
Introduction
Phosphatidylinositols species (PIXPs), derived by phosphoinositol (PI) phosphorylation at inositol position 3, 4, or 5 act as molecular adaptors, metabolic intermediates, or establish gradients in cargo transport. The interconverting kinases and phosphatases differ in specificity and tissue specific expression. Their unique subcellular distributions are regulated by a complex system of localization specific transporters1. PIP3 and its precursor PI(4,5)P2 have historically received the most attention due to their role in rapid signaling through Phospholipase C and class I PI3-kinase isoforms. Beyond rapid signaling, PI(4,5)P2 regulates talin and vinculin in focal adhesion sites (FA)2, is a cofactor in FA disassembly3and is critical for the determination of epithelial cell characteristics4. A distinct role in FA disassembly falls to the local synthesis of PI(3,4)P2 from PI4P, primarily by the class II kinase, PI3KC2B5 and the balance of PI(4,5)P2 versus PI(3,4)P2 regulates degradative versus recycling vesicular routing6,7. PI4P is the shared precursor of both PIP2 species and a protein binding tag in its own rights8. Multiple kinases contribute to PI4P synthesis. PI4KB (PI4Kb, PI4KIIIb) is the main source for the largest, cis-Golgi localized pool of PI4P9. Smaller, transient, but critical pools are generated by PI4KA (PI4Ka, PI4KIIIa) and PI4K2A (PI4KIIa) at varying locations, including the plasma membrane pool for PI(4,5)P2 synthesis10,11,12. The quantitative dominance of the cis-Golgi pool reflects the magnitude of its involvement in vesicular flow and secretion, including that of insulin13. It also plays a crucial role in intracellular transport, such as counter current cargo for cholesterol. Reverse transport of PI4P to the ER and hydrolysis by SAC1 phosphatase creates a chemical gradient with donor membranes14 that drives indirect active transport for cholesterol8,15. Inhibition of the responsible transporters, oxysterol binding proteins (OSBPs), results in a collapse of membrane fluidity control15. The associated PI4P dysregulation induces Golgi fragmentation and stress-induced cell death16. Thus, the overwhelming majority of cellular PI4P plays critical roles that are integral to complex metabolic equilibria but not rapid signaling events.
For PI(4,5)P2 and PIP3, and to a lesser extent PI4P, real time sensors based on FRET, BRET, or related principles such as dimerization dependent fluorescent proteins (ddFPs). As explored by our ddRFP based comparison sensor for PI4P, the draw-back of this class of rapid response sensors is the dependency on higher levels of sensor components due to the transient nature of signal-generating interactions. Thus, they are frequently deployed by transient overexpression and are difficult to apply to slower metabolic equilibria and drug or si/shRNA actions, or PIXP measurements in proliferation, migration, or invasion studies. More importantly, the required high expression levels sequester less abundant or locally limited PIXP species and interfere with cellular function, such as cell adhesion or vesicular transport. Alternatively, targeted fluorescent markers, such as mCherry-PH(FAPP1)17 or P4M(SidM)-GFP18, indirectly report on level changes by their relative degree of focused versus diffuse localization. However, beyond technically challenging quantification, the marker requires a narrowly defined expression range with just the right amount of excess free probe. Direct profiling of PIXPs by mass spectrometry19,20 is not widely accessible and the assessment of different PIXP species, especially isomers like PI3P, PI4P, PI5P or PI(4,5)P2 vs. PI(3,4)P2 or PI(3,5)P2, remains extremely challenging.
For PI4P, a large pool is critical to slow changing, non-signaling related functions while smaller and localized pools contribute critical signaling and adaptor functions. This places an emphasis on a robust signal from sensors that are closer to being biologically neutral and easily integrated into conventional assays. Various PIXPs are the target of novel inhibitors, but the determination of their specificities has largely been limited to cell free systems. The drug impact on complex PIXP metabolism in a cellular setting remains often elusive in the absence of robust and versatile readouts. For PI4P, we envisioned a biosensor for use on a single-cell and population scale and readout modalities available to most molecular biology laboratories. Since subcellular localization and organelle dynamics are key aspects, it should provide robust spatial assessment with minimal background and avoid technically more challenging readouts such as cell-based FRET. Most importantly, we wanted a sensor that provides continuous signal under conditions of low stable expression that approximate biological neutrality.
PIXP’s have been targeted through selective PH, FYVE or PX domains21. Of the over 200 Pleckstrin Homology (PH) domain containing proteins17 only 10% show high PIXP species affinity and selectivity22. Several studies23,24,25 identified PH-domains with sufficient selectivity for localization markers18,26 or FRET/BRET-based sensors10,27 that exploit PIXP-dependent local sensor crowding. BRET sensors stand out through high sensitivity suitable even for low abundance PIXP species, such as PI(3,5)P228, but are more difficult to use for quantification in a population of live cells. Compared to FRET or BRET methods, the readout of a PI(4,5)P2 sensor based on dimerization-dependent red fluorescent protein (ddRFP)29 is more direct. However, as a rapid response sensor based on transient interactions, it shares the need for higher concentrations of sensor components. To establish a comparable PI4P sensor as a refence point, we used the selectivity of the PH-domain of Golgi-resident FAPP1 23,24. We compare this ddRFP(PI4P) sensor to a conceptually different split GFP based sensor (sGFP(PI4P)). This sensor design is not expected to compete with existing modalities for single-cell, rapid response measurements. It is aimed instead at the study of biological processes on longer time scales, such as proliferation, migration, metabolic adaptation, or prolonged drug exposure. Ideally, this should be possible with stable expression of sensors at low levels to maximize biological neutrality.
Split fluorescent proteins (sFP) are well established in macro molecular interaction studies, combine high sensitivity and specificity in live-cell settings, are FACS-compatible, and expandable to multiplexed readouts30. However, their suitability as metabolic sensors, complicated by the fact that PIXPs are not merely metabolites, remains unknown. The unique thermodynamics of complementation and fluorophore maturation create readout dynamics that are fundamentally different from sensors based on proximity (e.g. FRET, BRET) or weak transient interactions (ddRFP). For the best studied sFP(1–10/11) design, the complementation of the 11th strand into strands 1–10 of the GFP b-barrel is energetically very unfavorable for soluble fragments. Refolding and association studies show that either joint refolding, thermal relaxation, or high energy photoconversion are needed to facilitate rapid reassembly in solution of fragments derived from nascent or previously matured FPs31. Otherwise, components need close proximity for extended time periods, and consequently the background signal from spontaneous complementation is extremely low. Combined with the need for fluorophore maturation, this establishes a very specific but slow ON-switch. Likewise, the high stability of the complemented FP, with a KD in the pico-molar range31, constitutes a very slow OFF-switch that is effectively driven by sFP turnover. Hence, PIXP mediated complementation should accumulate sFP signal in a cellular setting at a steady state level determined by FP turnover and PIXP abundance. One would therefore expect significantly slower response dynamics but more sensitivity compared to sensors dependent on transient interactions. Steady state signal levels should retain a correlation to PIXP levels, even at lower overall expression levels of sensor components, thus providing more sensor neutrality. In addition, we explored whether the nature of the sGFP signal can meet an urgent need for readout options that are more readily available to most molecular biology laboratories and experimental settings.
