) or IRDye 680RD Donkey anti-rabbit (926–68073, LI-COR, 1:5000 dilution) for 1 h at room temperature and visualized using an Odyssey scanner (LI-COR).
Protein extraction and Western blotting analysis
Frozen patient left ventricular tissue was lysed in radioimmunoprecipitation assay (RIPA) buffer containing 10 mM Tris-base pH 7.6, 150 mM NaCl, 0. 5% sodium deoxycholate, 1% SDS, 1% Triton X-100, and protease and phosphatase inhibitor cocktail. Eight microgram protein per lane was separated on a 4–12% NuPage gel (NP0323BOX, Invitrogen) and electrophoresed at 110 V for 30 min and then 150 V for 1 h in an XCell SureLock Mini cell. Proteins were transferred to a methanol-activated PVDF membrane at 300 mA for 90 min at room temperature. The membrane was washed with 1× TBS for 5 min, blocked with Intercept Blocking Buffer for 1 h at room temperature, and incubated with primary antibodies anti-OXPHOS (ab110413, Abcam, 1:1000 dilution) or anti-VDAC1 (ab235143, Abcam, 1:1000 dilution) overnight at 4 °C. The membrane was subsequently incubated with secondary antibodies IRDye 800CW Donkey anti-mouse (926–32212, LI-COR, 1:5000 dilution) or IRDye 680RD Donkey anti-goat (926–68074, LI-COR, 1:5000 dilution), respectively, for 1 h at room temperature and visualized using an Odyssey scanner.
Statistics and reproducibility
Oxygen concentration and oxygen flux were recorded and processed by DatLab software 7.4 (Oroboros Instruments). All the comparisons and graphs were made using the GraphPad Prism software (v9.5.1). To compare mitochondrial respiration, mitochondrial size and mass among ventricular and atrial tissues, as well as mitochondrial respiratory capacities in the NZO mouse model fed with and without HFD, one-way analysis of variance (ANOVA) followed with a post-hoc Fisher’s LSD test was used. To compare each mitochondrial complex-driven respiratory capacity between physiological or pathophysiological mouse and human samples and their respective controls, a two-tailed unpaired Student t-test was used. To correlate the mitochondrial respiratory capacities and relative complex protein level, Pearson correlation analysis was used. Error bars in all figures represent the standard error of the mean. The number of biological replicates (n) used in each experiment, along with the statistical test used to analyze the particular experiment, are provided in the figure legends. The statistical significance was shown as *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data supporting the findings of this study are available within the paper and its Supplementary Information file. The numerical source data can be found in Supplementary Data file.
References
-
Müller, M. J., Wang, Z., Heymsfield, S. B., Schautz, B. & Bosy-Westphal, A. Advances in the understanding of specific metabolic rates of major organs and tissues in humans. Curr. Opin. Clin. Nutr. Metab. Care 16, 501–508 (2013).
-
Parker, A. M. et al. Characterisation of the myocardial mitochondria structural and functional phenotype in a murine model of diabetic cardiomyopathy. Front Physiol. 12, 672252 (2021).
-
Hoeks, J., Hesselink, M., & Schrauwen, P. Mitochondrial respiration. In: Encyclopedia of Exercise Medicine in Health and Disease (ed Mooren F. C.) (Springer Berlin Heidelberg, 2012).
-
Kumar, A. A., Kelly, D. P. & Chirinos, J. A. Mitochondrial dysfunction in heart failure with preserved ejection fraction. Circulation 139, 1435–1450 (2019).
-
Nollet, E. E. et al. Mitochondrial dysfunction in human hypertrophic cardiomyopathy is linked to cardiomyocyte architecture disruption and corrected by improving NADH-driven mitochondrial respiration. Eur. Heart J. 44, 1170–1185 (2023).
-
Walters, A. M., Porter, G. A. & Brookes, P. S. Mitochondria as a drug target in ischemic heart disease and cardiomyopathy. Circ. Res. 111, 1222–1236 (2012).
