Background & Summary
With approximately 15,000 described species, Trichoptera (caddisfly) represents the second most diverse monophyletic group of aquatic insects1. The most species diversity for Trichoptera is found in the Indomalayan and Neotropical regions, with 47–77% of widespread genera recorded. Five families comprise 55% of the global Trichoptera species, while 19 families each contain fewer than 30 species2. Trichoptera larvae (or caddisfly larvae) build their protective cases using secreted silk combined with selected foreign materials, such as sand grains, mollusk shells, or plant fragments3,4,5. This case-building behavior has enabled their ecological diversification, allowing them to inhabit environments that are otherwise inaccessible to many other species3. Among caddisflies, Stenopsyche angustata stands out due to its large size and preference for fast-flowing water environments. Its larvae are typically dark brown, with long, narrow heads and short antennae6. S. angustata produces adhesive silk, which is used to construct intricate underwater composite structures7.
Advancements in sequencing technology have significantly enhanced our understanding of the genome, leading to the decoding of Trichoptera species, such as Himalopsyche anomala and Eubasilissa splendida8, as well as Cheumatopsyche charites9. However, the genome of S. angustata has not yet been sequenced. High-quality reference genomes are crucial for advancing genetic and evolutionary research on this species. In this study, we employed PacBio long-read sequencing and Hi-C techniques to achieve chromosome-level genome assembly for S. angustata. The final assembly totaled 510.47 Mb, with a scaffold N50 of 39.81 Mb. Hi-C scaffolding anchored 99.63% of the initial sequences to 13 pseudochromosomes. Repetitive elements accounted for 44.29% (226.08 Mb) of the genome, and a total of 10,699 protein-coding genes were identified. This high-quality genome facilitates our understanding of the adaptive evolution in Trichoptera.
Methods
Sample collection and sequencing
S. angustata larvae were collected form the Beilun River (21.80 N, 107.89E), Guangxi Zhuang Autonomous Region, China on November 15, 2022. Nine live larval individuals were immediately frozen in liquid nitrogen and stored at −80 °C until DNA extraction. Due to their small size, DNA was extracted from the whole bodies of four of the collected individuals, and prepared for both second-generation and third-generation sequencing using the classic phenol–chloroform method. The quality and quantity of the extracted DNA were assessed using an Agilent 2100 bioanalyzer (Agilent Technologies, Santa Clara, CA, USA), and integrity was evaluated using agarose gel electrophoresis with ethidium bromide staining. Second-generation sequencing was performed on Illumina platform conducted by Novogene Bioinformatics Technology (Novogene, Beijing, China) (Table 1).
For PacBio sequencing, high-quality DNA samples were randomly fragmented into smaller pieces using a Covaris ultrasonic disruptor (Covaris, Woburn, MA, USA). Large DNA fragments were enriched and purified with magnetic beads, after which they underwent damage repair and end repair. Adapters were then ligated to both ends of the DNA fragments, forming stem-loop structures. Unligated fragments were removed via exonuclease treatment. The constructed libraries were then sequenced using the PacBio Sequel system (PacBio, Menlo Park, CA, USA). Consensus sequences were generated by aligning subreads obtained from a single Zero-Mode Waveguide (ZMW), omitting the need for a reference genome. Circular Consensus Sequence (CCS) reads were obtained using the CCS algorithm, requiring at least two full-pass subreads from the insert. The raw sequencing data yielded 97.12 Gb (approximately 180.9 × coverage) with a scaffold N50 of 33.78 kb (Table 1).