Results
The sGFP(PI4P) sensor is functional and specific
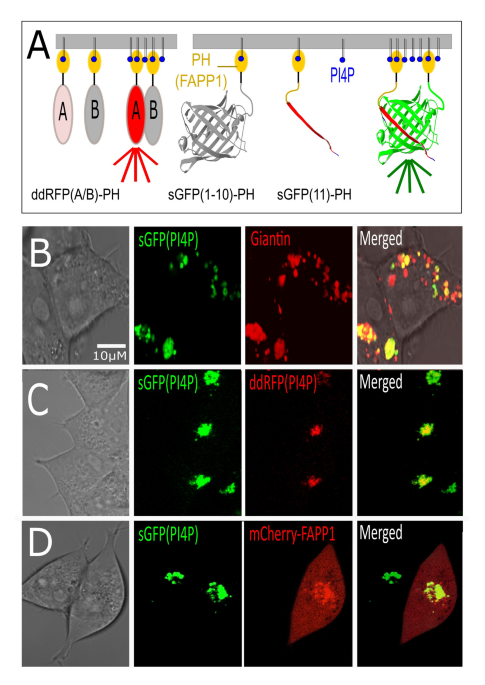
With a ddRFP and an sGFP-based sensor we generated two complementary sensor designs that are targeted by the PH domain of FAPP1. Since both sensors already involve transient or stable dimerization, each individual construct carries only a single PH domain (Fig. 1A). The ddRFP(PI4P) design is based on the existing sensor for PI(4,5)P229. Dimerization dependent RFP (ddRFP) is derived by interface mutagenesis-derived from dTomato with reduced affinity (KD 33µM) and 10-fold lower fluorescence when dissociated32. In sGFP(PI4P), the FAPP1-PH domains are placed at the N-termini of both fragments of split GFP b-strands 1–10 and 11 respectively. The (1–10,11) sGFP design is a derivative of super-folder GFP, and analogous split variants exist of BFP, CFP, and YFP. A comparison with the Golgi marker ScarlettGiantin (Fig. 1B) shows that the resulting signal for sGFP(PI4P) falls within portion of the areas that are marked by Giantin. This is consistent with the synthesis of the primary PI4P pool in the early/cis Golgi by PI4KB9close to ER/Golgi contact sites involved in cholesterol transport. The ddRFP(PI4P) and sGFP(PI4P) sensors colocalize, consistent with their shared targeting by the FAPP1-PH domain (Fig. 1C). A comparison of sGFP(PI4P) signal with a direct targeting of mCherry by the same FAPP1 module17 (Fig. 1D) not only confirms colocalization but highlights basic features of marker-based PIXP assessments. In the absence of alternatives for PIXP quantification, this widely used “fallback” method quantifies the shift in localization of signal from excess cytoplasmic probe towards membrane localized signal. By necessity, this requires an excess of “untargeted” cytoplasm localized probe.

Design and localization of sGFP(PI4P). A Design of sGFP based PI4P sensor and its ddRFP based counterpart. The ddRFP design is based on a previous PI(4,5)P2 rapid response sensor. Selective targeting of PI4P uses the PH domain of FAPP1, and signal creation depends on elevated local PI4P concentrations inducing either transient (ddRFP) or stable (sGFP) sensor dimers. B The localization of the sGFP(PI4P) sensor falls within the compartments identified by the Golgi marker Scarlett-Giantin. C In HEK293 cells that stably carry sGFP(PI4P), double transfected cells colocalize ddRFP(PI4P). D As a classic localization probe for PI4P, mCherry-FAPP1 colocalizes with stably expressed sGFP(PI4P) signal that is generated on target. The mCherry-FAPP1 displays the expected background signal from excess untargeted probe. Single-cell quantification of relative changes in localized versus diffuse probe signal is one of the currently employed methods of assessing changes in PI4P levels in the absence of suitable sensors.
The ddRFP sensor is more dynamic but the sGFP based sensor generates a significantly stronger signal that facilitates its use at low expression levels
The different underlying principles of the ddRFP and sGFP sensor would predict a more dynamic ddRFP signal at the expense of signal intensity. To evaluate “rapid” response capability versus signal intensity, we exploited the SAC1-driven hydrolysis of PI4P in conjunction with its use as counter current cargo in intracellular cholesterol transport. The inhibition of oxysterol binding protein (OSBP) by OSW-1 does not only cause a rapid breakdown of cholesterol homeostasis, but also an accumulation of PI4P in the Golgi as shown by the convergence of diffuse PI4P marker signal and associated increase of the total cellular PIP species by mass-spectrometry within an hour15. We therefore tracked individual cells for their relative change in ddRFP and sGFP sensor signal (Fig. 2A). After transient transfection with either sensor, we merged and normalized multiple single-cell time lapse measurements to address cell-to-cell variability. As expected, the ddRFP(PI4P) sensor shows a signal increase at the anticipated time scale while the sGFP(PI4P) sensor did not respond over the same time period. This lag in response is expected due to the requirement for fluorophore maturation of de-novo associated fragments of sGFP. In vitro studies have shown that assembled and matured sGFP can be disrupted, yielding pieces with very rapid signal response, but this disruption requires aggressive conditions31. The lack of a fast response that we observe confirms that despite a base level of PI4P mediated complementation, no significant pool of fragments derived by dissociation from previously matured sGFP exists in a biological setting. Besides technical limitations for single-cell tracking by microscopy, the duration of studies is also limited by a negative impact on cell adhesion, especially at the high OSW-1 concentrations needed to induce relatively rapid PI4P increases. In addition, scoring relative changes in normalized signal fails to properly reflect differences in signal intensity between both sensors in steady state. To quantify this difference, we used transient cotransfection conditions that establish equal protein levels of HA-epitope tagged species of both sensors (Fig. 2B). To compare signal intensity directly (Fig. 2C), data acquisition parameters were normalized using GFP and mRuby. The latter has matching fluorescence properties to dTomato, the parent construct of ddRFP. Fluorescence intensities were quantified for a larger population of cells to account for single cell variability (Fig. 2D). The median of the distribution of intensity ratios shows a 56-fold higher intensity for sGFP(PI4P) (Fig. 2E). While the lower intensity from ddRFP(PI4P) is expected for a real-time signal that is driven by transient dimerization, this has profound consequences for use at lower sensor expression levels that would be needed to improve biological neutrality. In order to demonstrate the limitations on biological neutrality that result from the use of either sensor at high, transient expression levels we transfected cells with sGFP(PI4P) or ddRFP(PI4P) and mCherry as a control and tracked proliferation and the fraction of non-adherent viable cells (Fig. 2F). The transient overexpression of both PH-domain guided sensors has pronounced negative impacts on proliferation and adhesion. The more pronounced impact of sGFP(PI4P) at these high expression levels is likely a reflection of the accumulation of complemented sensor with its dual PH-domain modules that provide more favorable targeting to PI4P-rich clusters. The selection of stable lines for both sensors strongly favors minimal sensor expression. For the resulting sGFP(PI4P) carrying line, the remaining signal was robust over multiple passages and the impairment in proliferation was minimal while no impact on cell adhesion was detected. For the corresponding ddRFP(PI4P) carrying lines, stable lines showed no or very marginal signal by microscopy, despite repeated attempts of enrichment by FACS. A stable selected line carrying low levels of sGFP(PI4P) was therefore used for all subsequent studies.

