
Introduction
The global surge in neurodegenerative disorders, projected to affect 139 million people by 20501, urgently demands innovative therapeutic approaches. Recent breakthroughs in drug development have highlighted natural biflavonoids as promising candidates, particularly for their unique ability to engage multiple therapeutic targets2,3. Among various biflavonoids, amentoflavone-type derivatives, characterized by a C3’, C8”-linked asymmetric skeleton, have emerged as leads due to their superior drug-like properties and robust target engagement2,4. Notably, these derivatives exhibited the most potent inhibitory activity against amyloid-β peptide (Aβ40) aggregation with IC50 values ranging from 5 to 10 μM2,5, a hallmark pathological feature of Alzheimer’s disease. Moreover, the regioselective formation of the intermolecular C–C bonds is crucial for achieving their distinct therapeutic benefits6.
A prominent clinical application of amentoflavone-type biflavonoids is EGb 761, a standardized Ginkgo biloba leaf extract known for its cognitive-enhancing and neuroprotective properties7. Its clinical efficacy in treating mild cognitive impairment was demonstrated in a 24-week randomized controlled trial, showing significant improvement in neuropsychiatric symptoms and cognitive abilities compared to placebo8. The key active components, including amentoflavone (1), bilobetin (2), ginkgetin (3), and isoginkgetin (4), account for 0.047% to 1.68% of the extract and are responsible for its multi-target therapeutic effects7. However, despite their promising potential, the broader clinical application of these compounds is severely limited by their scarce natural abundance (typical isolation yield <0.001% from plant sources)9, creating a critical bottleneck in drug development and highlighting the urgent need for efficient synthetic strategies.
To address such limitations, chemical synthesis of biflavonoids has been explored as a potential strategy. Nevertheless, these approaches require high temperatures, strong oxidants, and multi-step procedures, leading to low-to-moderate yields and limited practical applications10,11. In contrast, biosynthetic technology has emerged as a promising alternative, offering high efficiency and selectivity under mild reaction conditions for cost-effective biflavonoid production11,12. Although biflavonoids were discovered nearly a century ago, beginning with the isolation of 3 from G. biloba13, the enzymes responsible for flavonoid dimerization remain unknown, hindering further development and applications.
In the broader context of biaryl natural product biosynthesis6, various oxidative enzymes, including peroxidases14, laccases15, polyphenol oxidases (PPOs)16, and cytochromes P450 monooxygenases (CYPs)17, are known to catalyze cross-coupling reactions. Among them, peroxidases, laccases, and PPOs exhibit limited control over post-oxidation bond formation. In contrast, CYPs mediate highly selective oxidation reactions, including regio- and stereoselective dimerization18 in the biosynthesis of dinaphthalenes19, diketopiperazine alkaloids20, and bicoumarins21. Structural analyses of CYPs reveal diverse substrate-binding pockets crucial for defining substrate specificity22,23,24,25. This structural diversity enables CYPs to recognize specific substrates and catalyze dimeric reactions with precise connectivity, driven by steric and electronic biases and facilitated by radical-mediated mechanisms22,23,24,25. Although such catalytic activities for CYPs are exclusively identified in bacteria and fungi22,23,24,25, the widespread presence of biflavonoids in plants suggests that similar enzymatic processes may also operate in the plant kingdom.
Here, we report the identification of CYP90J and CYP90E subfamily members, which catalyze the regioselective C–C cross-coupling dimerization of apigenin (5) and its related compounds. Unlike previously known CYP90 family members that primarily catalyze the sterols hydroxylation, including canonical brassinosteroids26, our identified CYP90J and CYP90E members demonstrate distinct catalytic functions. Our findings link the presence of CYP90J orthologs to the biflavonoids in plants. Through comprehensive QM/MM calculations, we reveal that GbCYP90J6 catalyzes the intermolecular C–C bond formation via an energetically favorable diradical coupling mechanism, with key active site residues stabilizing intermediates and directing regioselective coupling. The subsequent methylation modification pathway of biflavonoids 2–4 and 8–10 is mediated by GbOMT1-GbOMT5. Based on these findings, we successfully engineer a synthetic pathway in Escherichia coli for de novo production of 1, achieving a titer of 4.75 mg/L. This study reveals unique plant-derived CYPs that expand our understanding of atypical P450 functions and establishes further insights into the biosynthetic pathways of biflavonoids, providing opportunities for the application of biflavonoids in biomanufacturing and therapeutic development.
Results
Unprecedented GbCYP90J6 drives the dimerization of biflavonoids
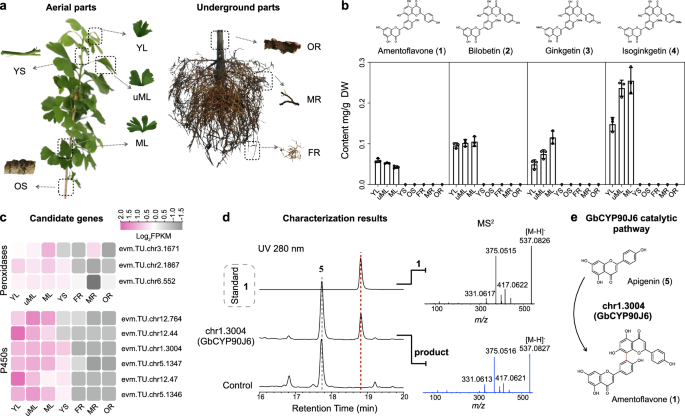
G. biloba is rich in 3’,8”-linked amentoflavone-type biflavonoids, including 1–427. The recent availability of its nearly complete genome28 makes it a perfect model for elucidating the molecular mechanism underlining the dimerization of biflavonoids. To identify candidate genes, we examined the tissue-specific distribution of biflavonoids, quantitatively assessing biflavonoids 1–4 across different tissues. Our results showed that biflavonoids predominantly accumulated in the leaves, regardless of developmental stage (Fig. 1a, b). Then, we conducted RNA-Seq analyses using total RNA from various tissues and annotated the transcriptome profiles with the reference genome28. Genes with high leaf-specific expression [fragments per kilo base of transcript per million mapped fragments (FPKM) > 20] were prioritized for further study. Giving the roles of CYPs, peroxidases, laccases, and PPOs in C–C oxidative cross-coupling in biaryl natural products14,15,16,17, we identified six CYPs (chr1.3004, chr5.1346, chr5.1347, chr12.44, chr12.47, and chr12.764) and three peroxidases (chr2.1867, chr6.552, and chr3.1671) as candidates for functional characterization, while no suitable laccases or PPOs were selected (Fig. 1c and Supplementary Fig. 1).

a Detailed schematic of plant tissue sampling. YL young leaf, uML unmature leaf, ML mature leaf, YS young shoot, OS old shoot, OR old root, MR main root, FR fibrous root. b Dry weight content of amentoflavone (1), bilobetin (2), ginkgetin (3), and isoginkgetin (4) in selected tissues of G. biloba. All data represent the means values ± SD; error bars show the standard deviation of three biological replicates. Source data are provided as a Source Data file. c Expression patterns of screened peroxidases and CYPs in different organs. The figure displays the values as log2(average FPKM of biological replicates). d LC-MS/MS analysis of tobacco leave crude extract expressing the chr1.3004 (GbCYP90J6) candidate with the filtration of substrate apigenin (5). The empty vector was used as a negative control. The MS2 spectra of the 1 standard (black) and the product from GbCYP90J6-expressing tobacco (blue) were shown on the right. e The dimeric reaction catalyzed by GbCYP90J6 with 5 as substrate.
Since 1 is the most primitive C–C type biflavonoid2 and 5 functions as the essential structural unit, we employed 5 as the substrate for our primary screening. Nicotiana benthamiana (tobacco) was chosen as the initial host due to its suitability for expressing functional CYPs and peroxidases. The full-length cDNAs of the candidate genes were individually cloned into the pEAQ-HT vector and expressed in tobacco leaves using Agrobacterium-mediated transformation. Upon the infiltration of the substrate 5, only the transient expression of the chr1.3004 protein, designated GbCYP90J6, produced a detectable product (Fig. 1d and Supplementary Fig. 1). Sequential liquid chromatography coupled with high-resolution mass spectrometry (LC-HRMS) analysis revealed that the newly formed product had a molecular weight 2 Da less than twice that of 5 (Fig. 1d), implying that GbCYP90J6 catalyzed the dimerization of two molecules of 5 to form bi-apigenin (Fig. 1e). This bi-apigenin exhibited identical retention time and MS2 spectra as the authentic standard 1 (Fig. 1d), thereby proving its identification. To eliminate the involvement of native enzymes in tobacco, we conducted assays with N-terminal modified GbCYP90J6 (trGbCYP90J6), heterologously expressed in E. coli, alongside its electron transport chain partner, N-terminal modified CPR from Arabidopsis thaliana (trAtCPR). As in tobacco, E. coli cells carrying trGbCYP90J6 yielded 1 when fed with 5 (Supplementary Fig. 2). Further kinetic analysis revealed that GbCYP90J6 had a high affinity for 5, with a Km value of 0.4925 μM (Supplementary Fig. 3), and remarkable regioselectivity, catalyzing the formation of 3’,8”-linked biflavonoids. To our knowledge, GbCYP90J6 represented a rare example of a plant-derived CYP enzyme that catalyzed the intermolecular C–C dimerization in the biosynthesis of biaryl natural products.
To further investigate the substrate specificity of GbCYP90J6, we designed synthetic routes to assess whether methylated monomers could serve as substrates for dimerization into putative methylated derivatives of 1 (Supplementary Fig. 4a). In an E. coli reaction system, incubation of 5 with acacetin (6) was intended to generate 2 and 4, and co-incubation with apigenin 7,4’-dtimethyl ether (7) aimed to produce 3. However, GbCYP90J6 exclusively catalyzed the dimerization of non-methylated 5, while methoxylated analogs 6 and 7 as substrates were not recognized (Supplementary Fig. 4b, c). This observation suggested that the dimerization preceded the methylation of phenol groups in 5. Moreover, 1 likely served as the key precursor for the biosynthesis of 2–4, consistent with its early evolutionary origin29.
To validate this, we conducted gene co-expression analysis using GbCYP90J6 as a bait gene, revealing candidates for O-methyltransferases (GbOMTs, Fig. 2a). The resulting 2, which was modified by O-methyltransferases GbOMT1 and GbOMT2 from 1, verified this hypothesis (Fig. 2b). Subsequently, GbOMT3 further modified 2 to produce 3, while GbOMT4 and GbOMT5 convert 2 into 4 (Fig. 2c). Beyond 2–4, we identified several naturally occurring O-methylated biflavonoids, including podocarpusflavone A (8)30, sequoiaflavone (9)31 (Fig. 2d), with their methylation sites confirmed by NOESY correlations and comparison to authentic standards (Supplementary Figs. 5–11). A low-yield product, tentatively identified as 4’,7”-O-methylamentoflavone (10)32 (Fig. 2e), was also detected. Based on unmodified 7-OH and 4’”-OH groups retained in 3 and 4, the methylation in 10 was assigned to the 7”-OH position (Fig. 2f). Interestingly, biochemical characterization revealed that each GbOMT exhibited distinct regioselectivity. GbOMT1 showed preference for the 4’-OH and 7”-OH positions adjacent to the intramolecular C3’-C8” linkage, indicating high activity toward 4’-OH but limited activity toward 7”-OH. GbOMT2 selectively methylated the 4’-OH position, while GbOMT3 selectively methylated the 7-OH position. Both GbOMT4 and GbOMT5 demonstrated selective methylation of the 4”’-OH position (Fig. 2f). Based on these findings, we concluded that the biosynthetic pathway of amentoflavone-type biflavonoids began with oxidative cross-coupling reactions, followed by subsequent modifications such as regioselective O-methylation mediated by specific GbOMTs (Fig. 2f).

