
Introduction
Pyroptosis is a unique form of inflammatory cell death executed by the pore-forming protein “gasdermin” [1]. The human gasdermin family comprises six members: GSDMA, GSDMB, GSDMC, GSDMD, GSDME and DFNB59. Mouse gasdermins are mostly identical to humans, except for the absence of GSDMB and the unique existence of GSDMA1-3 and GSDMC1-4 [2, 3]. To activate the pore-forming activity, gasdermins are recognized and cleaved by appropriate proteases to produce the free gasdermin N-terminal fragment. There are two main branches of the gasdermin activation pathway: canonical and non-canonical [3, 4]. In the canonical pathway, the recognition of pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) by pattern recognition receptors (PRRs) leads to the assembly of inflammasomes, which activate caspase-1 to cleave GSDMD [5]. Several non-canonical pathways are documented. In the presence of cytosolic lipopolysaccharide (LPS), caspase-4/5 (in humans) or caspase-11 (in mice) directly recognizes and cleaves GSDMD [5]. Chemotherapeutic agents, such as etoposide and cisplatin, activate caspase-3 to cleave GSDME in tumor cells [6]. With the aid of perforin, NK cells deliver granzyme A and granzyme B into tumor cells to cleave GSDMB and GSDME, respectively [7, 8]. In some conditions, such as hypoxia, activated caspase-8 also cleaves GSDMC [9]. These liberated gasdermin N-terminal fragments form pores in the plasma membrane and disrupt cellular integrity.
Pyroptosis was first observed in macrophages [10,11,12] and later in other immune cells and diverse non-immune cells. It not only contributes to defenses against pathogenic organisms [13], but also plays roles in other pathological conditions, such as diabetes [14], chronic inflammation, and cancer [15,16,17,18]. Thus, fine-tuning the extent of pyroptosis is valuable for potentially changing the outcome of pyroptosis-related diseases. As the key executioner of pyroptotic cell death, GSDMD has been proven to be a promising pharmacological target for this purpose [19].
To explore the mechanism underlying the low efficiency of GD-CT in blocking GD-NT-mediated pyroptosis, we developed a 293-tetO-GD-NT cell model, in which pyroptosis can be exclusively mediated by GD-NT in response to doxycycline. We found that the spatial separation and rapid turnover of GD-CT are the main factors that compromise its blocking efficacy. To overcome the weakness of GD-CT, we engineered the chimera protein FKBP-GD-CT, the structure and function of which are tuned by AP20187. We found that FKBP-GD-CT combined with AP20187 significantly blocked GD-NT-mediated pyroptosis.
Results
The pyroptosis model cell line 293-tetO-GD-NT is created to assess the blocking ability of GD-CT
Pyroptosis signaling pathways converge on the activation of specific proteases, which liberate the cytolytic gasdermin N-terminal domain from the inhibitory gasdermin C-terminal domain (Extended Fig. 1A, B). For the first identified pyroptosis executioner GSDMD, the separation of the N-terminal domain and C-terminal domain is achieved via caspase-1/11-mediated cleavage, allowing the manifestation of its pore-forming activity (Extended Fig. 1C, D). Interestingly, despite binding and inhibiting GD-NT after cleavage [5], GD-CT can no longer prevent the occurrence of pyroptosis. This phenomenon implies the low efficiency of GD-CT in blocking GD-NT-mediated pyroptosis.
To explore the biological characteristics of GD-CT underlying its limited blocking efficacy, we created a pyroptosis model cell line “293-tetO-GD-NT”, in which GD-NT expression is induced by Dox (Fig. 1A). Immunoblot showed rapid GD-NT expression, detectable at 2 h and peaking at 4 h post-Dox (Fig. 1B). Significant ATP decline occurred by 4 h (Fig. 1C), while LDH release was evident at 8 h (Fig. 1D). DAPI staining revealed obvious cell death at 8 h, with most cells dead by 24 h (Fig. 1E). Cell death was quantitatively analyzed using Annexin V-FITC/PI double-staining kit [20,21,22,23]. Flow cytometry revealed that 293-tetO-GD-NT underwent progressive pyroptosis over time (Fig. 1F). These data demonstrate the Dox-inducible pyroptosis in 293-tetO-GD-NT and suggest it is a suitable model for assessing GD-CT blockade.