The sGFP(PI4P) sensor lacks the rapid response capability of its ddRFP(PI4P) counterpart, but shows superior sensitivity. A Time course for the response of HEK293 cells transfected transiently with ddRFP(PI4P) or sGFP(PI4P) following stimulation with 500nM OSW-1. The relative changes for three individual time courses (3–4 cells per observation window and time course) were merged. Within the two hours of observation, overexpressed ddRFP(PI4P) shows a clear response while no clear signal change emerges from sGFP(PI4P) at this time scale, as expected due to folding and fluorophore maturation delay. B Design of HA-tagged sensor constructs. Under the selected cotransfection conditions, both sensors are expressed at comparable levels on a population scale. Traces of HA-signal with an apparent molecular weight close to 100 kDa (*) are found also in HA-ddRFP(PI4P) only transfections and represent traces of fusion proteins with T2A read-through. C) At matched expression levels, sGFP(PI4P) shows a significantly stronger signal at normalized fluorescence detection conditions. D + E A quantitative assessment of a larger double transfected cell population shows that, even under conditions of overexpression, the sGFP(PI4P) sensor shows an approximately 60x higher signal. F At the high levels of transient expression used for the assessment of relative signal intensity in C-E, both sensors impact proliferation and cell adhesion negatively compared to a control transfection with mCherry. The relative count of viable attached and detached cells was measured by alamarBlue at day four post transfection. The combined signal from attached and detached cells was normalized for the mCherry control.
Stable expression of the sGFP(PI4P) sensor provides a direct readout for PI4P accumulation by FACS and microscopy while retaining spatial information with low background signal
The titration of OSW-1 in a 24-hour treatment allows for sufficient time for sGFP sensor signal to reach an equilibrium that shows an increase in OSW-1 dependent signal intensity (Fig. 3A) albeit with varying degrees of drug induced changes in cell morphology and adhesion. For a direct quantitation of the dose response on a population scale, we measured the cellular sGFP signal by FACS. Standard cell-count versus GFP data were normalized for total cell-count across measurements and superimposed to demonstrate the dose dependent curve shift (Fig. 3B). At the same time, the sGFP signal with its on-site fluorophore synthesis and lack of excess untargeted GFP signal provides a localization signal with minimal background compared to classic markers (Fig. 1D). Confocal imaging therefore provides an assessment of the immediate response to OSW-1 with regard to signal localization when signal intensity does not yet respond. Within the time window in which the overexpressed ddRFP sensor reports quantitative increases (Fig. 2A), the stably expressed sGFP(PI4P) sensor documents the profound impact of OSW-1 on Golgi morphology (Fig. 3C). Previous studies had shown this disorganized or fragmented structure of the Golgi when cells were transfected with inactive PI4KB (D656A)33 or Golgi directed SAC1 phosphatase34when OSBP35 was targeted, or in response to OSW-1 treatment in Neuro2a cells36.

Stably expressed at low levels, the sGFP(PI4P) sensor combines a sensitive readout for OSW-1 dose response by FACS combined with high resolution spatial information. A Low magnification image of a population of HEK293 cells that stably express low levels of sGFP(PI4P) after exposure to various concentration of OSW-1 for 24 h. B The stability and intensity of the sGFP signal facilitates quantitative FACS analysis to obtain an OSW-1 dose response. To allow for a proper evaluation of shifts of the cell population based on GFP signal, standard plots that show absolute cell (event) numbers versus GFP signal were normalized (area under curve) to accommodate differences in total cell number before plot merger. The color coded OSW-1 concentrations are indicated C At an early time point, the average sGFP(PI4P) signal across a cell population does not provide a quantitative readout of PI4P increases (compare Fig. 2), but a confocal cross section view shows the onset of Golgi fragmentation. Classic markers (see mCherry-FAPP1 in Fig. 1D) show significant untargeted signal from excess, untargeted probe unless ideal expression levels are achieved. By comparison, the on-site sGFP signal creation for sGFP(PI4P) provides robust signal without significant untargeted GFP background.
The signal provided by the sGFP sensor is accessible through the analysis of cell lysate
Next, we wanted to explore the limits of fluorophore stability to expand the range of available signal readouts. Studies on the reassociation behavior of fragments obtained from previously assembled and matured sGFP demonstrated a need for very aggressive dissociation conditions37,38. We therefore tested how the sGFP derived signal would withstand different cell lysis conditions (Fig. 4A). As a benchmark, lysis in 6 M Guanidinium hydrochloride yields a baseline fluorescence that is the same with or without heat treatment. By contrast, lysis under various conditions showed high and comparable recovery of signal. Of those conditions, 8 M Urea provided the lysate that was the most amenable to processing. In all cases, heat treatment provides a measure of lysate background fluorescence. Using this approach, we reevaluated the response to 24 h of OSW-1 treatment (Fig. 4B). Triplicate measurements of sGFP signal in cell lysate are consistent with the data obtained by FACS analysis.