Using a modified standard protocol as described previously10, we constructed Hi-C libraries using the whole bodies of the remaining five S. angustata larvae individuals. Larval individuals were ground in liquid nitrogen and cross-linked with a 4% formaldehyde solution at room temperature under vacuum for 30 min. The crosslinking reaction was quenched by adding 2.5 M glycine and incubating for 5 min, followed by placing the sample on ice for 15 min. The samples were then centrifuged at 2500 rpm at 4 °C for 10 min, and the pellet was washed with 500 μl PBS and centrifuged again at 2500 rpm for 5 min. The pellet was resuspended in 20 μl of lysis buffer (1 M Tris-HCl, pH 8, 1 M NaCl, 10% CA-630, and 13 units of protease inhibitor) and centrifuged at 5000 rpm at room temperature for 10 min. The pellet was washed twice with 100 μl ice-cold 1x NEB buffer and centrifuged at 5000 rpm for 5 min. The nuclei were resuspended in 100 μl of NEB buffer, solubilized with dilute sodium dodecyl sulfate (SDS), and incubated at 65 °C for 10 min. SDS was neutralized with Triton X-100, and the solution was incubated overnight with the 4-cutter restriction enzyme MboI (400 units) at 37 °C on a rocking platform to digest the DNA into smaller fragments. The DNA ends were then labeled with biotin-14-dCTP, followed by blunt-end ligation of the cross-linked fragments. Proximal chromatin DNA was re-ligated using a ligation enzyme, and the nuclear complexes were reverse cross-linked by incubation with proteinase K at 65 °C. DNA was then purified through phenol-chloroform extraction, and biotin was removed from non-ligated fragment ends with T4 DNA polymerase. The ends of sonicated fragments (200–600 bp) were repaired with a mixture of T4 DNA polymerase, T4 polynucleotide kinase, and Klenow DNA polymerase. Biotin-labeled Hi-C samples were enriched with streptavidin C1 magnetic beads. After adding A-tails to the fragment ends and ligating Illumina paired-end (PE) sequencing adapters, Hi-C sequencing libraries were amplified by polymerase chain reaction (PCR) (12–14 cycles) and sequenced on an Illumina PE150 platform by Novogene Bioinformatics Technology (Novogene, Beijing, China) (Table 1).
Genome size estimation and assembly
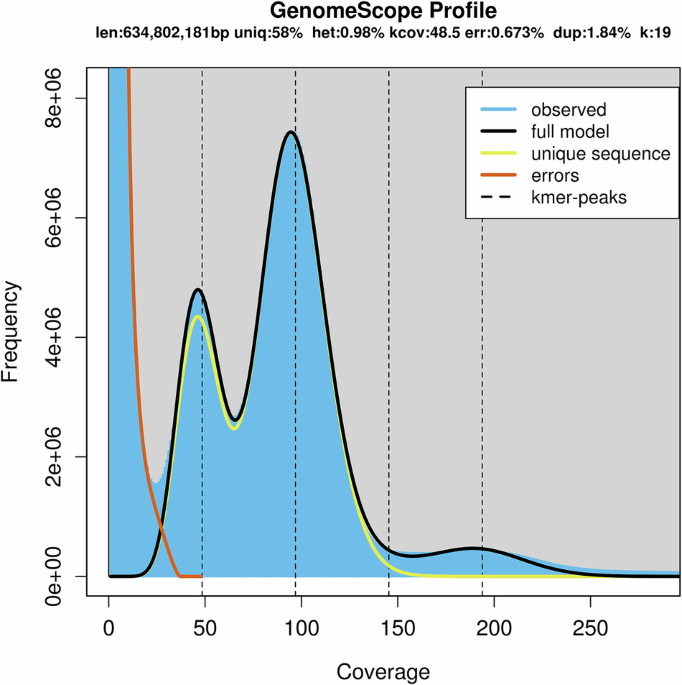
The genome size, heterozygosity rate, and repeat content of S. angustata were estimated through k-mer analysis using Jellyfish (v2.3.0)11. A total of 17, 977, 255, 521 17-mers with a depth peak of 33 were analyzed. Using the formula: genome size = K-num/K-depth, the genome size of S. angustata was estimated to be 544.77 Mb. After removing contaminated and erroneous sequences, the revised genome size was determined to be 536.86 Mb. Meanwhile, the estimated heterozygous ratio and repeat content were approximately 0.97% and 43.07%, respectively (Table 2).
PacBio subreads were used for de novo genome assembly using the wtdbg2 software12. Initially, DNA sequences were randomly sheared into 1,024 bp fragments for clone sequencing. Reads were then used to construct a vertex sequence based on their similar relationships. Sequencing reads were then analyzed to identify overlaps, and vertex sequences were constructed based on their similarity relationships. These vertex sequences were subsequently concatenated based on their position on the reads. Contigs were obtained by segmenting sequences at repeat region boundaries to improve assembly accuracy. Scaffold sequences were constructed using the paired-end relationships of large-fragment sequencing data. The chromosome number and ploidy were determined through our previous karyotype analysis. Chromosome-level genome assembly was achieved using the ALLHiC pipeline13, which consists of five steps: pruning, partition, rescue, optimization, and building. In the pruning step, crosslinking signals between homologous chromosomes were trimmed to separate alleles and homologous sequences into their respective haplotypes, enabling independent assembly and minimizing errors. During partition, Hi-C interaction signals between contigs were analyzed to cluster them into groups, effectively distinguishing homologous chromosomes. The rescue step addressed assembly inconsistencies by identifying collapsed regions, which are highly similar sequences that were not properly separated, and by detecting the strongest interaction signals between non-collapsed regions. Additionally, contigs that remained unassigned during pruning were reassessed and correctly placed. In the optimization step, genetic algorithms were used to iteratively refine the sorting and orientation of contigs within each chromosome group. Finally, in the building step, a chromosome-level assembly was generated and visualized using a contact map to assess Hi-C interaction patterns, confirming the accuracy of the final genome assembly.