For GbOMT characterization, crude enzymes generated from BL21(DE3) carrying the empty vector pET28a were used as a negative control. a Expression patterns of screened O-methyltransferases in different organs. The figure displayed the values as log2FPKM. b LC-MS/MS analysis of G. biloba OMTs chr7.385 (GbOMT1) and chr8.315 (GbOMT2) activities on substrates 1 in vitro. GbOMT1 and GbOMT2 catalyzed substrate 1 to produce bilobetin (2). MS2 fragmentation spectra for authentic 2 and products of GbOMT1 and GbOMT2 were shown on the right. c LC-MS/MS analysis of G. biloba OMTs chr1.879 (GbOMT3), chr10.1253 (GbOMT4) and chr12.613 (GbOMT5) activities on substrates 2 in vitro. GbOMT3 catalyzed 2 to produce ginkgetin (3), GbOMT4 and GbOMT5 catalyzed 2 to produce isoginkgetin (4). d LC-MS/MS analysis of G. biloba OMTs chr1.879 (GbOMT3), chr10.1253 (GbOMT4), and chr12.613 (GbOMT5) activities on substrate 1 in vitro. GbOMT4 and GbOMT5 catalyzed substrates 1 to podocarpusflavone A (8). GbOMT3 catalyzed 1 to sequoiaflavone (9). MS2 fragmentation spectra of products 8 and 9 were shown on the right side. e LC-MS/MS analysis of G. biloba OMTs chr7.385 (GbOMT1) activities on substrates 2 in vitro. GbOMT1 catalyzed 2 to 10, which was speculated as 4,7”-O-methylamentoflavone. f The biosynthetic pathway of biflavonoid 1 and its methylated derivatives 2–4 and 8–10 in G. biloba. GbCYP90J6 dimerized 5 to generate 1, which served as the common precursor for 2–4 and 8–10. GbOMT1 and GbOMT2 catalyze the conversion of 1 to 2. Subsequently, GbOMT3 can modify 2 to produce 3, while GbOMT4 and GbOMT5 convert 2 into 4. 8–10 are shunt products.
Additionally, we probed the substrate specificity of GbCYP90J6 toward a broader range of flavonoids, as hundreds of C–C linkage biflavonoids were reported to form via flavonoids dimerization12,33. This range extended beyond 5 and its methylated derivatives. To this end, we selected a series of unmodified aglycones, including flavanones, flavones, flavonols, and flavanonols, as substrates in the biosynthetic route of flavonoid (Supplementary Fig. 12a). LC-HRMS results revealed that GbCYP90J6 catalyzed the dimerization of (S)-naringenin (11), a precursor in the biosynthesis of 5, generating two bi-naringenins 11a and 11b (Supplementary Fig. 12b), albeit with a conversion rate of less than 3%. Additionally, a trace amount of bi-tricetin 12a was also detected when using tricetin (12) as the substrate (Supplementary Fig. 12c), although its exact structure also remains to be elucidated. Nevertheless, 5 remained the preferred substrate for GbCYP90J6. Thus, the CYP90J subfamily likely played a crucial role in flavonoid dimerization, contributing to the formation of biflavonoid scaffolds and sparking further interest in this unique CYP subfamily.
Gymnosperm-specific CYP90J subfamily enzymes specially catalyze the intermolecular C–C dimerization of flavonoids
Phylogenetically, CYP90 members were initially identified in lycophytes34 and later maintained their presence in ferns, gymnosperms, and angiosperms. To investigate the evolutionary origin of GbCYP90J6, we conducted a systematic phylogenetic analysis of CYP90 proteins across representative species of lycophytes35,36, ferns37, gymnosperms38,39,40,41,42,43, and angiosperms44 using available genome data (Supplementary Data 1). Additionally, we incorporated known angiosperm CYP90 proteins collected in the Plant Cytochrome P450 database45. Three additional G. biloba CYP90 family members: chr1.681 (GbCYP90B61), chr9.869 (GbCYP90A68), and chr10.96 (GbCYP90D59) were included, each sharing less than 45% protein sequence identity with GbCYP90J6 (Supplementary Fig. 1c). Notably, the three G. biloba CYP90 proteins were dispersed and clustered with functionally known AtCYP90, suggesting their potential typical role in the oxidation of polyhydroxylated steroids26 (Fig. 3a). In contrast, GbCYP90J6 clusters within the gymnosperms CYP90J subclade, closely related to lycophytes CYP90E subclade, with no angiosperm CYP90 members in this lineage (Fig. 3a). This suggested that the CYP90J subfamily may be unique to gymnosperms, and the proximity of lycophyte CYP90E members implied a potential evolutionary relationship between these two subclades. Furthermore, we constructed the phylogenetic tree of GbCYP90 subfamily orthologs in land plants, spanning from bryophytes to gymnosperms (Supplementary Fig. 13), to investigate the evolutionary distribution of GbCYP90 members. Based on this analysis, we extracted relevant orthologs distribution patterns and presented a simplified overview in Fig. 4. Notably, the phylogenetic relationships revealed a strong correlation between the presence of GbCYP90J6 orthologs and the reported occurrence of biflavonoids (Fig. 4). GbCYP90J6 orthologs were predominantly found in plant species documented to produce biflavonoids (Supplementary Data 2), while they were generally absent in plant species lacking such reports. For instance, biflavonoids and their corresponding orthologous genes are present in mosses but absent in hornworts and liverworts46. A similar pattern was observed in gymnosperms, such as Gnetales47,48 (including Ephedra, Gnetum, and Welwitschia) and Pinaceae48,49, which lacked both biflavonoids and orthologous genes. Although the catalytic function of each ortholog remained to be fully validated, their phylogenetic association with biflavonoid-producing species suggested a potentially conserved role in biflavonoid biosynthesis. We acknowledge, however, that some uncertainty persists in species lacking comprehensive phytochemical characterization. The co-distribution of biflavonoids and GbCYP90J6 orthologs suggested a plausible evolutionary trajectory from mosses to gymnosperms in biflavonoid biosynthesis, with the CYP90J playing a specialized role in biflavonoid synthesis within gymnosperms.

a CYP90s from representative species of lycophytes, ferns, gymnosperms, and angiosperms, along with known CYP90s from the Plant Cytochrome P450 database45, were selected to construct a phylogenetic tree of plant CYP90s. All sequences used in the phylogenetic analysis were listed in Supplementary Data 1. Protein sequences were aligned using MUSCLE86 and then used to construct a Maximum Likelihood (ML) tree with IQ-TREE68 with 1000 bootstrap replicates. The resulting tree was visualized using iTOL87. In the tree, the purple clade represented the CYP90J subfamily from gymnosperms, with GbCYP90J6 located within it. The pink clade represented the CYP90E subfamily from lycophytes, and the blue clade represented the known CYP90A-D subfamily that catalyzed the hydroxylation of steroids from angiosperms. Additionally, the green leaf represented three other CYP90 members in Ginkgo biloba: GbCYP90A68, GbCYP90B61, and GbCYP90D59, which were adjacent to known steroid hydroxylases. b LC-MS/MS results characterized the dimerase activity of additional CYP90Js from gymnosperms and SmCYP90E3v1 from lycophytes towards apigenin (5). Functional characterization was conducted in vivo in E. coli JM109(DE3), with the strain carrying the pFe00 vector used as a negative control. MS2 fragmentation spectra for authentic amentoflavone (1) and products of ChCYP90J7 and SmCYP90E3v1 were shown on the right. All these members were also tested for dimerase activity toward substrate (S)-naringenin (11) (Supplementary Fig. 14), with the results summarized on the right side of b.

Plants containing biflavonoids possess GbCYP90J6 orthologs. These include mosses (Sphagnum fallax, Sphagnum magellanicum, Physcomitrium patens, Ceratodon purpureus, and Pohlia nutans), the lycophyte Selaginella moellendorffii, ferns (Alsophila spinulosa and Adiantum capillus-veneris), and gymnosperms (Ginkgo biloba, Cycas panzhihuaensis, Taxus chinensis, Thuja plicata, Metasequoia glyptostroboides, and Sequoiadendron giganteum). Conversely, in plants lacking biflavonoids, GbCYP90J6 orthologs are absent. These include hornworts (Anthoceros angustus, Anthoceros agrestis, and Anthoceros punctatus), liverworts (Marchantia polymorpha), Pinaceae (Pseudotsuga menziesii, Picea babies, and Pinus teada), and Gnetales (Gnetum montanum and Welwitschia mirabilis). Thus, the distribution of biflavonoids is highly correlated with the presence of GbCYP90J6 orthologs, indicating the important role of these orthologs in the dimerization of biflavonoids. Additionally, we provide the detailed distribution of orthologs in plants in Supplementary Fig. 13 and detailed plant biflavonoids dispersed information in Supplementary Data 2.
To determine whether biflavonoid synthesis was common within CYP90J, we characterized additional gymnosperm CYP90J members: TcCYP90J7 from Taxus chinensis, CpCYP90J8 from Cycas panzhihuaensis, ChCYP90J7 from Cephalotaxus harringtonia, MgCYP90J1 from Metasequoia glyptostroboides, and TpCYP90J1v1, TpCYP90J9, and TpCYP90J10 from Thuja plicata. Despite their high sequence similarity (68–71%), their catalytic activities varied. Notably, ChCYP90J7, similar to GbCYP90J6, exhibited dimerization activities that were more inclined towards 5 than 11, as evidenced by its higher catalytic efficiency (Fig. 3b and Supplementary Fig. 14). In contrast, MgCYP90J1 catalyzed only the dimerization of 11 (Supplementary Fig. 14), while the other CYP90J members did not show these activities. Given the rich diversity of C–C type biflavonoids in gymnosperms12,50, it was plausible that these CYP90J enzymes participate in the dimerization of additional flavonoid substrates. Additionally, to further elucidate the evolutionary relationship, SmCYP90E3v1 from Selaginella moellendorffii in CYP90E subclade was also chosen for screening. It exhibited weak catalytic activity toward 5 and 11, producing trace amounts of 1 and 11a (Fig. 3b and Supplementary Fig. 14). Based on the above observations and the established plant phylogeny, gymnosperm CYP90J subfamily members likely evolved from ancestral lycophyte CYP90Es and subsequently diverged to specialize in biflavonoid synthase. Their substrate scope, however, required further investigation.