The sGFP(PI4P) sensor is compatible with quantification after cell lysis and provides insights into the dynamics of targeted inhibition of PI4P synthesis. (A) Comparison of the sGFP signal in cell lysate under different lysis conditions. The extent of unrelated fluorescence in the untreated cell lysate (green) is accessible through sGFP disruption by heat treatment (5 min (orange) or 10 min (red), 95Co). (B) Assessment of the OSW-1 dose response using a cell lysis protocol is consistent with visual observation (Fig. 3A) and FACS data (Fig. 3B) with p-values below 0.05 and 0.005 indicated with * and ** respectively. (C) Direct assessment of the impact of two alternative inhibitors of PI4P synthesis on HEK293 cell proliferation by alamarBlue viability assay. IN10 and PI273 target PI4KB and PI4K2A respectively. D + E: The combined assessment of protein content (D) and protein-normalized sGFP signal (E) shows a differential impact of IN10 (PI4KB) and PI273 (PI4K2A). The protein measurements of lysates mirror alamarBlue consistent with a primarily cytostatic impact of both drugs. When normalized for protein, IN10 and PI273 show an opposing impact on total PI4P levels with elevated levels of PI273 eliciting an unanticipated accumulation of PI4P. F) Microscopic analysis of representative cells prior to lysis at 10µM of either drug shows that the signal intensity increase after PI273 treatment occurs without significant changes in cellular distribution, suggesting PI4P accumulation primarily in the Golgi.
Having confirmed the functionality of cell lysate-based evaluation, we expanded our analysis to two pharmacological inhibitors of PI4P synthesis, IN10 and PI273, to demonstrate how the stably expressed sensor can advance the study of PI4P metabolism. Using these inhibitors highlights a very fundamental question in PI4P metabolism that is difficult to approach without a suitable sensor and underscores current challenges in drug development against PIXP kinases. While various isoforms of class I PI3-kinases have attracted the most attention, other PIXP kinases are increasingly being targeted. However, with close similarities between substrates and multiple alternative kinases and isoforms for almost all relevant conversion steps, the assessment of specificity is often limited to cell free assays. The cellular levels of specific PIXP species are the consequence of complex equilibria and compartmentalization in ways that are currently largely inaccessible for broader drug evaluation. In the case of PI4P, four different enzymes contribute to synthesis, PI4KA and PI4KB (historically also labeled as class III) and two class II kinases (PI4K2A and PI4K2B)39. Most of the cellular PI4P is synthesized by PI4KB in the cis Golgi9. The synthesis of the much smaller but signaling relevant plasma membrane pool of PI4P has been assigned to PI4KA and PI4K2A. However, the relative contributions of both PM enzymes remain a focus of investigation10,11,12.
Based on studies in cell free systems, two of the more selective PI4K synthesis inhibitors are PI4KIIIb-IN10 (IN10) against PI4KB and PI273 against PI4K2A. In addition to its impact on various vesicular trafficking events, PI273 was reported to induce apoptosis in the ER-positive MCF7 breast cancer cell line40 with an IC50 of 0.5µM. (27). IN10 inhibits PI4KB in cell free systems with a KD of 3.7nM41 and was the result of structure based drug design to minimize cross reactivity of earlier compounds, especially class I and III PI3Ks. In an alamarBlue assay of cell viability and proliferation, PI273 shows a more pronounced impact at lower concentrations (Fig. 4C). When stable sGFP(PI4P) transfected cells were treated with both drugs and instead evaluated using the urea lysis protocol, the total protein content is in good agreement with alamarBlue data (Fig. 4D) consistent with a primarily cytostatic impact of both drugs under the tested conditions. In addition to a surrogate proliferation readout when mechanistically applicable, the protein content allows for the normalization of fluorescent data for cell number (Fig. 4E). For IN10, the normalized sGFP signal shows a gradual decrease. The required concentration to significantly deplete the cellular PI4P pool in a three-day assay is higher than expected from the cell free system data and finally reaches 50% at 10µM, possibly reflecting drug stability or compensatory responses in PI4P utilization. The overall PI4P response correlates with the IN10 impact on proliferation. By contrast, at dosages when it already impacts proliferation, PI273 does not have a noticeable impact on the larger PI4P pool. This proliferation impact is consistent with the anticipated inhibition of its primary target, plasma membrane localized PI4K2A, and hence signaling relevant PI(4,5)P2 production. A marked increase in impact on viability occurs for higher concentrations, but this differs qualitatively from the PI4P signal which shows an increase. To evaluate whether this increase is likely to represent mainly Golgi localized PI4P, we acquired higher resolution images of select cells that were representative of the population average used in the lysis assay (Fig. 4E). The elevated signal displays after PI273 treatment shows the punctate signal characteristic of Golgi localized PI4P in previous assessments (Figs. 1, 2 and 3) as opposed to significant changes in both intensity and cellular distribution. This suggests that the accumulation does in fact reflect an increase in the Golgi pool.
Discussion
When studying the impact of metabolic reactions that intersect with PI4P, very divergent scales are at play. While smaller pools of PI4P at locations such as the plasma membrane may undergo relatively fast changes, the primary pool of PI4P at the Golgi is far larger and not subject to changes on a signaling time scale. Instead, it is closely integrated into broader metabolic regulation, such as the transport and homeostasis of cholesterol that changes slowly but consumes large quantities of PI4P. Existing sensor modalities are not suited to be applied to drug screens where the consequences are time delayed or for extended longitudinal studies of proliferation, migration or similar biological events. Likewise, batch level readouts such as lipid extraction followed by mass spectrometry or ELISA are technically an option but beyond issues of scale, they are very involved and limited in their implementation for broader studies beyond few select time points.
Split fluorescent proteins are well established for macro-molecular interaction studies. The simple direct readout and the excellent signal to noise ratio that results from the high activation barrier of complementation make sFPs ideal for this application. We demonstrate that in its implementation as a simple PI4P location marker, the obtained signal mirrors that of established PH-domain driven probes, such as mCherry-FAPP1, but without being limited to a narrow window of expression between low probe signal and high background from untargeted probe. The dominant signal from sGFP(PI4P) locates to a subsection of the Giantin-marked Golgi, consistent with primary production in the cis-Golgi. The signal obtained from other cell compartments is very low. This may reflect both low and transient PI4P levels, especially at the plasma membrane. However, we cannot exclude the possibility that the FAPP1-PH domain, while showing high specificity, also introduces location bias. The P4M domain of SidM from Legionella pneumophila was reported to improve identification of PI4P beyond the Golgi, although the Golgi pool remains by far dominant18. A systematic analysis of signal dynamics versus potential location bias may further explore this issue but goes beyond the initial assessment of metabolic sFP sensors.