-
Tatarková, Z. et al. Effects of aging on activities of mitochondrial electron transport chain complexes and oxidative damage in rat heart. Physiol. Res. 60, 281–289 (2011).
-
Tocchi, A., Quarles, E. K., Basisty, N., Gitari, L. & Rabinovitch, P. S. Mitochondrial dysfunction in cardiac aging. Biochim. Biophys. Acta 1847, 1424–1433 (2015).
-
Karamanlidis, G. et al. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab. 18, 239–250 (2013).
-
Bolea, I. et al. Defined neuronal populations drive fatal phenotype in a mouse model of Leigh syndrome. eLife 8, e47163 (2019).
-
Scheubel, R. J. et al. Dysfunction of mitochondrial respiratory chain complex I in human failing myocardium is not due to disturbed mitochondrial gene expression. J. Am. Coll. Cardiol. 40, 2174–2181 (2002).
-
Sharov, V. G., Todor, A. V., Silverman, N., Goldstein, S. & Sabbah, H. N. Abnormal mitochondrial respiration in failed human myocardium. J. Mol. Cell. Cardiol. 32, 2361–2367 (2000).
-
Lemieux, H., Semsroth, S., Antretter, H., Höfer, D. & Gnaiger, E. Mitochondrial respiratory control and early defects of oxidative phosphorylation in the failing human heart. Int. J. Biochem. Cell Biol. 43, 1729–1738 (2011).
-
Chen, Y., Liu, Y. & Dorn, G. W. 2nd. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ. Res. 109, 1327–1331 (2011).
-
Croston, T. L. et al. Functional deficiencies of subsarcolemmal mitochondria in the type 2 diabetic human heart. Am. J. Physiol. Heart Circ. Physiol. 307, H54–H65 (2014).
-
Montaigne, D. et al. Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation 130, 554–564 (2014).
-
Anderson, E. J. et al. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J. Am. Coll. Cardiol. 54, 1891–1898 (2009).
-
John, C. et al. Sex differences in cardiac mitochondria in the new zealand obese mouse. Front. Endocrinol. 9, 732 (2018).
-
Paradies, G., Petrosillo, G., Pistolese, M. & Ruggiero, F. M. The effect of reactive oxygen species generated from the mitochondrial electron transport chain on the cytochrome c oxidase activity and on the cardiolipin content in bovine heart submitochondrial particles. FEBS Lett. 466, 323–326 (2000).
-
Gnaiger, E. Mitochondrial pathways and respiratory control: an introduction to OXPHOS analysis. Bioenerg. Commun. 2020, 2–2 (2020).
-
Skladal, D., Sperl, W., Schranzhofer, R., Krismer, M. & Gnaiger, E. Preservation of mitochondrial functions in human skeletal muscle during storage in high energy preservation solution (HEPS). In: What is Controlling Life?. Mod. Trends BioThermoKinetics 3, 268–271 (1994).
-
Acin-Perez, R. et al. A novel approach to measure mitochondrial respiration in frozen biological samples. Embo J. 39, e104073 (2020).
-
Kuznetsov, A. V. et al. Cryopreservation of mitochondria and mitochondrial function in cardiac and skeletal muscle fibers. Anal. Biochem. 319, 296–303 (2003).
-
Yamaguchi, R. et al. Mitochondria frozen with trehalose retain a number of biological functions and preserve outer membrane integrity. Cell Death Differ. 14, 616–624 (2007).
-
Larsen, S., Wright-Paradis, C., Gnaiger, E., Helge, J. W. & Boushel, R. Cryopreservation of human skeletal muscle impairs mitochondrial function. Cryo Lett. 33, 170–176 (2012).
-
García-Roche, M. et al. Respiratory analysis of coupled mitochondria in cryopreserved liver biopsies. Redox Biol. 17, 207–212 (2018).