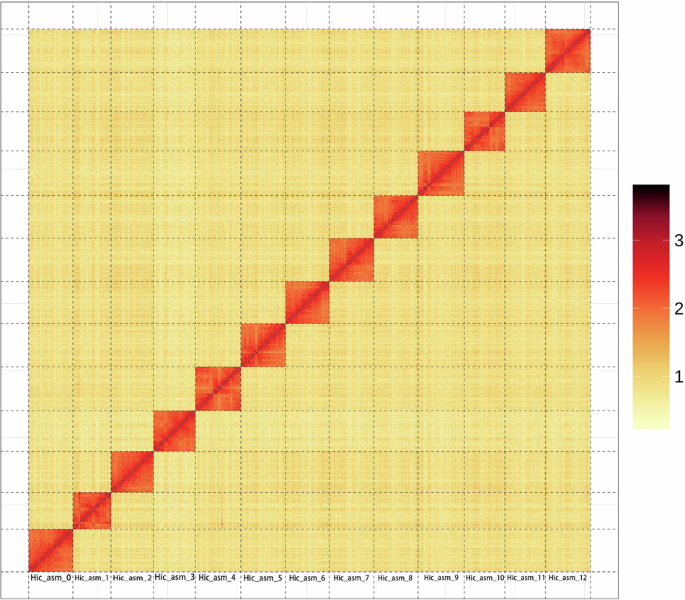
The assembly had a total contig length of 510,457,233 bp and a contig N50 length of 13,847,508 bp. The total scaffold length was 510,471,733 bp, and a scaffold N50 length of 39,811,520 bp (Table 3). The genome anchoring rate was 99.63% (Table 4). Based on the Hi-C contact map (Fig. 1), both the genome metrics and anchoring rate were excellent.

Hi-C contact map of the chromosome-level assembly of Stenopsyche angustata. The x- and y-axes represent the assembled chromosomes, arranged in numerical order. Red color indicates higher contact frequencies, while yellow color reflects lower frequencies. The strong diagonal pattern demonstrates predominant intra-chromosomal interactions, confirming accurate chromosome anchoring and assembly. The color scale bar shows the contact intensities (from 1 to 3).
Repeat annotation
A comprehensive repeat annotation pipeline was applied, integrating homology-based alignment and de novo search strategies to identify genome-wide repeats. Tandem repeats were detected using Tandem Repeats Finder (TRF)14 through ab initio prediction, identifying approximately 3.97 Mb of sequences. For homology-based repeat identification, the Repbase database15 was employed in conjunction with RepeatMasker16 and its in-house script RepeatProteinMask with default parameters. This approach identified 222.37 Mb and 27.76 Mb of repeat regions, respectively (Table 5). The ab initio prediction generated a de novo repetitive element database using LTR_FINDER17, RepeatScout18, and RepeatModeler19 with default parameters (Table 6). Repeat sequences longer than 100 bp and with less than 5% ambiguous nucleotides (‘N’) were retained to construct the raw transposable element (TE) library. A customized, non-redundant library was then constructed by merging the de novo TE library with existing Repbase data, removing duplicate sequences using the UCLUST algorithm20. The obtained library was subsequently used for DNA-level repeat identification with RepeatMasker.