Ligand conformation controlled by surrounding amino acids determines enzyme dimerization catalytic activity
Although the unique gymnosperms CYP90Js specialized in flavonoid dimerization, only GbCYP90J6 and ChCYP90J7 catalyzed the dimerization of 5 to 1 via an intermolecular 3’,8”-linkage (Figs. 1d and 3b). To explain this phenomenon, we investigated the unique function of GbCYP90J6. Using molecular docking, we constructed a structural model of the GbCYP90J6-apigenin (5D)-apigenin (5U) complex (Supplementary Fig. 15). The alanine scanning mutagenesis targeting residues within 5 Å of 5D and 5U in the active pocket revealed 31 residues that significantly impact activity (Fig. 5a). Residues A357 and A460 were excluded due to their native alanine identity, and T293 (in the conserved AGxxT motif) and the heme-coordinating C425 were not mutated due to their essential roles. Most deleterious mutations were clustered in the substrate-binding region, spanning M53-H60 in αA, H83-P88 in loop 1, Y113-L127 in loop 2-αB-loop 3, Y208-I216 in αC, L284-M296 in αD, N355-R362 in loop 4, and F458-V461 in loop 6 (Fig. 5a, b). Notably, alanine mutations at the tunnel entrance (M53, F458, P459, V461) and within the catalytic center (Y113, P114, F121, I126, F286, L361, R362) abolished or nearly abolished dimerization. In contrast, only V215A mutation in αC, located near the entrance, significantly enhanced the activity. To further probe the structure-function relationship, we examined the homologous mutants derived from inactive CYP90Js, as well as the selected mutations (V215T, F286Y, L382N) based on hydrophobicity and polarity differences between active and inactive enzymes (Fig. 5c and Supplementary Fig. 16a). We noticed that different homologous mutants at the same site often resulted in divergent functional outcomes, reinforcing the importance of spatial constraints in maintaining catalytic activity (Supplementary Fig. 16a). For instance, at position M287 in αD, M287A retained approximately 14% activity, M287L showed near wild-type activity, while M287Y and M287F nearly abolished function. Similarly, mutations at L382 in loop 5 at the bottom of the substrate tunnel revealed contrasting outcomes: L382N eliminated activity, whereas L382F doubled it. Comparable trends were observed at M53, L127, V215, L284, F286, and L361, further highlighting the critical role of spatial constraints in substrate positioning. Moreover, none of the single-site mutations introduced into the inactive TcCYP90J7 conferred dimeric activity (Supplementary Fig. 16b). Western blot analyses confirmed the comparable expression across all mutants, indicating that the observed activity differences were attributable to functional effects of the mutations rather than expression level (Supplementary Fig. 17). These mutation tests suggest that the steric hindrance within the substrate-binding cavity creates a sophisticated enzymatic environment crucial for substrate dimerization, governing substrate entry and arrangement.

a Results of alanine scanning within a 5 Å range of the 5 ligands of GbCYP90J6. The x-axis represented the mutagenesis sites of GbCYP90J6, and the y-axis showed the relative enzyme activity compared to the wild-type (WT). Each value was presented as mean ± S.D. (n = 3). Experiments were conducted in vivo using E. coli JM109(DE3). “nd” indicated no production of 1. All mutagenesis within a 5 Å range of the 5 ligands showed significant variation compared to the WT. Source data are provided as a Source Data file. b GbCYP90J6-apigenin (5D)-apigenin (5U) model generated by molecular docking and MD simulation, showing the spatial relationship between key residues identified by mutagenesis and the substrate molecules. Two ligands maintained a head-to-tail, anti-parallel configuration, with distance from C4’-OH of ligand 1 (5D) and C7”-OH of ligand 2 (5U) to the heme center of 2.2 Å and 4.9 Å, separately. The distance between C3’ of 5D and C8’ of 5U was 3.5 Å. Key sites from alanine scanning mutagenesis were highlighted in different colors. c Protein sequence alignment of gymnosperm CYP90J, ordered by the sequence of GbCYP90J6, revealing conservation patterns of functionally important residues across active and inactive enzymes.
To clarify the catalytic mechanism underlying biflavonoid formation, we conducted molecular dynamics (MD) simulations on the protein variants GbCYP90J6L382F and GbCYP90J6L382N due to their opposing catalytic activities at the same site (Supplementary Fig. 16a). Additionally, we included the active ChCYP90J7 and the inactive TcCYP90J7 for comparative analysis. Structural alignment revealed that even subtle sequence variations significantly alter the conformations of surrounding residues and bound ligands (Fig. 6a). While the specific locations of these conformational changes varied among variants, they consistently altered ligand positioning relative to the catalytic center. This tread was observed both in the single-site mutant comparison (Fig. 6a) and across active (GbCYP90J6, ChCYP90J7) versus inactive (TcCYP90J7) enzymes (Fig. 6b). These observations prompted further investigation into which specific conformational arrangements favor the regiospecific catalysis of C3’-C8” linked biflavonoids.

a Structural alignment of active sites among GbCYP90J6 wild-type, high-activity mutant GbCYP90J6L382F, and low-activity mutant GbCYP90J6L382N. Regions targeted in alanine scanning were shown as cartoon representations, with L382, L382F, and L382N displayed as sticks. Critical distances were indicated in the lower left corner of each structure panel: d1 (distance between Fe = O and 4’-OH of 5D), d2 (distance between Fe = O and 7”-OH of 5U), and d3 (distance between C3’of 5D and C8” of 5U). These results demonstrated how single-point variations in the active site significantly altered both protein pocket and substrate conformations. b Structural comparison of active sites between functional amentoflavone synthases (GbCYP90J6 and ChCYP90J7) and inactive TcCYP90J7. Regions corresponding to the alanine scanning area in GbCYP90J6 were shown as cartoon representations. The three critical distances (d1, d2, and d3) were displayed in the lower left corner of each structural panel, highlighting conformational differences between active and inactive enzymes. c Binding free energies (kcal/mol) of ligand 1 (5D) and ligand 2 (5U) in GbCYP90J6, GbCYP90J6L382F, GbCYP90J6L382N, TcCYP90J7, and ChCYP90J7 as determined by gmx_MMPBSA analysis.
Our analysis revealed a critical correlation between enzyme activity and substrate positioning. In active enzyme models (Fig. 6), the two apigenin molecules (5U and 5D), were positioned closer to the heme center than in inactive variants (Fig. 6 and Supplementary Fig. 18). This proximity directly correlated with enhanced dimerization activity, as exemplified by GbCYP90J6L382F with shorter critical distances (d1 = 2.0 Å, d2 = 3.3 Å, d3 = 3.1 Å) showing higher activity than wild-type GbCYP90J6 (d1 = 2.2 Å, d2 = 4.9 Å, d3 = 3.5 Å) (Fig. 6a). Conversely, increased distances from the heme center in GbCYP90J6L382N (d1 = 4.2 Å, d2 = 4.0 Å, d3 = 5.1 Å) correlated with substantially reduced activity (Fig. 6a).
To elucidate the molecular basis underlying substrate positioning, we conducted detailed protein-ligand interaction analysis using PLIP51 (Supplementary Fig. 18), complementing the structural comparisons in Fig. 6. In wide-type GbCYP90J6, residues S358 formed hydrogen bonds with substrates and L117, V215, I216, T293, and A357 contributed hydrophobic interactions (Supplementary Fig. 18a). The crucial π–π stacking interaction between 5U and 5D, stabilized the substrates in an almost head-tail anti-parallel configuration52, positioning C3’ of 5D close to C8” of 5U (d3 = 3.5 Å) (Supplementary Fig. 18a). The enhanced activity of GbCYP90J6L382F resulted from the larger hydrophobic side chain of F382 disrupting the original hydrogen bonding network, accompanied by new hydrogen bonds forming between A357 and 5D, and between H84 and 5U. These altered interactions, along with hydrophobic contacts from P114, F121, V215, and A357 (Supplementary Fig. 18b), effectively positioned the substrates closer to the heme center, enhancing catalytic efficiency. In contrast, the introduction of a polar asparagine in GbCYP90J6L382N destabilized the substrate-binding network. N382 and H84 formed new hydrogen bonds with 5U and 5D, respectively, disrupting the original interaction pattern. Hydrophobic interactions between L117, V215, and A357 with 5U and 5D. While under these interactions, the inappropriate π–π stacking between the ligands pulled 5D away from 5U, increasing d3 to 5.1 Å and diminishing activity (Supplementary Fig. 18c). A similar ligand arrangement was observed in the active ChCYP90J7, though driven by a distinct set of residues (Supplementary Fig. 18d). In contrast, the inactive TcCYP90J7 failed to support π–π stacking between 5D and 5U, resulting in the displacement of 5D away from both 5U and the heme center (d1 = 30.3 Å, d2 = 6.2 Å, d3 = 24.5 Å) (Supplementary Fig. 18e). Therefore, suitable protein-ligand interactions were crucial for positioning the ligands near the heme center. Changes in surrounding amino acid residues will disrupt the original interactions and form new networks, leading to alterations in dimerization activity.
Based on the analysis of protein-ligand models and experimental results, we concluded that the key factors influencing CYP90J dimerization activity are as follows: (1) Non-covalent interactions between amino acid residues in the active pocket and ligands create a spatial constraint that brings the ligands close to the heme center, which is essential for dimerization activity; (2) In addition to constraints from surrounding amino acid residues, π-stacking between 5D and 5U in a head-tail manner stabilizes their anti-parallel alignment, positioning C3’ near C8”. This arrangement prevents random ligand coupling and prompts the regiospecific formation of 1 (3’,8”-biapigenin); (3) Mutations within the pocket disrupt the existing interaction network. A new network is formed, changing the distance between the ligands and the heme center. These alterations in conformation and interactions indicate that a tight steric hindrance between the substrates and pocket amino acids determines the dimerization activity. This altered distance affects dimerization activity. Regiospecificity in biflavonoids biosynthesis is intricately tied to the conformational orientation and relative positioning of the substrates within the active pocket.
Dimerization mechanism validated by QM/MM calculation
After confirming that the stable conformation of ligands close to the heme center determined GbCYP90J6 dimerization activity, we proposed two plausible C–C bond formation mechanisms: a radical addition pathway (A) and a diradical coupling pathway (B), inspired by mechanical insights from fungal and bacterial P450s20,22,23,24. As illustrated in Fig. 7a, both pathways began with a hydrogen atom transfer (HAT) from the C4’-OH of the proximal unit (5D) (Fig. 6a) to the highly reactive Cpd I, producing a delocalized apigenin radical R5d. In pathway A, R5d attacked the C8” position of a distal apigenin (5U), forming a C3’-C8” linked dimeric radical intermediate (R5du), which was subsequently oxidized by Cpd II via HAT from C7”-OH, yielding the keto form of 1. Alternatively, in pathway B, a second HAT from the C7”-OH of 5U, also mediated by Cpd II, generated a second delocalized radical (R5u). The resulting radicals (R5d and R5u) then underwent intermolecular coupling to form the same C–C linked keto intermediate. This species spontaneously tautomerized to the final enol form of 153.