A Tetracycline on (TetOn) system uses tetracycline (or one of its analogs like doxycycline) as a regulator of gene expression. Tetracycline-dependent promoter is created by placing a TRE upstream of a minimal promoter. TRE is 7 repeats of tetracycline operator (tetO) sequence and is recognized by reverse tetracycline-controlled transactivator (rtTA). In the presence of tetracycline or one of its analogs like doxycycline (Dox), rtTA binds to the TRE and tetracycline, permitting transcription. To create 293-tetO-GD-NT cells, HEK293 cells were transduced with lentivirus to express Dox-inducible GD-NT. B Immunoblot (IB) analysis of 293-tetO-GD-NT cells. The cells were treated with Dox (2 µg/mL) and harvested at different time points to obtain whole cell lysate (WCL). IB analysis of WCL was performed with different antibodies. β-actin served as an internal control. C Similar to (B), except that the cells were subjected to ATP-based Cell Viability Assay. D Similar to (B), except that the cells were subjected to LDH-based Cytotoxicity Assay. E Similar to (B), except that the morphological changes were observed using phase-contrast imaging (upper panel). Dying cells were stained with DAPI for fluorescent microscope imaging (lower panel). F Similar to (B), except that cell death was analyzed by flow cytometry using Annexin V/PI staining kit. In C and D, differences among groups were analyzed by two-tailed Student’s t-test (means ± s.e.m). Error bars represent the variation range of duplicated experiments. Data are representative of at least two independent experiments.
GD-CT blocks pyroptosis in a dose-dependent but inefficient manner
First, we transiently transfected empty vector (EV) or GD-CT into 293-tetO-GD-NT cells and verified their expression by immunoblotting (Fig. 2A). To assess GD-CT’s blocking ability, Dox was added after transfection to induce the expression of GD-NT and subsequent pyroptosis. 14 hours post-Dox, cells and culture media were collected for Cell cytotoxicity/viability assays, phase-contrast and fluorescence microscopy, and flow cytometry analysis. Our results showed that GD-CT reduced LDH release and restored ATP production in a dose-dependent manner (Fig. 2B, C). Consistently, GD-CT significantly increased cell viability (Fig. 2D, E). Compared with the ~20% survival rate in the EV-control group, GD-CT dose-dependently increased survival to a maximum of ~40% (Fig. 2F, G). These findings indicate that GD-CT incompletely but dose-dependently blocks pyroptosis.

A 293T-tetO-GD cells were transfected with different doses of GD-CT. 48 hours post-transfection, the cells were treated with Dox (2 µg/mL) to induce GD-NT expression. 4 h post-Dox, the cells were harvested for IB analysis with indicated antibody. B Similar to (A), except that the cells were harvested 24 h post-Dox and subjected to LDH-based Cytotoxicity Assay. C Similar to (A), except that the cells were harvested 24 h post-Dox and subjected to ATP-based Cell Viability Assay. D Similar to (A), except that, 24 h post-Dox, the cells floating in the culture medium were removed, and the adherent cells (considered as living cells) were subjected to phase-contrast imaging. E Similar to (A), except that, 4 h post-Dox, the cells were stained with PI and analyzed under immunofluorescence microscope. PI-positive cells were considered pyroptotic cells in this context. F. Similar to (A), except that, 14 h post-Dox, cell death in 293-tetO-GD was analyzed via flow cytometry and Annexin V/PI staining kit. G The proportion of surviving cells in F was statistically analyzed. In (B, C, and G,) differences among groups were analyzed by two-tailed Student’s t-test (means ± s.e.m). Error bars represent the variation range of duplicated experiments. Data are representative of at least two independent experiments.