Denovo + Repbase transposable elements (TEs) were predicted using de novo tools (RepeatModeler, RepeatScout, Piler21, and LTR_FINDER) and combined with the RepBase nucleic acid data. The results were integrated using UCLUST following the 80-80-80 rule to ensure high-confidence matches, and were finally annotated using RepeatMasker. TE proteins were identified by annotating the genome with the RepBase protein data using the RepeatProteinMask software. Combined TEs represent the results obtained by integrating the two aforementioned methods and removing redundancy. The ‘Unknown’ category includes repeat sequences that could not be classified by RepeatMasker. The ‘Total’ category represents the non-redundant result obtained after removing overlaps between the different classifications. DNA: DNA transposons; LINE: long interspersed nuclear elements; SINE: short interspersed nuclear elements; LTR: long terminal repeat.
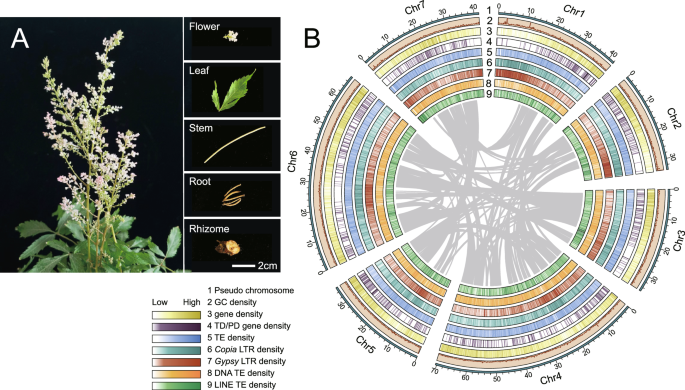
The genome of S. angustata exhibited notable variations across chromosomes. GC content varied distinctly across the chromosomes, with a relatively higher level on chromosomes 12, potentially associated with gene enrichment and transcriptional activity in this region. Moreover, the distribution of interspersed repeats also showed clear chromosome-specific patterns, particularly on chromosomes 8 and 12, where an increased density of these sequences may suggest frequent replication or insertion events. In contrast, long repeats were predominantly concentrated on chromosomes 12 and 13, indicating the potential importance of these regions in maintaining genome structure and regulating chromosomal conformation. Additionally, we observed higher gene density on chromosome 13 (Fig. 2), which may contain a large number of functional genes or active transcription units. These findings provide valuable insights into the structural and functional characteristics of the S. angustata genome, offering a foundation for future research on functional gene characterization.

Circos plot illustrating the genomic features of S. angustata. From the innermost to the outermost ring, the plot shows GC content, interspersed repeats, long repeats and gene density, highlighting variations in these figures across chromosomes. Chromosomes are labeled around the outermost ring.
Gene structure and functional annotation
Homologous protein sequences were obtained from Ensembl22 and NCBI23. These sequences were aligned to the genome using TblastN (v2.2.26) with E-value ≤ 1e−5. Subsequently, GeneWise (v2.4.1) was used to align the matching proteins to the corresponding genome sequences, ensuring accurate spliced alignments and gene structure prediction of the identified protein regions. For ab initio gene prediction, an automated pipeline was employed, incorporating Augustus (v3.2.3)24, Geneid (v1.4)25, Genescan (v1.0)26, GlimmerHMM (v3.04)27, and SNAP (2013-11-29)28. The genome annotation was further refined using transcriptome read assemblies generated by Trinity (v2.1.1)29.
Gene functions were assigned by aligning the predicted protein sequences to the Swiss-Prot database30 using Blastp (E-value ≤ 1e−5). Motifs and domains were annotated using InterProScan70 (v5.31) against publicly available databases, including Swiss-Prot, Nr31, Interpro32, Pfam33, etc. Gene Ontology (GO) terms were assigned based on the corresponding InterPro entries. Protein functions were predicted by transferring annotations from the closest BLAST hit (E-value < 1e−5) in the Swiss-Prot database and DIAMOND (v0.8.22) or BLAST hits (E-value < 1e−5) in the NR20 database. Additionally, the gene set was mapped to Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways to identify the best match for each gene. The protein sequences derived from gene structure prediction were aligned to known protein databases, allowing functional prediction for 10,699 encoding genes, which represent 97.7% of the total genes (Table 7).
Non-coding RNA annotation
Non-coding RNA (ncRNA) annotation was performed to identify tRNAs, rRNAs, miRNAs, and snRNAs. tRNA genes were predicted using tRNAscan-SE34. Due to the high conservation of rRNA sequences, rRNAs were identified by aligning reference sequences from related species to the genome using BLAST. Other ncRNAs, including miRNAs and snRNAs, were detected by searching against the Rfam database35 using Infernal36 (http://infernal.janelia.org/) with default parameters (Table 8).