a Two proposed dimerization pathways (A and B) for the formation of 1. Pathway A represented a radical addition mechanism, initiated by formation of the apigenin radical R5d. This radical underwent rearrangement and attacked C8” of 5U, forming the C3’-C8” linked dimeric radical intermediate R5du. Cpd II then abstracted hydrogen from C7”-OH, generating 1 (keto form), which underwent tautomerization to produce 1 (enol form). Pathway B described a diradical coupling pathway. Specifically, apigenin radicals (R5d and R5u) were generated via heme-mediated sequential hydrogen extraction reactions. These radicals underwent rearrangement, intermolecular radical-radical coupling, and aromatized to produce 1. b QM/MM calculated energy profiles for the formation of 1 catalyzed by GbCYP90J6 via two proposed pathways. The relative Gibbs free energy changes (ΔG, kcal/mol) during product 1 formation were shown with pathway A in red and pathway B in blue. Key points along the reaction coordinate included: RC (reactant complex), TS1–5 (transition states 1–5), IM1–4 (intermediates 1–4), and PC (product complex). The QM/MM optimazed structures were shown in Supplementary Fig. 19.
To assess the feasibility of both pathways, we performed QM/MM calculations based on a representative conformation derived from 200 ns MD simulation. The QM region was carefully defined to include key residues involved in hydrogen bonding and steric interactions near the reactive center. The computed energy profiles, the optimized structures of key intermediates and transition states were shown in Fig. 7b and Supplementary Fig. 19. The initial HAT step from the C4’-OH of 5D to Cpd I proceeded with a low activation barrier of 12.5 kcal/mol, generating intermediate IM1, which was 7.9 kcal/mol above the reactant complex (RC). Spin population analysis revealed that the radical in IM1 was delocalized across the aromatic ring of R5d54, with a high spin densities at the C-3’/C-5’ (−0.27/−0.25), consistent with its potential as a radical reaction site. In pathway A, the subsequent radical addition (IM1 → IM2) and HAT from C7”-OH (IM2 → IM3) catalyzed by Cpd II proceeds via a high energy barrier of 32.8 kcal/mol (TS3 relative to RC), attributed to the suboptimal geometry and the distal positioning of the hydroxyl group. This renders the pathway kinetically unfavorable. In contrast, pathway B involved a second HAT (IM1 → IM4) with a lower barrier of 22.5 kcal/mol compared to TS3. This was followed by a radical-radical coupling (IM4 → IM3) via TS5, which required only 4.6 kcal/mol from IM4 to TS5. Notably, this C–C formation barrier was substantially lower than that in pathway A (15.5 kcal/mol from IM1 to TS2). The final keto-enol tautomerization (IM3 → PC) occured spontaneously, driven by a sharp energy drop of over 42 kcal/mol (from 18.2 to −24.2 kcal/mol), thereby stabilizing the coupled product 1 (Fig. 7b)53. These results strongly supported the diradical coupling mechanism (pathway B) as the thermodynamically preferred pathway for GbCYP90J6-catalyzed dimerization.
To further elucidate the conformational changes required for C–C bond formation, we analyzed the structural transition from IM1 through the pre-coupling intermediate IM4 to the transition state TS5. QM/MM-optimized structures (Supplementary Fig. 19) revealed a progressive decrease in the C3’-C8” distance, from 4.7 Å in IM1 to 1.8 Å in IM4 and 1.7 Å in TS5. This reorganization was primarily driven by counterclockwise rotation and reorientation of R5d, which brought its C3’ atom into close proximity with the C8” atom of R5u, thereby enabling selective C3’-C8” coupling. QM/MM calculations further identified residues V215, I216, E292, and G359 were crucial for stabilizing high-energy transition states and intermediates via hydrogen bonding interaction (Supplementary Fig. 19). While mutagenesis and MD simulations revealed additional residues essential for catalytic activity, these QM/MM-identified residues specifically exemplified how the active site environment precisely orchestrates substrate alignment, thereby supporting the central role of the conformational control in the GbCYP90J6-catalyzed dimerization.
De novo biosynthesis of amentoflavone in E. coli
Based on our discovery of biflavonoid dimerases, we established a synthetic pathway for producing 1 in E. coli. To this end, we first engineered a L-tyrosine (L-Tyr) over-producing strain GJA3, as L-Tyr is the key precursor of apigenin (5). The engineered E. coli GJA3 was derived from JM109(DE3) by deleting tyrR and pheA genes, followed by integration of aroGfbr, tyrAfbr, ydiB, tktA, and ppsA genes into the tyrR locus (Fig. 8a)55. This engineered GJA3 produced 1.89 g/L L-Tyr after 72 h cultivation, reaching an OD600 of 15.5 (Fig. 8b).

a Metabolic engineering strategy for L-Tyr over-production in E. coli JM109(DE3). b L-Tyr production by the engineered strain GJA3 after 72 h fermentation. Each value represents mean ± S.D. (n = 3). c Biosynthetic modules for 1 production from L-Tyr in GJA3 strain. d Titers of 1 and its precursor 5. Each value represents mean ± S.D. (n = 3). “nd”: not detected. Source data are provided as a Source Data file.
We then assembled two functional modules for 1 production: an apigenin monomer generation module and a dimerization synthesis module. The apigenin generation module, carried on plasmid pYH5756, includes four known functional proteins (RtPAL from Rhodotorula toruloides, Pc4CL from Petroselinum crispum, PhCHS from Petunia hybrida, and MsCHI from Medicago sativa) to produce 5 from L-Tyr in E. coli (Fig. 8c). Strain sXH138 (GJA3 carrying pYH57) produced 236.93 mg/L 5 after 72 h cultivation, reaching an OD600 of 18.8 (Fig. 8d). The dimerization synthesis module was implemented through plasmid pBIF21, carrying the apigenin dimerase variant trGbCYP90J6L382F along with its electron transfer partners AtCPR2 and RsCYB5, required for CYP-catalyzed oxidation reaction (Fig. 8c). Finally, strain sXH140 (GJA3 carrying pYH57 and pBIF21) successfully produced 1 at the titer of 4.75 mg/L after 72 h cultivation in shake flasks (Fig. 8d).
Discussion
Plant CYP enzymes are crucial for synthesizing and modifying natural compounds, engaging in typical monooxygenation and hydroxylation reactions, as well as unique processes like oxidative rearrangements, oxidative C–C bond cleavage, and phenol coupling57. While intramolecular C–C and intermolecular C-O phenol coupling reactions have been previously documented58,59, intermolecular C–C phenol coupling remains largely unexplored. C–C linkages in biflavonoids, which are widespread in plants, play an essential role in conferring distinct biological activities, differentiating them from mono-flavonoids60,61. Despite their significance, the enzymes responsible for biflavonoid biosynthesis have long been unknown.
In this work, we identified key enzymes involved in this pathway, especially focusing on the synthesis of the biflavonoid skeleton amentoflavone (1), which is produced by the action of GbCYP90J6, ChCYP90J7, and SmCYP90E3v1. Beyond native substrate apigenin (5), these enzymes, together with MgCYP90J1, also catalyzed the oxidative dimerization of (S)-naringenin (11) to form products 11a and 11b, with SmCYP90E3v1 producing only 11a. Although low conversion efficiency prevented precise structural characterization, MD simulations suggested these dimers may arise from C3’-C8”, C3-C8”, or C7-O-C3” coupling modes, depending on substrate’s conformational alignment (Supplementary Fig. 20). In G. biloba, 1 underwent successive methylation by GbOMT1-GbOMT5 to yield prominent biflavonoids including bilobetin (2), ginkgetin (3), and isoginkgetin (4), podocarpusflavone A (8), sequoiaflavone (9), and likely 4’,7”-O-methylamentoflavone (10) (Fig. 2).
Unlike classical CYP90 enzymes known for catalyzing the steroid hydroxylation45, these CYP90J subfamily enzymes facilitate the formation of an intermolecular C–C bond, a function previously attributed solely to microorganisms CYPs22,23,24,25. This represents a rare identification of CYP enzymes in plants capable of catalyzing intermolecular C–C coupling, marking a significant advance in plant biochemistry. The distinctive functional divergence of CYP90J enzymes prompted us to investigate their evolutionary origins.
Given the unique role of GbCYP90J6 compared to other CYP90 members in G. biloba (Supplementary Fig. 1d), we investigated the origin and distribution of CYP90J to understand its evolutionary background. Phylogenetic analysis of plant-derived CYP90s (Fig. 3), together with the known evolutionary history of plant lineages, suggests that CYP90Js in gymnosperms likely evolved from the CYP90E family in lycophytes. This hypothesis was supported by our observation that SmCYP90E3v1 also catalyzes the dimerization of apigenin (5) and (S)-naringenin (11) (Fig. 3 and Supplementary Fig. 14). While this result provided functional support for the proposed evolutionary link, we noted that only one CYP90E paralog from Selaginella was tested in this study. Additional biochemical validation of closely related CYP90E members was considered necessary to determine whether this activity is broadly conserved within the subclade. In contrast, no CYP90J-like members were found in angiosperms, demonstrating the uniqueness of CYP90J in gymnosperms (Fig. 3). Further characterization of other gymnosperm-specific CYP90J subfamily members confirmed their specialized in flavonoid dimerization (Fig. 3b and Supplementary Fig. 14), but their varying catalytic activities suggested the need for further investigation into suitable natural substrates, especially considering the divers subtypes and C–C connection patterners of biflavonoids found in gymnosperms33,50. Moreover, our findings reveal that CYP90J exists exclusively in gymnosperms, suggesting that different enzymes may be responsible for biflavonoid skeleton synthesis in other plants, such as angiosperms. Additionally, while previous studies suggested that the emergence of CYP90 in lycophytes marks the advent of brassinosteroid biosynthesis34, our analysis reveals that the distribution of CYP90J in gymnosperms aligns with that of biflavonoids, enhancing our understanding of the functional role of the CYP90 family (Fig. 4). The biosynthesis of brassinosteroids and biflavonoid skeletons shares a common CYP ancestor, suggesting that ancestral subfamilies such as CYP85 and CYP763 likely possessed bifunctional activities in the biosynthesis of both biflavonoids and brassinosteroids34. However, the evolutionary divergence of these bifunctional activities remains unclear.