-
Barrientos, A., Fontanesi, F., & Díaz, F. Evaluation of the mitochondrial respiratory chain and oxidative phosphorylation system using polarography and spectrophotometric enzyme assays. Curr. Protoc. Hum. Genet. Chapter 19, Unit 19.13 (2009).
-
Mühleip, A. et al. Structural basis of mitochondrial membrane bending by the I–II–III2–IV2 supercomplex. Nature 615, 934–938 (2023).
-
Jang, D. H., Greenwood, J. C., Spyres, M. B. & Eckmann, D. M. Measurement of mitochondrial respiration and motility in acute care: sepsis, trauma, and poisoning. J. Intensive Care Med. 32, 86–94 (2017).
-
Zuccolotto-Dos-Reis, F. H. et al. Acetyl-CoA-driven respiration in frozen muscle contributes to the diagnosis of mitochondrial disease. Eur. J. Clin. Investig. 51, e13574 (2021).
-
Enríquez, J. A. Supramolecular organization of respiratory complexes. Annu. Rev. Physiol. 78, 533–561 (2016).
-
Lenaz, G. & Genova, M. L. Supramolecular organisation of the mitochondrial respiratory chain: a new challenge for the mechanism and control of oxidative phosphorylation. Adv. Exp. Med. Biol. 748, 107–144 (2012).
-
Novack, G. V., Galeano, P., Castaño, E. M. & Morelli, L. Mitochondrial supercomplexes: physiological organization and dysregulation in age-related neurodegenerative disorders. Front. Endocrinol.11, 600 (2020).
-
Wu, F. & Minteer, S. Krebs cycle metabolon: structural evidence of substrate channeling revealed by cross-linking and mass spectrometry. Angew. Chem. Int. Ed. 54, 1851–1854 (2015).
-
Bulutoglu, B., Garcia, K. E., Wu, F., Minteer, S. D. & Banta, S. Direct evidence for metabolon formation and substrate channeling in recombinant TCA cycle enzymes. ACS Chem. Biol. 11, 2847–2853 (2016).
-
Xu, X. et al. Effect of downregulated citrate synthase on oxidative phosphorylation signaling pathway in HEI-OC1 cells. Proteome Sci. 20, 14 (2022).
-
Eisenberg, T. et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat. Med. 22, 1428–1438 (2016).
-
Jedlička, J. et al. Impact of aging on mitochondrial respiration in various organs. Physiol. Res. 71, S227–s236 (2022).
-
Bowman, A. & Birch-Machin, M. A. Age-dependent decrease of mitochondrial complex II activity in human skin fibroblasts. J. Investig. Dermatol. 136, 912–919 (2016).
-
Kuznetsov, A. V. et al. The role of mitochondria in the mechanisms of cardiac ischemia-reperfusion injury. Antioxidants 8, 454 (2019).
-
Marin, W., Marin, D., Ao, X. & Liu, Y. Mitochondria as a therapeutic target for cardiac ischemia‑reperfusion injury (Review). Int. J. Mol. Med. 47, 485–499 (2021).
-
Leistner, M., Sommer, S., Kanofsky, P., Leyh, R. & Sommer, S.-P. Ischemia time impacts on respiratory chain functions and Ca2+-handling of cardiac subsarcolemmal mitochondria subjected to ischemia-reperfusion injury. J. Cardiothorac. Surg. 14, 92 (2019).
-
Duicu, O. M. et al. Ageing-induced decrease in cardiac mitochondrial function in healthy rats. Can. J. Physiol. Pharmacol. 91, 593–600 (2013).
-
Lemieux, H., Vazquez, E. J., Fujioka, H. & Hoppel, C. L. Decrease in mitochondrial function in rat cardiac permeabilized fibers correlates with the aging phenotype. J. Gerontol. A Biol. Sci. Med. Sci. 65, 1157–1164 (2010).
-
Boardman, N. T., Pedersen, T. M., Rossvoll, L., Hafstad, A. D. & Aasum, E. Diet-induced obese mouse hearts tolerate an acute high-fatty acid exposure that also increases ischemic tolerance. Am. J. Physiol. Heart Circ. Physiol. 319, H682–h693 (2020).