Data Records
This Whole Genome Shotgun project has been deposited at GenBank under the accession JBPJGE00000000037. Besides, the genome and raw sequencing data are publicly accessible in China National Gene Bank (https://db.cngb.org/) with the accession number CNP000649038. The genome assembly data and annotations have also been deposited at Figshare39. The PacBio reads are available in the NCBI SRA database under accession number SRR3208962140, while the Hi-C reads can be accessed under SRR3208962041.
Technical Validation
The integrity and accuracy of S. angustata genome assembly were evaluated through multiple approaches. First, the Hi-C contact map revealed strong intra-chromosomal interaction signals along the diagonal (Fig. 1), confirming the integrity of the genome structure. Second, the distribution of GC content demonstrated that there was no significant contamination in the assembly sequence (Fig. 2). To further assess genomic integrity, a BUSCO42 analysis was performed, showing that 98.8% of the complete single copy genes were assembled from a set of 1,013 single-copy orthologous genes (C: 98.8% [S: 98.0%, D: 0.8%], F: 0.4%, M: 0.8%, n: 1,013). At the same time, CEGMA43 was used to evaluate the completeness of the S. angustata genome. The results showed that 230 of the 248 full-length genes in the core gene set were included, achieving a 92.74% coverage. For accuracy assessment, small fragment library reads were mapped to the assembled genome using the BWA software44. The mapping rate and genome coverage rate were found to be 99.02% and 99.53%, respectively. Additionally, 10,456 (97.7%) gene models were successfully annotated in databases such as NR, KEGG, GO, Pfam and Interpro. Taken together, these results provide strong evidence that the obtained de novo S. angustata genome is of high quality.
Code availability
No specific code or script was used in this work. Commands used for data processing were all executed according to the manuals and protocols of the corresponding software.
References
-
Malm, T., Johanson, K. A. & Wahlberg, N. The evolutionary history of Trichoptera (Insecta): A case of successful adaptation to life in freshwater. Systematic Entomology 38, 459–473, https://doi.org/10.1111/syen.12016 (2013).
-
de Moor, F. C. & Ivanov, V. D. in Freshwater Animal Diversity Assessment (eds E. V. Balian, C. Lévêque, H. Segers, & K. Martens) 393-407 (Springer Netherlands, 2008).
-
Mouro, L. D., Zatoń, M., Fernandes, A. C. S. & Waichel, B. L. Larval cases of caddisfly (Insecta: Trichoptera) affinity in Early Permian marine environments of Gondwana. Scientific Reports 6, 19215, https://doi.org/10.1038/srep19215 (2016).
-
Gaino, E., Cianficconi, F., Rebora, M. & Todini, B. Case‐building of some Trichoptera larvae in experimental conditions: Selectivity for calcareous and siliceous grains. Italian Journal of Zoology 69, 141–145 (2002).
-
Stewart, R. J. & Wang, C. S. Adaptation of caddisfly larval silks to aquatic habitats by phosphorylation of h-fibroin serines. Biomacromolecules 11, 969–974, https://doi.org/10.1021/bm901426d (2010).
-
Huang, J.-C. et al. Characterization of the complete mitochondrial genome of Stenopsyche angustata (Trichoptera, Stenopsychidae). Mitochondrial DNA Part B 5, 3114–3115 (2020).
-
Wang, Y. J. et al. The silk gland proteome of Stenopsyche angustata provides insights into the underwater silk secretion. Insect Molecular Biology 33, 41–54 (2024).
-
Ge, X. et al. Chromosome-scale genome assemblies of Himalopsyche anomala and Eubasilissa splendida (Insecta: Trichoptera). Scientific Data 11, 267 (2024).
-
Ge, X. et al. The first chromosome-level genome assembly of Cheumatopsyche charites Malicky and Chantaramongkol, 1997 (Trichoptera: Hydropsychidae) reveals how it responds to pollution. Genome biology and evolution 14, evac136 (2022).
-
Belton, J. M. et al. Hi-C: a comprehensive technique to capture the conformation of genomes. Methods 58, 268–276, https://doi.org/10.1016/j.ymeth.2012.05.001 (2012).
-
Marcais, G. & Kingsford, C. Jellyfish: A fast k-mer counter. Tutorialis e Manuais 1, 1038 (2012).