Rapid changes of small pools of PI4P in select subcellular locations may be best accessed in single cell microscopy studies that measure relative changes of transient interaction based sensors such as ddRFP, FRET or BRET readouts. In simple technical terms, any single cell based measurement of relative changes in sensor signal or intracellular probe localization may be acceptable for most rapid signaling oriented studies but faces severe limitations for extended longitudinal studies. Beyond this simple but very real practical limitation in tracking PIXP level changes, any expression of PH-domain guided sensors at high levels significantly compromise biological neutrality, which. Our sGFP(PI4P) sensor is not exempt from this limitation. The most important and distinguishing factor is the significantly lower level of sensor expression needed to obtain a robust readout. Besides maturation related delays, it is especially the slow response to PI4P decreases that is limited by sensor turn-over that drives slow sensor dynamics. However, the resulting signal accumulation remains proportional to complementation-driving PI4P levels, and this translates into a steady-state signal enhancement. For high levels of overexpression at which the matching ddRFP(PI4P) sensor is accessible for analysis, we measured a median 54-fold intensity advantage of sGFP(PI4P). At these high expression levels, neither sensor is close to biological neutrality. However, this intrinsic signal enhancement for sGFP(PI4P) allows for the creation of stable cell lines with low sensor levels and robust signal. It is this feature that fundamentally changes the kind of biological questions that can be addressed. Despite, or in fact because of its slow response dynamics, the sGFP(PI4P) sensor design therefore meets a critical need.
Beyond robust signal in a low stable expression setting, sGFP(PI4P) provides several complementary readout options that are easy to implement in most laboratory settings. FACS sorting is facilitated by the slow changes in sGFP(PI4P) signal that shield the readout from the sensitivity of cellular PI4P to the cell adhesion state that would impact any rapid response readout (Fig. 3A). Even when ddRFP is transiently overexpressed to obtain sufficient signal, a real-time readout of the PI4P signal is difficult to preserve during cell processing for FACS, yielding poor quantitative reproducibility (negative data on high variability not shown). By contrast, the intrinsic lag time of the sGFP(PI4P) sensor makes it immune to most changes induced by processing of samples. Unless interventions that cause PI4P accumulations are allowed for extended time periods (such as the extended treatment with high concentrations of PI273), we observed very little freely diffusible sGFP(PI4P) signal, compared to the default scenario for conventional PH-domain-guided probes (Fig. 1D). This greatly facilitates high-resolution imaging (Fig. 3C). Assay compatibility is further expanded by the fact that the sGFP(PI4P) signal is retained after cell lysis. This approach allows for the processing of larger data sets while protein determination of the same lysate provides for signal normalization.
In order to apply the sGFP(PI4P) sensor to a problem set that is technically challenging for existing methods, we evaluated two alternative inhibitors of PI4P synthesis in the context of standard three-day proliferation assays, across a concentration range, in triplicate (Fig. 4D + E). The difficulty associated with such a standard drug evaluation has been one of the most striking deficiencies that prompted the design of our sensor. With different PI4-kinases contributing to distinct cellular pools, and PI4P also impacting vesicular flow, a direct assessment of the impact on the total cellular pool of PI4P is essential. For IN10, the dose response curve for proliferation and PI4P levels largely coincide. The dominant sGFP(PI4P) signal remains in the Golgi, and the observed response deviates from expectations only in the required concentrations in a three-day proliferation assay. For PI273, the data interpretation is more complex. At lower concentrations its impact on proliferation is consistent with the reported induction of apoptosis and disruption of PIP3 signaling in MCF7 breast cancer cells. However, this inhibition had been attributed to the depletion of the total cellular PI4P pool40. A systemic PI4P depletion would be inconsistent with the dominant role of PI4KB in cellular PI4P synthesis. Indeed, we observe no significant decrease of total PI4P. In addition, accumulation of PI4P at higher concentrations of PI273 points towards a secondary impact on the utilization of Golgi PI4P. The current data speak also to the degree of compartmentalization of the cellular PI4P pools and their contribution to PI(4,5)P2 synthesis. Studies relying on the depletion of TGN PI4P through recruitment of SAC1 from the ER show a drop in the recovery of PI(4,5)P2 after receptor activation42. This favored of a model in which the PM pool of PI4P is supported by the larger Golgi pool. Studies using multiple redirected phosphatases concluded an observable but more limited contribution of the Golgi pool43. Our direct observation of total PI4P suggests that the Golgi pool cannot rescue the PI273 induced sufficiency that manifests in its impact on proliferation at lower concentrations. The fact that at higher concentrations of PI273 the Golgi pool of PI4P does in fact accumulate points towards an unanticipated secondary impact on Golgi PI4P utilization, possibly through inhibition of its PM directed flow. This crosstalk cannot be derived from the determination of drug specificity in cell free systems and underscores the need for an easy to deploy and robust sensor of PI4P levels that can be integrated in standard dose response and proliferation assays.
With respect to the general assessment of split fluorescent protein sensors for the detection of metabolites, this study provides an early step in the general exploration of this sensor type. Sensor selectivity is linked to the guiding PH domain and as such can be redirected or improved along with these modules. For PI4P this could involve the evaluation of a SidM-derived domain instead of FAPP1 18. Targeted evolution of sGFP complementation has recently resulted in sGFP mutants with significantly improved complementation kinetics which could improve the response dynamics for PI4P increases44. Studies aimed at destabilizing full size GFP resulted in species with faster turnover45. In its current form, the balance of dynamics, sensitivity, and stability creates a sensor type with unique properties that complement existing rapid response sensors in ways that open up a wider range of biological studies, with readout options that are readily accessible to most laboratories, and while minimizing interference with cellular behavior.
Materials and methods
Cell culture and maintenance
HEK293 (ATCC) were maintained at 37 °C in DMEM (with 4.5% g/L glucose, L-glutamine, sodium pyruvate) (Corning) supplemented with 10% fetal bovine serum (FBS) (SeraPrime) and 1% Antibiotics-Antimycotics (Gibco) in a humidified atmosphere at 5% CO2 unless indicated differently. Cells were maintained at medium confluence and treated with OSW-1 (Cayman Chemical Company 30310), IN10 (PI4KIIIbeta-IN10; MedChemExpress HY-100198) or PI273 (PI4KIIα inhibitor; MedChemExpress HY-103489) at the indicated concentrations.