GD-CT blocks GD-NT in a unique way distinct from its precursor “C-terminal domain of GSDMD”
Interestingly, despite interacting with GD-NT and inhibiting its release into the culture medium (Fig. 2F, G, and Fig. 3A–C), GD-CT failed to block pyroptosis as efficiently as the intramolecular inhibition mechanism within full-length GSDMD (GD-FL), in which the C-terminal domain completely blocks the pore-forming ability of the N-terminal domain and renders GD-FL noncytolytic. We hypothesized that the low efficiency of GD-CT arises from structural changes induced by proteolytic cleavage at the interdomain linker region of GD-FL. To test this hypothesis, 3D structural prediction was performed using AlphaFold 3 [24]. The predicted 3D structures were visualized using PyMOL and ChimeraX. We found that the structure of the GD-CT-GD-NT complex was conformationally distinct from that of GD-FL (Fig. 3D–F). These structural differences may explain the divergences in their biological activities and, consequently, blocking efficiency in GD-NT-mediated pyroptosis. Next, we conducted experiments to investigate the molecular properties of GD-CT, including its subcellular localization and protein stability.

A, B HEK293 cells were co-transfected with Flag-GD-FL, Flag-GD-NT and HA-GD-CT. 16 h later, the cells were harvested for anti-Flag (A) or anti-HA (B) IP assay. The precipitants and WCL were immunoblotted with indicated antibodies. C Subcellular localization of GD-NT and GD-CT. 293T-tetO-GD cells were transfected with different doses of GD-CT. 48 h post-transfection, the cells were treated with Dox (2 µg/mL) to induce GD-NT expression. 16 h post-Dox, the cells were fractionated separately. Proteins in culture medium, cytoplasm, membrane, or WCL were subjected to IB analysis with the indicated antibodies. StrataClean beads were used to precipitate and enrich proteins from the culture medium. D Experimentally determined murine GSDMD structure (Method X-RAY DIFFRACTION; Resolution 3.30 Å) was downloaded from RCSB PDB (PDB code: 6N9N) and visualized via PyMOL. E The structures of GD-FL, NT, and CT, the interaction between the free GD-NT and the free GD-CT protein were predicted via AlphaFold3 and then visualized via PyMOL (3D structure) and ChimeraX (PAE plot). N/A: not applicable. F Structure Analysis via ChimeraX. Hydrogen bonds (H-bonds, Blue) and contacts (Purple) were identified between the free GD-NT (Red) and the free GD-CT (Gray). Data are representative of at least two independent experiments.
The restriction of GD-CT to the cytoplasm causes spatial isolation
To assess the subcellular localization of GD-CT, we co-transfected Flag-tagged GD-NT and HA-tagged GD-CT into HeLa cells and performed confocal microscopy. It showed that GD-CT remained in the cytoplasm, and GD-NT was primarily translocated to the plasma membrane (Fig. 4A and Extended Fig. 2). To precisely determine their distribution, we transfected these GSDMD constructs into HEK293 cells and subjected them to subcellular fractionation, which separates cellular components into five compartments: soluble cytoplasmic, membrane, soluble nuclear, chromatin-bound nuclear, and insoluble cytoskeletal. Immunoblot analysis revealed that GD-CT was exclusively localized in the cytoplasm, whereas GD-NT was detected in the cytoplasm, plasma membrane, and cytoskeleton (Fig. 4B).