-
Ruan, J. & Li, H. Fast and accurate long-read assembly with wtdbg2. Nature methods 17, 155–158 (2020).
-
Zhang, X., Zhang, S., Zhao, Q., Ming, R. & Tang, H. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nature Plants 5, 833–845, https://doi.org/10.1038/s41477-019-0487-8 (2019).
-
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic acids research 27, 573–580 (1999).
-
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenetic and genome research 110, 462–467 (2005).
-
Chen, N. Using Repeat Masker to identify repetitive elements in genomic sequences. Current protocols in bioinformatics 5, 4.10. 11–14.10. 14 (2004).
-
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic acids research 35, W265–W268 (2007).
-
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358 (2005).
-
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences 117, 9451–9457 (2020).
-
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010).
-
Edgar, R. C. & Myers, E. W. PILER: identification and classification of genomic repeats. Bioinformatics-Oxford 21, i152 (2005).
-
Hubbard, T. et al. The Ensembl genome database project. Nucleic acids research 30, 38–41 (2002).
-
Pruitt, K. D., Tatusova, T. & Maglott, D. R. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic acids research 35, D61–D65 (2007).
-
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic acids research 34, W435–W439 (2006).
-
Blanco, E., Parra, G. & Guigó, R. Using geneid to identify genes. Current protocols in bioinformatics 18, 4.3. 1–4.3. 28 (2007).
-
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. Journal of molecular biology 268, 78–94 (1997).
-
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
-
Korf, I. Gene finding in novel genomes. BMC bioinformatics 5, 1–9 (2004).
-
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nature protocols 8, 1494–1512 (2013).
-
Gasteiger, E., Jung, E. & Bairoch, A. SWISS-PROT: connecting biomolecular knowledge via a protein database. Current issues in molecular biology 3, 47–55 (2001).
-
Yu, K. & Zhang, T. Construction of customized sub-databases from NCBI-nr database for rapid annotation of huge metagenomic datasets using a combined BLAST and MEGAN approach. PLoS One 8, e59831 (2013).
-
Hunter, S. et al. InterPro: the integrative protein signature database. Nucleic acids research 37, D211–D215 (2009).
-
Mistry, J. et al. Pfam: The protein families database in 2021. Nucleic acids research 49, D412–D419 (2021).
-
Chan, P. P., Lin, B. Y., Mak, A. J. & Lowe, T. M. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic acids research 49, 9077–9096 (2021).
-
Nawrocki, E. P. et al. Rfam 12.0: updates to the RNA families database. Nucleic acids research 43, D130–D137 (2015).
-
Nawrocki, E. P. Annotating functional RNAs in genomes using Infernal. RNA sequence, structure, and function: computational and bioinformatic methods, 163-197 (2014).
-
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_051167455.1 (2025).
-
NCBI Sequence Read Archive, https://db.cngb.org/search/project/CNP0006490/ (2024).
-
Wang, Y. chromosome-level genome assemblies and annotation of Stenopsyche angustata, https://doi.org/10.6084/m9.figshare.28200614.v2 (2025).
-
Sericulture, I. o. Stenopsyche angustata PacBio sequences. NCBI Sequence Read Archive <https://identifiers.org/ncbi/insdc.sra:SRR32089621 (2025).
-
Sericulture, I. o. Stenopsyche angustata Hi-C sequences. NCBI Sequence Read Archive, https://identifiers.org/ncbi/insdc.sra:SRR32089620 (2025).
-
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
-
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067 (2007).
-
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. bioinformatics 25, 1754–1760 (2009).
Acknowledgements
This study was supported by Natural Science Foundation of China (32470554, 42266004, 31972873), Hebei Natural Science Foundation (C2024406007), and the Scientific Research Foundation Project of GuangXi (2021JJD130022, 2025GXNSFAA069988). We would like to thank Yinshan Cui and Pulis Biotechnological Company (Kunming, Yunnan, China) for their support in data analysis. We also appreciate their valuable insights and assistance throughout this project.
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, Y., Liu, X., Qin, H. et al. Chromosome-level genome assembly of the caddisfly Stenopsyche angustata (Insecta: Trichoptera). Sci Data 12, 1523 (2025). https://doi.org/10.1038/s41597-025-05602-8
-
Received:
-
Accepted:
-
Published:
-
DOI: https://doi.org/10.1038/s41597-025-05602-8