Plasmids and creation of stable lines
Transfections were performed according to manufacturers’ instructions (Lipofectamine 2000, ThermoFisher). Constructs pPHT-PIP2 (plasmid #6093746,, pmScarlet3-Giantin_C1 (plasmid #18977347, and Tet-pLKO-Puro (plasmid #2191548, were obtained from Addgene and source preferred references are added. Two synthetically created FAPP1 PH domains were fused to the respective N or C-termini of ddRFP or the 1–10 and 11 fragments of sGFP with spacers. The individual PH-domain boundaries were MEGVLYKW … KASLTDTRT. For the comparison of sensor expression levels in double transfection studies, an HA-epitope tag was added as indicated in Fig. 2B. A stable cell line with sGFP(PI4P) was generated by clonal selection and expansion of GFP positive cells after transfection followed by FACS enrichment for robust GFP signal once expression levels had stabilized. The resulting cells were used to create frozen stocks and displayed a stable sGFP signal over multiple passages. Cells used in experiments were refreshed from frozen stock after approximately ten passages or when signal drift and significant loss of uniformity was observed. Transfection with the corresponding ddRFP(PI4P) construct yielded stable lines but consistently resulted in extremely low fluorescence signal despite clonal selection of cells and multiple attempts to enrich for cells that displayed marginal signal above background in FACS.
AlamarBlue assays
Freshly transfected cells carrying either ddRFP or sGFP sensor and untargeted mCherry as control were trypsinized, recounted and seeded in triplicate at 100,000 cells/well in a 6 well plate format. On day two, transfection efficiency was confirmed by fluorescence to be > 95%. On day four, plates were subjected to 15 min of agitation on a platform shaker to dislodge loosely attached cells. The supernatant with detached cells was transferred to a new plate and attached cells received fresh media. Both samples were subsequently incubated with 1/10 volume of alamarBlue reagent for 3 h in the cell culture incubator to determine the relative viable cell count. Dye conversion was measured by fluorescence at 560/590 nm (excitation/emission).
FACS
After treatments as indicated, cells were harvested in 1X L-15 (1mM EDTA) for analysis on the Becton Dickinson FACS-Aria Fusion equipped with 4 lasers, 2 light scatter parameters, housed in an integrated biosafety cabinet. The cells were identified using forward and side scatter parameters. Then the GFP signal was visualized by the blue laser excitation wavelength (488 nm) and the 530/30 emission wavelength filter. For a better graphic representation of shifts in the population when data form multiple experiments were superimposed, data for the cell count versus GFP fluorescence were normalized for total cell number (area under curve).
Image analysis
For the analysis of transiently transfected cells, cells were seeded into specialized µ-slide 8 well tissue culture treated polymer coverslips (ibiTreat, FisherScientific, 50305795), 6 h post transfection. Stably sensor expressing cells were seeded accordingly and confocal images of cells were acquired at least 24 h post seeding on a Nikon A1R scanning laser confocal microscope with a 60X oil immersion objective (N.A. 1.4) at an acquisition rate of 2 s per frame.
Quantification by microscopy
For the quantification of fluorescence intensity by standard fluorescence microscopy and statistical processing, multi-fluorescence image acquisition was obtained on a Zeiss Axiovert 200 M under brightfield and fluorescent conditions using MicroManager data acquisition software49,50. To ensure that the excitation/emission conditions allowed for comparable detection of red and green fluorescence, we used a mRuby/EGFP double expression setup since mRUBY has comparable excitation/emission and fluorescence intensity characteristics to dTomato, the parent construct of ddRFP. Data analysis was carried out immediately on settled cells within 30 min of trypsinization as longer delays resulted in a drift for ddRFP sensor signal. Objects were identified in a brightfield setting using the automated mask creation and watershed filter processing options for object separation in Image J, Fiji51. Identified objects were quantified for their red and green fluorescent signal with each object having its immediately adjacent area set as background. Statistics were carried out on several hundred fluorescent cells per assessment.
Spectrophotometry
The sGFP signal in was accessible by spectroscopy by two means. For direct non-destructive assessment, cells seeded in 96 well cell culture dishes and fluorescence were measured on a CLARIOStar microplate reader at standard GFP excitation/emission (470–15 nm – 515–20 nm) setting with 6 mm orbital well scan and 32 flashes per well. For the assessment of protein-normalized sGFP signal in lysate, cells were seeded in 6 well plates in triplicate. Media was aspirated with a 26G needle and cells were lysed with 400 µl 8 M Urea. The lysate was cleared by centrifugation at 18,000 x g for 5 min and 200 µl were transferred to 96 well plates for direct assessment of sGFP signal and 100 µl were diluted with 3 volumes of 6 M GuaHCl, heated for 10 min at 95Co and 200 µl were used to calculate the control for heat treated lysate fluorescent background. The corresponding protein concentration was measured by adding 5 µl of cleared Urea lysate to 195ul of Coomassie Plus Protein assay reagent (ThermoScientific) and measured at 600 nm absorbance. Other lysis buffers for the assessment of lysis conditions in Fig. 4a were: 6 M Guanidinium hydrochloride, 1% Triton X-100, 1% SDS, MLB (20mM Tris, 137mM NaCl, 1% v/v Triton X-100, 10% v/v glycerol, 5mM EDTA, 1mM Sodium Orthovanadate).
Cell lysate and immunoblot analysis
Cells were harvested for immunoblot analysis in 1X SDS PAGE loading buffer (3X buffer = 171.4 mM Tris HCl, 30% v/v glycerol, 346.8 mM SDS, 746.3 mM Bromophenol blue, 324.1 mM DTT) and immediately heated at 95 °C for 15 min followed by SDS PAGE and immunoblotting. HA-tag (Rb) antibody (1:1000, Cell signaling technology) was used overnight at 4Co.
Data availability
This study does not contain data sets that require processing of the graphically presented results beyond data normalization (min/max values) and averaging. Raw data are available upon request from the corresponding author.
References
-
Lipp, N. F., Ikhlef, S., Milanini, J. & Drin, G. Lipid exchangers: cellular functions and mechanistic links with phosphoinositide metabolism. Front. Cell. Dev. Biol. 8, 663. https://doi.org/10.3389/fcell.2020.00663 (2020).
-
Di Paolo, G. et al. Recruitment and regulation of phosphatidylinositol phosphate kinase type 1 gamma by the FERM domain of Talin. Nature 420, 85–89. https://doi.org/10.1038/nature01147 (2002).
-
Franco, S. J. et al. Calpain-mediated proteolysis of Talin regulates adhesion dynamics. Nat. Cell Biol. 6, 977–983. https://doi.org/10.1038/ncb1175 (2004).