A Different distribution of NT and CT in cells. HeLa cells were co-transfected with Flag-GD-NT and HA-GD-CT. 18 hours later, the cells were fixed with methanol, stained with anti-Flag antibody conjugated to Alexa Fluor 594 (AF594, red) and anti-HA antibody conjugated to FITC (green), and counterstained with Hoechst. Subcellular localization of GD-NT and GD-CT was analyzed by confocal microscopy. B HEK293 cells were transfected with the indicated constructs. 16 h later, the cells were harvested and subjected to sample preparation using the Thermo Fisher Subcellular Fractionation Kit. Different fractions were subjected to IB analysis with the indicated antibodies. GAPDH, NA/K-ATPase α1, and LSD1 served as internal controls. C Design of nmFlag-GD-FL. In the schematic illustrative cartoon, GSDMD is Flag-tagged on both N-terminal of GD-NT domain and N-terminal domain of GD-CT domain. Cleavage of nmFlag-GD-FL by caspase-1/11 produces nFlag-GD-NT and nFlag-GD-CT at a ratio of 1:1. This allows the simultaneous detection of GD-NT, GD-CT with a unique Flag-antibody. D The structures of GD-FL, nmFlag-GD-FL were predicted via AlphaFold3 and then visualized via PyMOL (3D structure) and ChimeraX (PAE plot). N/A: not applicable. E HEK293 cells were transfected with nmFlag-GD-FL and/or GST-caspase-11 GD-NT. 24 h later, the proteins in cells and culture media were harvested, respectively, and subjected to immunoblot analysis with indicated antibodies. Data are representative of at least two independent experiments.
To imitate the dynamic changes of GSDMD localization during caspase-11 cleavage, we engineered a construct, “nmFlag-GD-FL” (Fig. 4C), with an N-terminal Flag-tag on both GD-NT and GD-CT for simultaneous detection using the same antibody. 3D structural prediction with AlphaFold 3 confirmed that the Flag-tags did not artificially disrupt GSDMD structure (Fig. 4D). We then transfected HEK293 cells with nmFlag-GD-FL and caspase-11 and separately harvested proteins from cells and culture medium. Immunoblot analysis showed that significantly less GD-CT was retained in cells, and no GD-CT was detected in culture medium. (Fig. 4E). Thus, our findings indicate that spatial isolation underlies the inefficiency of GD-CT in blocking GD-NT-mediated pyroptosis: The cytoplasmic GD-NT is bound and inhibited by co-localized GD-CT, while the GD-NT in the plasma membrane or culture medium remains active.
The short half-life of GD-CT contributes to the liberation of GD-NT
To assess the stability of GD-CT, HEK293 cells were transfected with Flag-GD-CT or Flag-GD-FL and then treated with CHX, a commonly used protein synthesis inhibitor. We found that GD-CT had a much shorter half-life and became undetectable within 4 h of CHX treatment, whereas GD-FL showed no degradation during the observation period (Fig. 5A, B). To identify its degradation pathways, we used several inhibitors to target specific processes: chloroquine (autophagy), calpeptin (calpain activity), Z-VAD-FMK (caspase activity), and MG132 (proteasome pathway). Our results showed that only MG132 completely blocked GD-CT degradation, indicating rapid turnover via the ubiquitin-proteasome system (Fig. 5C, D).

A HEK293 cells were transfected with Flag-GD-FL, GD-NT, or GD-CT, respectively. 16 h later, CHX (50 µg/mL) was used to block protein synthesis. The cells were harvested at different time points post-CHX and subjected to IB analysis with the indicated antibodies. β-actin served as an internal control. B Similar to (A), except that different concentrations of CHX were used to block protein synthesis, and the cells were harvested at 4 h post-CHX. C. Similar to (A), except that the cells were treated with CHX (50 µg/mL) and different protease inhibitors including Chloroquine (100 µM), Calpeptin (50 µM), Z-VAD-FMK (50 µM), and MG132 (20 µM). D Similar to (C), except that the cells were treated with CHX (50 µg/mL), and different concentrations of MG132 were used. E HEK293 cells were transfected with nmFlag-GD-FL and/or HA-caspase-11. 24 h later, the cells were harvested for IB analysis with the indicated antibodies. F,G HEK293 cells were transfected with nmFlag-GD-FL and/or GST-caspase-11, and, at the same time, treated with one of the following chemicals to block the activities of diverse proteases: Chloroquine (100 µM), Calpeptin (50 µM), Z-VAD-FMK (50 µM), MG132 (20 µM). 24 hours later, the cells were harvested for IB analysis with the indicated antibodies. H HEK293 cells were transfected with nmFlag-GD-FL and/or HA-caspase-11. 20 h later, CHX (50 µg/mL) was used to block protein synthesis. The cells were harvested at different time points post-CHX and subjected to IB analysis with the indicated antibodies. I Schematic illustration of GD-CT degradation. When cut by caspase-11, GD-FL produces GD-NT and GD-CT. The inhibitory GD-CT undergoes a fast 26S protease-dependent degradation; thus, the redundant cytolytic GD-NTs form pores on the plasma membrane and cause pyroptosis. Data are representative of at least two independent experiments.