Although CYP90J subfamily members catalyzed flavonoids dimerization, they exhibit varying catalytic abilities (Fig. 3 and Supplementary Fig. 14). To investigate the reasons behind these differences, we built representative active models GbCYP90J6-apigenin (5D)-apigenin (5U) for analysis (Fig. 6). Using these models, we performed alanine scanning and single-site mutation analysis based on the sequences of the inactive CpCYP90J8, MgCYP90J1 and TcCYP90J7, within a 5 Å range of the 5D and 5U ligands. These analyses reveal the critical roles of each residue in dimerization activity (Fig. 5 and Supplementary Fig. 16) and indicate the importance of residues surrounding the ligand in determining dimerization activity. Understanding how single-site mutations affect dimerization activity can also uncover the mechanism behind CYP90J enzymes activity or inactivity. Therefore, we compared several complex models—GbCYP90J6, GbCYP90J6L382F GbCYP90J6L382N, TcCYP90J7, and ChCYP90J7—based on their minimal differences in active site amino acid residues. From a structural alignment perspective, single-site differences could significantly impact the conformation of residues within the active sites and their interaction with ligands (Fig. 6). Consequently, this affects the distance between the ligands and the heme center, leading to variations in enzyme activity. The regioselectivity and catalytic efficiency of the dimerization reaction are primarily determined by the substrate conformational orientation and spatial positioning during the formation of the reactive Fe(IV)-oxo intermediate, as demonstrated in microbial CYPs such as NasbB, aryC, and AtuP45022,23,24,25. At the molecular level, our analysis of protein-ligand interactions revealed that active enzymes establish tight interactions between surrounding residues and ligands through hydrogen bonds and hydrophobic interactions. Additionally, π–π stacking between the two apigenin (5D and 5U) ligands helps stabilize their relative positions52, bringing C3’ and C8” into close proximity. Minor changes in this balance could disrupt the molecular conformation, resulting in different interactions and conformations (Fig. 6 and Supplementary Fig. 18). These observations indicated that active enzymes, such as GbCYP90J6, create tight steric constraints between the substrates and pocket residues, which determines dimerization activity. As minor changes in the surrounding residues significantly affect both the protein-substrates conformations and their interactions, such tight steric hindrance typically plays a decisive role in CYP-catalyzed dimerization22,23,24.
To further elucidate the molecular mechanism underlying GbCYP90J6, we performed QM/MM calculations to evaluate the C–C bond formation pathway and to exclude the radical addition mechanism, which was kinetically inaccessible due to a high energy barrier of 32.8 kcal/mol. Instead, we proposed a more feasible diradical coupling mechanism in which residues V215, I216, E292, and G359 play key roles in stabilizing the conformations of intermediates and transition states through hydrogen bonding interactions (Supplementary Fig. 19). Alongside steric constraints imposed by surrounding residues, these interactions guide the reactive radicals into productive orientations, thereby conferring regioselectivity to the C3’-C8” coupling. Notably, while the intermolecular C–C formation often contributes to the rate-limiting step in previously characterized fungal and bacterial-derived CYPs20,25, GbCYP90J6 facilitates this step via a diradical coupling mechanism with a comparatively low energry barrier of only 4.6 kcal/mol from IM4 to TS5. In contrast, the second HAT step to Cpd II represents the rate-limiting step, with a barrier of 22.5 kcal/mol relative to RC, thereby distinguishing the catalytic profile of GbCYP90J6 from microbial CYPs (Fig. 7). In addition, the requirement for radical formation also explains the lack of GbCYP90J6 activity toward methylated apigenin substrates such as acacetin (6) and apigenin 7,4’-dimethyl ether (7) (Supplementary Fig. 4). Methylation at the C3’-OH and C7-OH positions blocks the initial hydrogen abstraction by the CYP heme center, preventing radical intermediate formation. Additionally, the binding free energy calculations revealed weaker substrate affinity for methylated compounds, with ΔG values of 13.25 and 18.39 kcal/mol for acacetin-containing complexes compared to apigenin (−12.06 kcal/mol) (Supplementary Fig. 21 and Supplementary Table 1). These findings demonstrate that successful intermolecular coupling requires both accessible hydroxyl groups for radical generation and optimal substrate positioning within the enzyme active site.
Building on the discovery of crucial biflavonoid dimerases responsible for oxidative C–C cross-coupling formation, we successfully engineered an E. coli strain for the de novo production of amentoflavone (1), achieving a titer of 4.75 mg/L through the sequential conversion of L-Tyr (1.89 g/L) to apigenin (5, 236.93 mg/L), followed by its subsequent dimerization (Fig. 8d). Notably, this microbial production system proved more efficient than natural sourcing (0.059 mg/g DW, Fig. 1b). This heterologous biosynthetic platform represents a promising alternative for efficient biflavonoid production and holds significant potential for pharmaceutical manufacturing.
In summary, we uncovered the function of CYP90J subfamily members that catalyze the specific C2 unsymmetrical dimerization of biflavonoids with distinct C3’-C8” linkages. These gymnosperm-specific CYP90J enzymes, which our analyses indicate evolved from CYP90E in lycophytes, are essential for C–C type biflavonoid synthesis. Additionally, we elucidated the methylation modification pathway for key ginkgo biflavonoid biosynthesis, including bilobetin, ginkgetin, and isoginkgetin, mediated by GbOMT1 to GbOMT5. Through MD simulations and QM/MM calculations, we discovered that CYP90Js share catalytic mechanisms with bacteria and fungi CYPs, controlling the regiospecificity of C–C cross-coupling reactions. Importantly, GbCYP90J6 catalyzes a diradical coupling reaction, where the steric hindrance from the enzyme pocket and π–π stacking interactions between two apigenin molecules guide their head-tail, anti-parallel alignment, regioselectivity yielding amentoflavone. Finally, building on these mechanistic insights, we successfully engineered a microbial platform for the de novo synthesis of amentoflavone, which holds significant promise for biotechnological applications in the production of valuable biflavonoids.
Methods
Plant materials and reagent
The G. biloba L. trees (3 years old) used in this study were collected from a cultivated population in Shanghai, China, under open-air conditions. The main roots (MR), old roots (OR), fibrous roots (FR), and young stems (YS) were collected in March, June, and August 2022, ensuring consistent growth conditions and ages across different trees. Young leaves (YL) and semi-mature leaves (uML) were collected in June 2022, and mature old leaves (ML) were collected in August 2022. The specific appearance of each tissue type is shown in Fig. 1a. Each tissue type was collected in three biological replicates for transcriptome sequencing and content determination. Tissues used for sequencing were immediately frozen in liquid nitrogen and stored at −80 °C until use. Tissues for content determination were dehydrated in a 37 °C oven until completely dry, followed by extraction and quantification.
Biflavonoid standards, including amentoflavone (catalog: FY1576), ginkgetin (catalog: FY1965), and isoginkgetin (catalog: FY3037), were purchased from Feiyubio (Nantong, China). Bilobetin (catalog: B20029) was obtained from Yuanye (Shanghai, China). Additionally, various mono-flavonoids, such as apigenin (catalog: CFN98843), acacetin (catalog: CFN98744), apigenin 7,4’-dimethyl (catalog: CFN98819), (S)-naringenin (catalog: CFN98742), tricetin (catalog: CFN70392), luteolin (catalog: CFN98784), eriodictyol (catalog: CFN99719), aromadedrin (catalog: CFN98736), kaempferol (catalog: CFN98838), quercetin (catalog: CFN99272), myricetin (catalog: CFN98877), taxifolin (catalog: CFN98734), and ampelopsin (catalog: CFN98326) were sourced from ChemFaces (Wuhan, China). All flavonoids were dissolved in DMSO with a concentration of 50 mM. The stock solutions of all reagents are stored at −20 °C.
Extraction and quantification of biflavonoids from G. biloba
Biflavonoid extraction from G. biloba tissues was performed according to Mistry et al.62. In brief, plant tissues were ground into powder using a high-throughput tissue grinder, and 0.1 g of the powder was transferred to a 2 mL microcentrifuge tube. Then, 1 mL of 70% methanol was added, followed by ultrasound treatment for 30 min. The supernatant was collected, and the extraction process was repeated twice. The solvent was evaporated using a vacuum centrifugal concentrator, and the residue was re-dissolved in 1 mL of 70% (v/v) methanol. Detection and quantification were performed using HPLC (Thermo, UltiMate 3000, USA). Three biological replicates were performed for each sample group.
Transcriptome analysis
G. biloba tissues from three different trees were used to prepare the Illumina sequence library. Total RNA was isolated using the mirVanaTM RNA Isolation Kit (Ambion, USA, catalog AM1560). The sequencing library was constructed from total RNA samples using a TruSeq Stranded mRNA LT Sample Prep Kit (Illumina, San Diego, CA, USA, catalog RS-122-9004DOC). The high-quality library was sequenced with Illumina HiSeqTM 2500. The transcriptome sequencing was conducted by OE Biotech Co., Ltd. (Shanghai, China). The genome of G. biloba, published in 202128, was used to establish the reference transcriptome.
Candidates screening and gene cloning
Candidate genes were initially identified by extracting peroxidases, PPOs, laccases, and CYPs from the G. biloba genome and transcriptome annotations. Additional candidates were identified using Hmmer (version 3.4)63 (E < 1 × 10−10) with Pfam identifiers: peroxidase (PF00141), PPO (PF12143), laccase (PF02578), and CYP (PF00067). Candidates with complete domains and high leaf-specific expression were selected for functional characterization. Expression levels of candidate biflavonoid synthase genes are provided in Supplementary Data 3.
For transient expression in tobacco, candidate genes were cloned from leaf cDNA using 2 × Phanta Flash Master Mix enzyme (Vazyme, Nanjing, China, catalog P520) and subcloned into the SalI and XhoI sites in the pEAQ-HT vector using In-Fusion cloning (ClonExpress II One Step Cloning Kit, Vazyme, Nanjing, China, catalog C112). The resulting vectors were transformed into E. coli DH10B. After sequencing by Sangon Biotech (Shanghai, China), the correct recombinant plasmids were transformed into A. tumefaciens GV3101.
For the plasmid used in the E. coli expression system, trGbCYP90J6 was subcloned into the NcoI and BamHI sites of pFe00. The pFe00 (pCDFduet-AtCPR2-RsCYB5) was constructed from pCDFDuet by inserting the AtCPR2-RBS-RsCYB5 fragment into the SalI and XhoI sites. RsCYB5 was amplified from pCZ44164, and AtCPR2 was cloned from A. thaliana (accession number: NM_179141). TcCYP90J7, ChCYP90J7, CpCYP90J8, MgCYP90J1, TpCYP90J1v1, TpCYP90J9, TpCYP90J10, and SmCYP90E3v1 were synthesized by General Biol (Anhui, China) after codon optimization for E. coli and inserted into pFe00 between the NcoI and BamHI sites. All CYPs and AtCPR2 transmembrane sequences were predicted using TMHMM-2.0 and replaced with a synthetic 17α-tag (MALLLAVF)65.
Mutations were introduced by using mutant-specific primers to amplify plasmids containing the wild-type gene, which served as the template. The amplified fragments were digested with DpnI and then transformed into DH10B. Correct clones were selected by Sanger sequencing.
The O-methyltransferases (OMTs) were screened based on transcriptome annotation and subcloned into pET28a between NdeI and EcoRI. Details of primers and plasmids used in this research are listed in Supplementary Data 4 and 5.