-
Kanemaru, K. et al. Plasma membrane phosphatidylinositol (4,5)-bisphosphate is critical for determination of epithelial characteristics. Nat. Commun. 13, 2347. https://doi.org/10.1038/s41467-022-30061-9 (2022).
-
Posor, Y. et al. Local synthesis of the phosphatidylinositol-3,4-bisphosphate lipid drives focal adhesion turnover. Dev Cell 57, 1694-1711.e1697. https://doi.org/10.1016/j.devcel.2022.06.011 (2022).
-
Schink, K. O., Tan, K. W. & Stenmark, H. Phosphoinositides in control of membrane dynamics. Annu. Rev. Cell. Dev. Biol. 32, 143–171. https://doi.org/10.1146/annurev-cellbio-111315-125349 (2016).
-
Ketel, K. et al. A phosphoinositide conversion mechanism for exit from endosomes. Nature 529, 408–412. https://doi.org/10.1038/nature16516 (2016).
-
Mesmin, B. et al. A four-step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER-Golgi tether OSBP. Cell 155, 830–843. https://doi.org/10.1016/j.cell.2013.09.056 (2013).
-
Weixel, K. M., Blumental-Perry, A., Watkins, S. C., Aridor, M. & Weisz, O. A. Distinct golgi populations of phosphatidylinositol 4-phosphate regulated by phosphatidylinositol 4-kinases. J. Biol. Chem. 280, 10501–10508. https://doi.org/10.1074/jbc.M414304200 (2005).
-
Toth, J. T. et al. BRET-monitoring of the dynamic changes of inositol lipid pools in living cells reveals a PKC-dependent PtdIns4P increase upon EGF and M3 receptor activation. Biochim. Biophys. Acta. 1861, 177–187. https://doi.org/10.1016/j.bbalip.2015.12.005 (2016).
-
Balla, A. et al. Maintenance of hormone-sensitive phosphoinositide pools in the plasma membrane requires phosphatidylinositol 4-kinase IIIalpha. Mol. Biol. Cell. 19, 711–721. https://doi.org/10.1091/mbc.e07-07-0713 (2008).
-
Balla, A., Tuymetova, G., Tsiomenko, A., Varnai, P. & Balla, T. A plasma membrane pool of phosphatidylinositol 4-phosphate is generated by phosphatidylinositol 4-kinase type-III alpha: studies with the PH domains of the oxysterol binding protein and FAPP1. Mol. Biol. Cell. 16, 1282–1295. https://doi.org/10.1091/mbc.e04-07-0578 (2005).
-
Panagiotou, S. et al. OSBP-mediated PI(4)P-cholesterol exchange at Endoplasmic reticulum-secretory granule contact sites controls insulin secretion. Cell. Rep. 43, 113992. https://doi.org/10.1016/j.celrep.2024.113992 (2024).
-
Zewe, J. P., Wills, R. C., Sangappa, S., Goulden, B. D. & Hammond, G. R. SAC1 degrades its lipid substrate PtdIns4P in the endoplasmic reticulum to maintain a steep chemical gradient with donor membranes. Elife https://doi.org/10.7554/eLife.35588 (2018).
-
Mesmin, B. et al. Sterol transfer, PI4P consumption, and control of membrane lipid order by endogenous OSBP. EMBO J. 36, 3156–3174. https://doi.org/10.15252/embj.201796687 (2017).
-
Sasaki, K. et al. Dysregulation of PI4P in the trans golgi regions activates the mammalian golgi stress response. J. Biol. Chem. 301, 108075. https://doi.org/10.1016/j.jbc.2024.108075 (2025).
-
Lenoir, M., Kufareva, I., Abagyan, R. & Overduin, M. Membrane and protein interactions of the pleckstrin homology domain superfamily. Membr. (Basel). 5, 646–663. https://doi.org/10.3390/membranes5040646 (2015).
-
Hammond, G. R., Machner, M. P. & Balla, T. A novel probe for phosphatidylinositol 4-phosphate reveals multiple pools beyond the golgi. J. Cell Biol. 205, 113–126. https://doi.org/10.1083/jcb.201312072 (2014).
-
Qiu, D. et al. Analysis of inositol phosphate metabolism by capillary electrophoresis electrospray ionization mass spectrometry. Nat. Commun. 11, 6035. https://doi.org/10.1038/s41467-020-19928-x (2020).
-
Morioka, S. et al. A mass spectrometric method for in-depth profiling of phosphoinositide regioisomers and their disease-associated regulation. Nat. Commun. 13, 83. https://doi.org/10.1038/s41467-021-27648-z (2022).
-
Overduin, M. & Kervin, T. A. The phosphoinositide code is read by a plethora of protein domains. Expert Rev. Proteom. 18, 483–502. https://doi.org/10.1080/14789450.2021.1962302 (2021).
-
Lemmon, M. A. Pleckstrin homology (PH) domains and phosphoinositides. Biochem. Soc. Symp. 81–93. https://doi.org/10.1042/BSS0740081 (2007).
-
Dowler, S. et al. Identification of pleckstrin-homology-domain-containing proteins with novel phosphoinositide-binding specificities. Biochem. J. 351, 19–31. https://doi.org/10.1042/0264-6021:3510019 (2000).
-
Furutani, M., Itoh, T., Ijuin, T., Tsujita, K. & Takenawa, T. Thin layer chromatography-blotting, a novel method for the detection of phosphoinositides. J. BioChem. 139, 663–670. https://doi.org/10.1093/jb/mvj076 (2006).
-
Furutani, M., Tsujita, K., Itoh, T., Ijuin, T. & Takenawa, T. Application of phosphoinositide-binding domains for the detection and quantification of specific phosphoinositides. Anal. Biochem. 355, 8–18. https://doi.org/10.1016/j.ab.2006.05.014 (2006).
-
Mizuno-Yamasaki, E., Medkova, M., Coleman, J. & Novick, P. Phosphatidylinositol 4-phosphate controls both membrane recruitment and a regulatory switch of the Rab GEF Sec2p. Dev. Cell. 18, 828–840. https://doi.org/10.1016/j.devcel.2010.03.016 (2010).
-
Gulyas, G. et al. Measurement of inositol 1,4,5-trisphosphate in living cells using an improved set of resonance energy transfer-based biosensors. PloS One. 10, e0125601. https://doi.org/10.1371/journal.pone.0125601 (2015).
-
Pemberton, J. G. et al. An advanced toolset to manipulate and monitor subcellular phosphatidylinositol 3,5-bisphosphate. J. Cell Biol. https://doi.org/10.1083/jcb.202408158 (2025).