-
Guarini, G. et al. Impaired coronary metabolic dilation in the metabolic syndrome is linked to mitochondrial dysfunction and mitochondrial DNA damage. Basic Res. Cardiol. 111, 29 (2016).
-
Tompkins, A. J. et al. Mitochondrial dysfunction in cardiac ischemia–reperfusion injury: ROS from complex I, without inhibition. Biochim. Biophys. Acta Mol. Basis Dis. 1762, 223–231 (2006).
-
Kruse, S. E. et al. Mice with mitochondrial complex I deficiency develop a fatal encephalomyopathy. Cell Metab. 7, 312–320 (2008).
-
Quintana, A., Kruse, S. E., Kapur, R. P., Sanz, E. & Palmiter, R. D. Complex I deficiency due to loss of Ndufs4 in the brain results in progressive encephalopathy resembling Leigh syndrome. Proc. Natl. Acad. Sci. USA 107, 10996–11001 (2010).
-
Verma, S. K., Garikipati, V. N. S. & Kishore, R. Mitochondrial dysfunction and its impact on diabetic heart. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 1098–1105 (2017).
-
Acín-Pérez, R., Fernández-Silva, P., Peleato, M. L., Pérez-Martos, A. & Enriquez, J. A. Respiratory active mitochondrial supercomplexes. Mol. Cell 32, 529–539 (2008).
-
Schon, E. A. & Dencher, N. A. Heavy breathing: energy conversion by mitochondrial respiratory supercomplexes. Cell Metab. 9, 1–3 (2009).
-
Gnaiger, E. Complex II ambiguities—FADH2 in the electron transfer system. J. Biol. Chem. 300, 105470 (2024).
-
Wojtovich, A. P. & Brookes, P. S. The endogenous mitochondrial complex II inhibitor malonate regulates mitochondrial ATP-sensitive potassium channels: implications for ischemic preconditioning. Biochim. Biophys. Acta 1777, 882–889 (2008).
-
Doerrier, C. et al. High-resolution Fluorespirometry and OXPHOS protocols for human cells, permeabilized fibers from small biopsies of muscle, and isolated mitochondria. Methods Mol. Biol. 1782, 31–70 (2018).
-
Calvaruso, M. A. et al. Mitochondrial complex III stabilizes complex I in the absence of NDUFS4 to provide partial activity. Hum. Mol. Genet. 21, 115–120 (2011).
-
Gladka, M. M. et al. Cardiomyocytes stimulate angiogenesis after ischemic injury in a ZEB2-dependent manner. Nat. Commun. 12, 84 (2021).
-
Costanzini, A. et al. Mitochondrial mass assessment in a selected cell line under different metabolic conditions. Cells 8, 1454 (2019).
Acknowledgements
We appreciate the technical assistance from Soumi Ganguli, Haomiao Wang, Rijk de Jong, Daan Hanegraaf from Human and Animal Physiology, and Jelmer Vroom from the Laboratory of Virology. This work was supported in part by the Netherlands Research Council VENI talent grant (NO. 09150161910179), the starter and incentive grant of Wageningen University, and the Dutch Heart Foundation Dekker senior scientist grant (03-004-2024-0156) to D.Z. L.H. is a recipient of the China Scholarship Council (CSC) grant to be trained at Wageningen University (NO. 202008320323).
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Gisela Beutner and the other, anonymous, reviewers for their contribution to the peer review of this work. Primary Handling Editors: Shona Mookerjee and Dario Ummarino. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hu, L., van Rinsum, A., Caliandro, R. et al. A robust method for assessing mitochondrial function in healthy and diseased frozen cardiac tissue. Commun Biol 8, 1249 (2025). https://doi.org/10.1038/s42003-025-08608-5
-
Received:
-
Accepted:
-
Published:
-
DOI: https://doi.org/10.1038/s42003-025-08608-5