Phylogenetic analyses
To infer the evolutionary history of the CYP90 family, we conducted phylogenetic analysis based on the existence of the CYP90 family in plants, including representative genomes of lycophytes, ferns, gymnosperms, and angiosperms. Additionally, this analysis incorporated known functional CYP90 members collected from the plant P450 database45. On the other hand, we used Orthofinder (version 2.5.4)66 to identify GbCYP90J6 orthologs in bryophytes, lycophytes, ferns, and gymnosperms, analyzing the relationship between the presence of orthologs and biflavonoid compounds. All sequences used in these analysis are listed in Supplementary Data 1 and 2. Sequences were aligned using MUSCLE (version 5.1) multiple sequence alignment, and phylogenetic analyses were conducted with IQ-tree (version 3.0)67 using the maximum likelihood method with bootstrap support and 1000 replications (Fig. 3 and Supplementary Fig. 13).
Transient expression in N. benthamiana
To screen functional genes, A. tumefaciens carrying candidate genes was used for transient infiltration of tobacco plants. Positive strains were cultured in LB medium supplemented with kanamycin (50 μg·mL−1), rifampicin (50 μg·mL−1), and gentamycin (50 μg·mL−1) at 28 °C with shaking until they reached an OD600 of 1. The culture was then centrifuged at 4629 g for 5 min, and the resulting pellet was resuspended in infiltration buffer (10 mM MES,10 mM MgCl2, 150 μM acetosyringone, pH 5.6) to achieve an OD600 of 0.8. The resuspended cultures were incubated at room temperature for 2–3 h before being infiltrated into the leaves of 4-week-old tobacco plants. Each sample was infiltrated into three different plants as biological replicates, with A. tumefaciens carrying the empty vector pEAQ-HT serving as a negative control. Three days post-infiltration, the leaves were infiltrated with 100 μM apigenin (dissolved in 4.75 mM methyl-β-cyclodextrin (MCD) solution) and further cultivated for two days. Leaf tissues (0.3 g) from each treatment were frozen in liquid nitrogen, ground into fine powder, and subjected to triple extraction with 1 mL of water-saturated n-butanol. After extraction, the resulting extracts were evaporated to dryness and reconstituted in 200 μL of methanol for subsequent analysis using HPLC and Q-Exactive mass spectrometry.
Characterization of CYPs in E. coli
E. coli JM109(DE3) (Promega, USA) was used for the characterization of CYPs in vivo. Plasmids utilized in these experiments are described in Supplementary Data 5. The starter cultures of strains carrying plasmids were precultured at 37 °C in LB medium overnight and then inoculated (1:100) into 10 mL of TB medium (with 20 g·L−1 glycerol), with three replicates for each experiment. After reaching an OD600 of 0.6–0.8, isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to the cooled cultures to a final concentration of 0.1 mM. Concurrently, flavonoid substrates dissolved in 50 mM MCD were added to a final concentration of 100 μM (with a final concentration of 4.75 mM MCD). Control cultures containing plasmid pFe00 were run in parallel. The cultures were shaken at 22 °C for 72 h. Afterward, 1 mL of fermentation broth was ultrasonicated and extracted three times with 1 mL of ethyl acetate. All extracts were evaporated to dryness and re-dissolved in 200 μL of methanol for subsequent analysis.
Kinetic analysis
The coding region of GbCYP90J6 was amplified from the vector pEAQ-1.3004 by PCR using primer pair ESC-GbJ6-F/-R (Supplementary Data 4). The PCR fragments were ligated into a predigested pESC/-HIS vector using EcoRI and NotI (NEB, U.S.A.), yielding pESC-GbCYP90J6. The correct plasmids were transformed into the Saccharomyces cerevisiae WAT11. The recombinant yeast strains grew at 30 °C on a synthetic drop-out medium without histidine (SD/-His, containing 2% glucose) plate for 48 h. A single clone was cultured in 200 mL SD/-His (containing 2% glucose) medium until an OD600 reached 1, after which the cells were transferred to SD/-His (containing 2% galactose) for an additional 12 h. Microsomes were prepared as previously described68.
For kinetic analyses, in vitro reactions were conducted in a total volume of 1 mL. The reaction mixtures consist of 500 μg microsomal protein, 50 mM Tris-HCl (pH 7.5), 1 mM NADPH, 5 μM FAD, 5 μg FMN, 4 mM glucose-6-phosphate, 1 unit glucose-6-phosphate dehydrogenase and apigenin ranging from 0.1 to 30 μM. Three biological replicates were set up for analysis. After incubating the mixtures at 30 °C for 1 h, the reactions were quenched by extracting three times with 1 mL of ethyl acetate. The supernatants were evaporated to dryness, resuspended in 100 μL methanol, and the products were quantified by HPLC. Curve fitting was performed using Michaelis–Menten model.
Additionally, for the GbCYP90J6 in vitro flavonoids multi-substrates characterization, substrates were added to 25 μM separately and extracted three times with 1 mL water-saturated n-butanol. The extractions were resuspended in 100 μL of methanol and analyzed by LC-MS/MS.
Characterization of OMTs in vitro
The pET28a vectors carrying the OMTs open reading frame were transformed into E. coli BL21(DE3) for protein expression. The starter cultures of strains harboring the plasmids were grown overnight at 37 °C in an LB medium containing 50 μg/mL kanamycin. These cultures were then inoculated (1:100) into 20 mL of LB medium (containing 50 μg/mL kanamycin). Upon reaching an OD600 of 0.4, IPTG was added to the cooled cultures to a final concentration of 0.1 mM. The expression system was incubated at 22 °C and 220 rpm for 20 h. Subsequently, the cell pellets were collected and resuspended in 1 mL of resuspension buffer (50 mM Tris-HCl, pH 7.5, 50 mM NaCl, 10% glycerol, 5 mM DTT). After adding protease inhibitor (1% v/v Cocktail, Sangon, China), the cells were lysed using an ultrasonic homogenizer at 30 W power. The resulting crude enzymes were used for in vitro enzyme reactions, while crude enzymes from recombinant strains harboring the pET28a empty vector were used as negative controls.
For the active analyses, in vitro reactions were conducted in a total volume of 100 μL. The reaction mixtures consist of 98 μL crude enzymes, 50 μM substrates (amentoflavone), and 0.1 mM S-adenosyl-L-methionine. After incubating the mixtures at 37 °C for 1 h, the reactions were quenched by extracting three times with 100 μL of ethyl acetate. The supernatants were evaporated to dryness and resuspended in 100 μL methanol, and the products were detected by HPLC and LC-MS/MS.
HPLC and LC-MS/MS analyses
For the biflavonoids analysis using the HPLC system, a Dionex Ultimate 3000 HPLC instrument (Thermo Fisher Scientific, USA) equipped with an Athena C18 column (4.6 × 250 mm) (ANPEL Laboratory Technologies, China) was utilized. The mobile phase consisted of solvent A (pure water with 0.1% formic acid) and solvent B (acetonitrile with 0.1% formic acid). Gradient elution began with 95% solvent A and 5% solvent B maintained for 5 min. Subsequently, solvent B increased from 5% to 100% over 15 min and maintained at 100% for 10 min. Thereafter, solvent B was reduced to 5% over 5 min. The flow rate was set at 1 mL·min−1, and UV absorbance was monitored at 280 nm. Quantification of biflavonoids was performed by measuring the peak areas in the UV absorbance at 280 nm, followed by calculation using a standard curve. For the catalytic activity analysis of mutants, the peak area of the wild-type was standardized to 100%, and the relative peak areas of mutants were calculated for comparison. All calculations were based on three biological replicates.
For L-Tyr detection, HPLC analysis was performed using the same column and mobile phase as for biflavonoids. The gradient elution program was as follows: 5–15% B (0–12 min), 15–95% B (12–12.1 min), held at 95% B (12.1–15.1 min), followed by re-equilibration at 5% B for 5 min. The flow rate was maintained at 1 mL·min−1, with UV detection at 278 nm.
Samples prepared as described above were subjected to qualitative analysis by LC-MS/MS (n = 1). Characteristic fragment ions of the target compounds were extracted and analyzed using Xcalibur (version 4.4) software. For the LC-MS/MS analyses of flavonoids, a Dionex Ultimate 3000 HPLC instrument (Thermo Fisher Scientific, USA) equipped with a 00D-4462-AN C18 column (2.1 × 100 mm, 2.6 μm) (Phenomenex, USA) coupled to a Q Exactive Hybrid Quadrupole-Orbitrap mass spectrometer (Thermo Scientific, USA) was employed. The mobile phase consists of H2O (0.1% formic acid, solvent A) and acetonitrile (solvent B). The elution gradient was as follows: 0–1 min 10% solvent B, 1–15 min with a linear increase from 10% to 99% solvent B, 15–18 min 99% solvent B, and 18–21 min returning to 10% solvent B. The flow was set at 0.3 mL·min−1. Electrospray ionization was used as the ion source in negative mode, with full-scan mass spectrometry set with a range of m/z 100–1500 Da.
NMR spectroscopy
The 1H NMR and NOESY spectra were recorded on a Bruker AVANCE III HD 500 MHz spectrometer. Lyophilized samples (1–2 mg) were dissolved in 450 μL of CD3OD (Cambridge Isotope Laboratories), and all spectra were acquired at 25 °C. 1H chemical shifts were referenced to the residual solvent signal of CD3OD at 3.31 ppm. NMR data were processed using Bruker TopSpin (version 3.2) and analyzed with MestReNova (version 16.0.0).
Protein structure prediction, molecular docking, and site-directed mutagenesis
The CYP protein structures containing heme used in this study were predicted by AlphaFold 2, as provided by the tools in PCPD database69. AutoDockTools (version 1.5.7) was employed to prepare the proteins and ligands70. Subsequently, the ligands were docked into the pocket near the heme using AutoDock Vina (version 1.1.2)71. The relative conformations of the two substrates were determined through sequential docking. First, apigenin (5D) was docked to identify its optimal binding pose. Then, the second apigenin (5U) was docked while keeping the first substrate fixed. Models were selected based on specific criteria: proximity of reactive sites to heme iron, proper orientation of C4’-OH in 5D and C7”-OH in 5U toward the catalytic center, compatible positioning for C3’-C8” coupling, and favorable interaction energy scores. No artificial constraints were applied during this process, allowing substrate conformations to emerge naturally from protein-substrate interactions. Amino acids residues within 5 Å of the apigenin ligands were chosen for alanine scanning mutagenesis. ChimeraX (version 1.8)72 was used for protein-ligand model analysis and visualization.