-
Ding, Y. et al. Ratiometric biosensors based on dimerization-dependent fluorescent protein exchange. Nat. Methods. 12, 195–198. https://doi.org/10.1038/nmeth.3261 (2015).
-
Tamura, R., Jiang, F., Xie, J. & Kamiyama, D. Multiplexed labeling of cellular proteins with split fluorescent protein tags. Commun. Biol. 4, 257. https://doi.org/10.1038/s42003-021-01780-4 (2021).
-
Kent, K. P. & Boxer, S. G. Light-activated reassembly of split green fluorescent protein. J. Am. Chem. Soc. 133, 4046–4052. https://doi.org/10.1021/ja110256c (2011).
-
Alford, S. C., Abdelfattah, A. S., Ding, Y. & Campbell, R. E. A fluorogenic red fluorescent protein heterodimer. Chem. Biol. 19, 353–360. https://doi.org/10.1016/j.chembiol.2012.01.006 (2012).
-
Godi, A. et al. ARF mediates recruitment of PtdIns-4-OH kinase-beta and stimulates synthesis of PtdIns(4,5)P2 on the golgi complex. Nat. Cell Biol. 1, 280–287. https://doi.org/10.1038/12993 (1999).
-
Liu, Y. et al. The Sac1 phosphoinositide phosphatase regulates golgi membrane morphology and mitotic spindle organization in mammals. Mol. Biol. Cell. 19, 3080–3096. https://doi.org/10.1091/mbc.e07-12-1290 (2008).
-
Nishimura, T. et al. Oxysterol-binding protein (OSBP) is required for the perinuclear localization of intra-Golgi v-SNAREs. Mol. Biol. Cell. 24, 3534–3544. https://doi.org/10.1091/mbc.E13-05-0250 (2013).
-
Oh-Hashi, K., Nakamura, H., Ogawa, H., Hirata, Y. & Sakurai, K. Elucidation of OSW-1-induced stress responses in Neuro2a cells. Int. J. Mol. Sci. https://doi.org/10.3390/ijms24065787 (2023).
-
Do, K. & Boxer, S. G. Thermodynamics, kinetics, and photochemistry of beta-strand association and dissociation in a split-GFP system. J. Am. Chem. Soc. 133, 18078–18081. https://doi.org/10.1021/ja207985w (2011).
-
Lin, C. Y., Both, J., Do, K. & Boxer, S. G. Mechanism and bottlenecks in strand photodissociation of split green fluorescent proteins (GFPs). Proc. Natl. Acad. Sci. U.S.A. 114, E2146–E2155. https://doi.org/10.1073/pnas.1618087114 (2017).
-
Balla, T. Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol. Rev. 93, 1019–1137. https://doi.org/10.1152/physrev.00028.2012 (2013).
-
Li, J. et al. PI-273, a Substrate-Competitive, specific Small-Molecule inhibitor of PI4KIIalpha, inhibits the growth of breast cancer cells. Cancer Res. 77, 6253–6266. https://doi.org/10.1158/0008-5472.CAN-17-0484 (2017).
-
Rutaganira, F. U. et al. Design and structural characterization of potent and selective inhibitors of phosphatidylinositol 4 kinase IIIbeta. J. Med. Chem. 59, 1830–1839. https://doi.org/10.1021/acs.jmedchem.5b01311 (2016).
-
Szentpetery, Z., Varnai, P. & Balla, T. Acute manipulation of golgi phosphoinositides to assess their importance in cellular trafficking and signaling. Proc. Natl. Acad. Sci. U.S.A. 107, 8225–8230. https://doi.org/10.1073/pnas.1000157107 (2010).
-
Dickson, E. J., Jensen, J. B. & Hille, B. Golgi and plasma membrane pools of PI(4)P contribute to plasma membrane PI(4,5)P2 and maintenance of KCNQ2/3 ion channel current. Proc. Natl. Acad. Sci. U.S.A. 111, E2281–2290. https://doi.org/10.1073/pnas.1407133111 (2014).
-
Pham, T. D. et al. FAST, a method based on split-GFP for the detection in solution of proteins synthesized in cell-free expression systems. Sci. Rep. 14, 8042. https://doi.org/10.1038/s41598-024-58588-5 (2024).
-
Li, X. et al. Generation of destabilized green fluorescent protein as a transcription reporter. J. Biol. Chem. 273, 34970–34975. https://doi.org/10.1074/jbc.273.52.34970 (1998).
-
De Wulf, H. & Keppens, S. Proceedings: is calcium the second messenger in liver for Cyclic AMP-independent glycogenolytic hormones? Arch. Int. Physiol. Biochim. 84, 159–160 (1976).
-
Gadella, T. W. J. Jr. et al. mScarlet3: a brilliant and fast-maturing red fluorescent protein. Nat. Methods. 20, 541–545. https://doi.org/10.1038/s41592-023-01809-y (2023).
-
Wiederschain, D. et al. Single-vector inducible lentiviral RNAi system for oncology target validation. Cell. Cycle. 8, 498–504. https://doi.org/10.4161/cc.8.3.7701 (2009).
-
Edelstein, A., Amodaj, N., Hoover, K., Vale, R. & Stuurman, N. Computer control of microscopes using microManager. Curr. Protoc. Mol. Biol. Chap https://doi.org/10.1002/0471142727.mb1420s92 (2010).
-
Edelstein, A. D. et al. Advanced methods of microscope control using muManager software. J. Biol. Methods https://doi.org/10.14440/jbm.2014.36 (2014).
-
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 9, 676–682. https://doi.org/10.1038/nmeth.2019 (2012).
Acknowledgements
This work was supported by NCI R01CA222918 and funding by the Sylvester Cancer Center as well as the Sylvester Cancer Center Tumor Biology program to R.L. We thank T.K Landgraf for the optimization and testing of the image processing pipeline used in sensor quantification by microscopy and the Daunert/Deo Lab for access to the CLARIOStar microplate reader. FACS data were obtained at the Flow Cytometry Shared Resource (FCSR) of the Sylvester Comprehensive Cancer Center at the University of Miami, RRID: SCR022501, which is supported by the National Cancer Institute (NCI) of the National Institutes of Health (NIH) under award number P30CA240139. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.P30CA240139. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Manuel, S.K.A., Bilenka, E., Gupta, M. et al. Dynamic assessment of PI4P metabolism using split GFP based sensors. Sci Rep 15, 30805 (2025). https://doi.org/10.1038/s41598-025-15745-8
-
Received:
-
Accepted:
-
Published:
-
DOI: https://doi.org/10.1038/s41598-025-15745-8