Classical MD
Using GROMACS (version 2021.3) software, a 100–200 ns MD simulation of the protein-ligand complex was conducted73. The AMBER99SB-ILDN force field was selected to generate the protein topology file, and the small molecule ligand topology file was generated using the MCPY module from Amber20 software and the GAFF force field74. A truncated octahedral TIP3P solvent box was applied to the system with a 10 nm distance, and Na+/Cl− ions were added to neutralize the system charge. Energy minimization was performed with 2500 steps of steepest descent followed by 2500 steps of conjugate gradient75. The system was then equilibrated using a 100 ps NVT ensemble simulation and a 100 ps NPT ensemble simulation at a temperature of 298.15 K76. A 100 ns MD ensemble simulation under periodic boundary conditions was subsequently carried out. The Particle Mesh Ewald method was employed for long-range electrostatic interactions, using a 1 nm cutoff for non-bonded interactions, a collision frequency of 2 ps, system pressure set to 101.325 kPa, an integration step of 2 s, and trajectory recording every 10 ps77. Stable molecular conformations from the last 20 ns of the equilibrium trajectory were extracted for the analysis of the model and the strength of interactions between the protein and the ligand.
Quantum mechanics/molecular mechanics (QM/MM) calculation
QM/MM calculations were performed to investigate the reaction mechanism at the active site of GbCYP90J6. Representative snapshots were extracted from the MD trajectory by selecting frames that showed both a minimal distance between the oxo group of Cpd I and the target hydroxyl proton, and a close spatial proximity between the C3’ and C8” carbons of the substrates. All QM/MM calculations were conducted using the ChemShell (version 23.0.3) software package78. The QM/MM system included all amino acid residues within 12 Å of the protein. The QM region comprised the apigenin substrates, a heme iron porphyrin axially coordinated by C425, and residues V215, I216, E291, E292, T293, S294, P295, G359, and V360 (see Supplementary Fig. 22). These residues were selected based on the criterion that they formed hydrogen bonds or hydrophobic interactions with the substrates or facilitated substrates positioning within the active site. The MM region was treated with the AMBER force field79. The active region during QM/MM calculations included all the residues and solvent molecules within 12 Å from the Fe center. During the QM/MM geometry optimization, the QM region was treated with the hybrid UB3LYP80 density functional. For geometry optimization and scanning calculations, the def2-SVP81 basis set was used. Then, the single-point calculations were conducted with the higher-level basis set def2-TZVP82. The D3(BJ) dispersion83 correction was employed for all QM/MM computations. The coordinates of all geometries obtained from the QM/MM calculations are provided in Supplementary Data 6.
De novo biosynthesis of amentoflavone in engineered E. coli
The E. coli GJA3, engineered for high L-Tyr production, was constructed from JM109(DE3) by knocking out tyrR and pheA genes, followed by inserting aroGfbr, tyrAfbr, ydiB, tktA, and ppsA genes into the tyrR locus using CRISPR-Cas9-assisted genome editing according to the protocol described by Yang et al.84. The CRISPR-related platmids pQZ26185 and pQZ26585, as well as plasmid pYH5756 harboring apigenin monomer generation module, were previously constructed in our laboratory.
The starter cultures of engineered strains were precultured at 37 °C in LB medium supplemented with appropriate antibiotics overnight and then inoculated (1:100) into 10 mL of TB medium containing 20 g·L−1 glycerol, with three replicates for each experiment. After reaching an OD600 of 0.6–0.8, 0.1 mM IPTG was added to the cooled cultures. The cultures were fermented at 22 °C, 220 rpm for 72 h.
For L-Tyr quantification, the culture supernatant was acidified with 1 N HCl and analyzed using HPLC. For flavonoid quantification, 1 mL of fermentation broth was ultrasonicated and extracted three times with 1 mL of ethyl acetate. All extracts were evaporated to dryness and re-dissolved in 200 μL of methanol for subsequent analysis.
Statistics & reproducibility
Quantitative experiments were conducted with three independent biological replicates unless otherwise stated. Results, including enzyme activity, metabolite concentrations, and product yields, are presented as mean ± standard deviation (s.d.). No statistical methods were used to predetermine sample size. No data were excluded from the analysis. The experiments were not randomized, and investigators were not blinded to group allocation or outcome assessment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data supporting the findings of this study are available within the paper and its supplementary information files. The raw transcriptome data of G. biloba has been deposited in the Genome Sequence Archive and is accessible under accession number CRA019958. The sequences of GbCYP90s reported in this study have been submitted to the NCBI GenBank under accession numbers GbCYP90J6, PV862377; GbCYP90D59, PV862378; GbCYP90A68, PV862379; GbCYP90B61, PV862380. Additional CYP90 sequences of this study are provided in Supplementary Data 1. The mass spectrometry data generated in this study are available in Figshare [https://doi.org/10.6084/m9.figshare.29552828.v1]. Source data are provided with this paper.
References
-
World Health Organization. Global status report on the public health response to dementia (World Health Organization, Geneva, 2021). https://www.who.int/publications/i/item/9789240033245.
-
Menezes, J. C. J. M. D. S. & Diederich, M. F. Bioactivity of natural biflavonoids in metabolism-related disease and cancer therapies. Pharmacol. Res. 167, 105525 (2021).
-
Uddin, M. S. et al. Emerging signal regulating potential of small molecule biflavonoids to combat neuropathological insults of Alzheimer’s disease. Sci. Total Environ. 700, 134836 (2020).
-
Zhang, Z. et al. Amentoflavone protects hippocampal neurons: anti-inflammatory, antioxidative, and antiapoptotic effects. Neural Regen. Res. 10, 1125 (2015).
-
Tatlı Çankaya, İİ, Devkota, H. P., Zengin, G. & Šamec, D. Neuroprotective potential of biflavone ginkgetin: a review. Life 13, 562 (2023).
-
Zetzsche, L. E. et al. Biocatalytic oxidative cross-coupling reactions for biaryl bond formation. Nature 603, 79–85 (2022).
-
Ihl, R. Gingko biloba extract EGb 761®: clinical data in dementia. Int. Psychogeriatr. 24, S35–S40 (2012).
-
Gavrilova, S. I. et al. Efficacy and safety of Ginkgo biloba extract EGb 761® in mild cognitive impairment with neuropsychiatric symptoms: a randomized, placebo-controlled, double-blind, multi-center trial. Int. J. Geriatr. Psychiatry 29, 1087–1095 (2014).
-
Šalić, A. et al. Extraction of polyphenolic compounds from ginkgo leaves using deep eutectic solvents: a potential solution for the sustainable and environmentally friendly isolation of biflavonoids. Ind. Crops Prod. 219, 119068 (2024).
-
Rahman, M., Riaz, M. & Desai, U. R. Synthesis of biologically relevant biflavanoids–a review. Chem. Biodivers. 4, 2495–2527 (2007).
-
Fregoso-López, D. & Miranda, L. D. Visible-light mediated radical alkylation of flavones: a modular access to nonsymmetrical 3,3″-biflavones. Org. Lett. 24, 8615–8620 (2022).
-
He, X., Yang, F. & Huang, X. Proceedings of chemistry, pharmacology, pharmacokinetics and synthesis of biflavonoids. Molecules 26, 6088 (2021).
-
Baker, W. & Simmonds, W. H. C. 258. Derivatives of 5: 6: 4′-and 5: 8: 4′-trihydroxyflavones, and a note on the structure of ginkgetin. J. Chem. Soc. (Resumed) 1370–1374 https://doi.org/10.1039/JR9400001370 (1940).
-
Sih, C. J. & Malnar, I. Peroxidase-catalyzed oxidative phenolic coupling of some tripeptides containing tyrosine and hydroxyphenylglycine residues. Biocatal. Biotransfor. 18, 301–310 (2000).
-
Kudanga, T., Nemadziva, B. & Le Roes-Hill, M. Laccase catalysis for the synthesis of bioactive compounds. Appl. Microbiol. Biotechnol. 101, 13–33 (2017).
-
Guyot, S., Vercauteren, J. & Cheynier, V. Structural determination of colourless and yellow dimers resulting from (+)-catechin coupling catalysed by grape polyphenoloxidase. Phytochemistry 42, 1279–1288 (1996).
-
Hüttel, W. & Müller, M. Regio- and stereoselective intermolecular phenol coupling enzymes in secondary metabolite biosynthesis. Nat. Prod. Rep. 38, 1011–1043 (2021).
-
Liu, J., Liu, A. & Hu, Y. Enzymatic dimerization in the biosynthetic pathway of microbial natural products. Nat. Prod. Rep. 38, 1469–1505 (2021).
-
Funa, N., Funabashi, M., Ohnishi, Y. & Horinouchi, S. Biosynthesis of hexahydroxyperylenequinone melanin via oxidative aryl coupling by cytochrome P-450 in Streptomyces griseus. J. Bacteriol. 187, 8149–8155 (2005).
-
Shende, V. V. et al. Structure and function of NzeB, a versatile C–C and C–N bond-forming diketopiperazine dimerase. J. Am. Chem. Soc. 142, 17413–17424 (2020).
-
Mazzaferro, L. S., Hüttel, W., Fries, A. & Müller, M. Cytochrome P450-catalyzed regio-and stereoselective phenol coupling of fungal natural products. J. Am. Chem. Soc. 137, 12289–12295 (2015).
-
Sun, C. et al. Molecular basis of regio- and stereo-specificity in biosynthesis of bacterial heterodimeric diketopiperazines. Nat. Commun. 11, 6251 (2020).
-
Ma, C. et al. Exploring the diverse landscape of fungal cytochrome P450-catalyzed regio- and stereoselective dimerization of diketopiperazines. Adv. Sci. 11, 2310018 (2024).
-
Shende, V. V. et al. Molecular dynamics simulations guide chimeragenesis and engineered control of chemoselectivity in diketopiperazine dimerases. Angew. Chem. Int. Ed. 62, e202210254 (2023).
-
Kardam, V., Bhatt, V. & Dubey, K. D. Structural and mechanistic insights into oxidative biaryl coupling to form the arylomycin core by an engineered CYP450. Dalton Trans. 54, 754–763 (2025).
-
Fujiyama, K. et al. Structural insights into a key step of brassinosteroid biosynthesis and its inhibition. Nat. Plants 5, 589–594 (2019).
-
Mohanta, T. K., Tamboli, Y. & Zubaidha, P. K. Phytochemical and medicinal importance of Ginkgo biloba L. Nat. Prod. Res. 28, 746–752 (2014).
-
Liu, H. et al. The nearly complete genome of Ginkgo biloba illuminates gymnosperm evolution. Nat. Plants 7, 748–756 (2021).
-
Geiger, H. Biflavones from some mosses. Z. Naturforsch. C. 43, 1–4 (1988).
-
Suárez, A. I., Diaz, M. B., Delle Monache, F. & Compagnone, R. S. Biflavonoids from Podocalyx loranthoides. Fitoterapia 74, 473–475 (2003).
-
Leong, K. I., Alviarez, P. F., Compagnone, R. S. & Suárez, A. I. Isolation and structural elucidation of chemical constituents of Amanoa almerindae. Pharm. Biol. 47, 496–499 (2009).
-
Li, X.-C. et al. Fatty acid synthase inhibitors from plants: isolation, structure elucidation, and SAR studies. J. Nat. Prod. 65, 1909–1914 (2002).
-
Šamec, D. et al. 3′-8″- Biflavones: a review of their structural diversity, natural occurrence, role in plants, extraction and identification. Molecules 29, 4634 (2024).
-
Nelson, D. & Werck-Reichhart, D. A P450-centric view of plant evolution. Plant J. 66, 194–211 (2011).
-
Banks, J. A. et al. The Selaginella genome identifies genetic changes associated with the evolution of vascular plants. Science 332, 960–963 (2011).
-
Li, C. et al. Extraordinary preservation of gene collinearity over three hundred million years revealed in homosporous lycophytes. Proc. Natl. Acad. Sci. 121, e2312607121 (2024).
-
Fang, Y. et al. The genome of homosporous maidenhair fern sheds light on the euphyllophyte evolution and defences. Nat. Plants 8, 1024–1037 (2022).
-
Liu, Y. et al. The Cycas genome and the early evolution of seed plants. Nat. Plants 8, 389–401 (2022).
-
Wan, T. et al. A genome for gnetophytes and early evolution of seed plants. Nat. Plants 4, 82–89 (2018).
-
Xiong, X. et al. The Taxus genome provides insights into paclitaxel biosynthesis. Nat. Plants 7, 1026–1036 (2021).
-
Wan, T. et al. The Welwitschia genome reveals a unique biology underpinning extreme longevity in deserts. Nat. Commun. 12, 4247 (2021).
-
Shalev, T. J. et al. The western redcedar genome reveals low genetic diversity in a self-compatible conifer. Genome Res. 32, 1952–1964 (2022).
-
Fu, F. et al. The Metasequoia genome and evolutionary relationships among redwoods. Plant Commun. 4, 100643 (2023).
-
Wang, B. et al. High-quality Arabidopsis Thaliana genome assembly with nanopore and HiFi long reads. Genomics Proteom. Bioinforma. 20, 4–13 (2022).
-
Hansen, C. C., Nelson, D. R., Møller, B. L. & Werck-Reichhart, D. Plant cytochrome P450 plasticity and evolution. Mol. Plant 14, 1244–1265 (2021).
-
Commisso, M. et al. Bryo-Activities: a review on how bryophytes are contributing to the arsenal of natural bioactive compounds against fungi. Plants 10, 203 (2021).
-
Wallace, J. W. C-Glycosylflavones in the Gnetopsida: a preliminary report. Am. J. Bot. 66, 343–346 (1979).
-
Geiger, H. & Quinn, C. Biflavonoids. In The Flavonoids 692–742 https://doi.org/10.1007/978-1-4899-2909-9_13 (Springer US, Boston, MA, 1975).
-
Kariyone, T. & Sawada, T. Studies on flavonoids of the leaves of coniferae and allied plants. I: on the flavonoid from the leaves of Torreya nucifera SIEB. et ZUCC. J. Pharm. Soc. Jpn. 78, 1010–1013 (1958).
-
Anand, A. V., Arinchedathu Surendran, V. & Sukumaran, S. T. Bioactive metabolites in gymnosperms. In Plant Metabolites: Methods, Applications and Prospects 317–346 https://doi.org/10.1007/978-981-15-5136-9_14 (Springer, Singapore, 2020).
-
Adasme, M. F. et al. PLIP 2021: expanding the scope of the protein–ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 49, W530–W534 (2021).
-
Moreno, A. G. et al. Structure, isomerization and dimerization processes of naringenin flavonoids. Phys. Chem. Chem. Phys. 23, 18068–18077 (2021).
-
Yu, X. et al. Biosynthesis of strained piperazine alkaloids: uncovering the concise pathway of herquline A. J. Am. Chem. Soc. 138, 13529–13532 (2016).
-
Li, S.-H. et al. Chemoenzymatic synthesis of fluorinated mycocyclosin enabled by the engineered cytochrome P450-catalyzed biaryl coupling reaction. J. Am. Chem. Soc. 146, 19962–19973 (2024).
-
Nakagawa, A. et al. A bacterial platform for fermentative production of plant alkaloids. Nat. Commun. 2, 326 (2011).
-
Li, J. et al. Production of plant-specific flavones baicalein and scutellarein in an engineered E. coli from available phenylalanine and tyrosine. Metab. Eng. 52, 124–133 (2019).
-
Mizutani, M. & Sato, F. Unusual P450 reactions in plant secondary metabolism. Arch. Biochem. Biophys. 507, 194–203 (2011).
-
An, Z. et al. Lineage-specific CYP80 expansion and benzylisoquinoline alkaloid diversity in early-diverging eudicots. Adv. Sci. 11, 2309990 (2024).
-
Meng, F. et al. Characterization of two CYP80 enzymes provides insights into aporphine alkaloid skeleton formation in Aristolochia contorta. Plant J. 118, 1439–1454 (2024).
-
Liu, Y. et al. Ginkgetin alleviates inflammation and senescence by targeting STING. Adv. Sci. 12, 2407222 (2025).
-
Smer-Barreto, V. et al. Discovery of senolytics using machine learning. Nat. Commun. 14, 3445 (2023).
-
Guo, J., Wu, Y., Jiang, M., Wu, C. & Wang, G. An LC–MS-based metabolomic approach provides insights into the metabolite profiles of Ginkgo biloba L. at different developmental stages and in various organs. Food Res. Int. 159, 111644 (2022).
-
Potter, S. C. et al. HMMER web server: 2018 update. Nucleic Acids Res. 46, W200–W204 (2018).
-
Sun, Y. et al. De novo production of versatile oxidized kaurene diterpenes in Escherichia coli. Metab. Eng. 73, 201–213 (2022).
-
Barnes, H. J., Arlotto, M. P. & Waterman, M. R. Expression and enzymatic activity of recombinant cytochrome P450 17 alpha-hydroxylase in Escherichia coli. Proc. Natl. Acad. Sci. USA 88, 5597–5601 (1991).
-
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238 (2019).
-
Trifinopoulos, J., Nguyen, L.-T., von Haeseler, A. & Minh, B. Q. W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44, W232–W235 (2016).
-
Pompon, D., Louerat, B., Bronine, A. & Urban, P. Yeast expression of animal and plant P450s in optimized redox environments. In Methods in Enzymology (eds Abelson, J. N., Simon, M. I., Johnson, E. F. & Waterman, M. R.) 272, 51–64 (Academic Press, 1996).
-
Wang, H. et al. PCPD: plant cytochrome P450 database and web-based tools for structural construction and ligand docking. Synth. Syst. Biotechnol. 6, 102–109 (2021).
-
Forli, S. et al. Computational protein–ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 11, 905–919 (2016).
-
Eberhardt, J., Santos-Martins, D., Tillack, A. F. & Forli, S. AutoDock Vina 1.2.0: new docking methods, expanded force field, and Python bindings. J. Chem. Inf. Model. 61, 3891–3898 (2021).
-
Meng, E. C. et al. UCSF ChimeraX: tools for structure building and analysis. Protein Sci. 32, e4792 (2023).
-
da Silva, T. U. et al. Development of parameters compatible with the CHARMM36 force field for [Fe4S4]2+ clusters and molecular dynamics simulations of adenosine-5’-phosphosulfate reductase in GROMACS 2019. J. Biomol. Struct. Dyn. 40, 3481–3491 (2022).
-
Man, V. H., Nguyen, P. H. & Derreumaux, P. High-resolution structures of the amyloid-β 1–42 dimers from the comparison of four atomistic force fields. J. Phys. Chem. B 121, 5977–5987 (2017).
-
Ren, J., Vaid, T. M., Lee, H., Ojeda, I. & Johnson, M. E. Evaluation of interactions between the hepatitis C virus NS3/4A and sulfonamidobenzamide based molecules using molecular docking, molecular dynamics simulations and binding free energy calculations. J. Comput. Aided Mol. Des. 37, 53–65 (2023).
-
Suleiman, M. R. et al. Discovery of small molecule inhibitors through pharmacophore modeling, molecular docking, molecular dynamics simulation and experimental validation against myeloid cell leukemia-1 (Mcl-1). J. Biomol. Struct. Dyn. 39, 2512–2525 (2021).
-
Ulutürk, M., Karabacak Atay, Ç., Dede, B. & Tilki, T. Potentially bioactive novel isophthalic acid based azo molecules: synthesis, characterization, quantum chemical calculations, ADMET properties, molecular docking and molecular dynamics simulations. Polycycl. Aromat. Compd. 1–22 https://doi.org/10.1080/10406638.2023.2284797 (2023).
-
Metz, S., Kästner, J., Sokol, A. A., Keal, T. W. & Sherwood, P. ChemShell—a modular software package for QM/MM simulations. WIREs Comput. Mol. Sci. 4, 101–110 (2014).
-
Zhang, J. & Liu, Y. A QM/MM study of the catalytic mechanism of aspartate ammonia lyase. J. Mol. Graph. Model. 51, 113–119 (2014).
-
Gritsenko, O. V., Cordero, N. A., Rubio, A. & Alonso, J. A. Gradient correction to the exchange pair-correlation function of the weighted spin-density approximation in the density functional formalism. Chem. Phys. Lett. 296, 307–312 (1998).
-
Bayach, I., Sancho-García, J. C., Di Meo, F., Weber, J.-F. F. & Trouillas, P. π-Stacked polyphenolic dimers: a case study using dispersion-corrected methods. Chem. Phys. Lett. 578, 120–125 (2013).
-
Weigend, F. & Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305 (2005).
-
Zheng, D., Yuan, Y. & Wang, F. Determining the hydration free energies of selected small molecules with MP2 and local MP2 through adaptive force matching. J. Chem. Phys. 154, 104113 (2021).
-
Li, Q. et al. A modified pCas/pTargetF system for CRISPR-Cas9-assisted genome editing in Escherichia coli. Acta Biochim. Biophys. Sin. 53, 620–627 (2021).
-
Zhang, Q., Wu, Y.-H., Huang, X.-S., Liu, H.-L. & Wang, Y. Design and optimization for efficient production of (S)-canadine in Escherichia coli. ACS Sustain. Chem. Eng. 12, 6941–6951 (2024).
-
Madeira, F. et al. Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res. 50, W276–W279 (2022).
-
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v6: recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. 52, W78–W82 (2024).
Acknowledgements
This work is financially supported by the National Key R&D Program of China (No. 2024YFA0919900) to Y.W. and H.-L.L., the State Key Laboratory of Plant Trait Design to Y.W., the Key Laboratory of Plant Carbon Capture, Chinese Academy of Sciences to Y.W., and the Shanghai Municipal Science and Technology Major Project to Y.W. We thank Prof. David R. Nelson (University of Tennessee Health Science Center) for his assistance on the classification of the CYP90J subfamily referenced in this study.
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Lorenzo Caputi, who co-reviewed with Qi Ding and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. [A peer review file is available].
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Dai, XH., Zhu, JM., Wang, GY. et al. Gymnosperm-specific CYP90Js enable biflavonoid biosynthesis and microbial production of amentoflavone. Nat Commun 16, 7792 (2025). https://doi.org/10.1038/s41467-025-62990-6
-
Received:
-
Accepted:
-
Published:
-
DOI: https://doi.org/10.1038/s41467-025-62990-6