As shown in Fig. 4C, the construct nmFlag-GD-FL enables the simultaneous assessment of GD-CT and GD-NT turnover using the same antibody, making it an ideal model for the concurrent comparative evaluation. Since cleavage occurs at a specific site, GD-CT and GD-NT should theoretically be produced in equimolar amounts (in a 1:1 ratio). Surprisingly, GD-CT levels were significantly lower than GD-NT in cells (Fig. 5E–H). Therefore, we conclude that GD-FL is stable, but upon caspase-11 cleavage, the produced GD-CT undergoes faster degradation than GD-NT, reducing its capacity to block GD-NT-mediated pyroptosis (Fig. 5I).
The design of the chimera protein FKBP-GD-CT
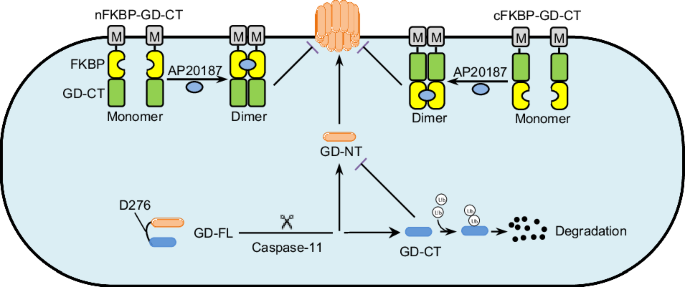
Blocking pathological pyroptosis, such as in sepsis, holds significant clinical importance, making the exploration of pyroptosis inhibitors an active research focus. GD-CT is a natural inhibitor of GD-NT-mediated pyroptosis, but its low efficiency hinders its potential in biomedical research and clinical applications. Given that GD-CT’s inefficiency results from its cytosolic retention and rapid degradation, we tried to engineer a chimeric GD-CT to address these limitations.
First, GD-NT oligomerizes and forms pores at the plasma membrane, membrane targeting is essential to enable the engineered chimera to directly interact with membrane-bound GD-NT. Myristoylation (Myr) is a vital lipid modification that facilitates protein targeting to membranes [25]. Based on vertical-scanning of sequence requirements for N-myristoylation [26], we fused a common Myr motif “GSSKSKPKDPSQR” to GD-CT to confer membrane translocation capability. Second, dimerization significantly influences protein conformation and stability [27,28,29,30]. For instance, cytosolic disulfide bridge-supported dimerization is crucial for the stability and cellular distribution of Coxsackievirus B3 protein 3 A [31]. FK506 binding protein 12 (FKBP12) is typically monomeric but dimerizes in the presence of ligands such as FK506 (for FKBP12WT) and AP20187 (for FKBP12F36V) [32]. As genetically engineering of proteins using this chemically induced dimerization (CID) system enables manipulation of protein localization, signaling pathways, and activation [33,34,35,36,37,38], we fused two copies of FKBP12F36V to GD-CT to confer a prolonged half-life upon AP20187 treatment. These designs yielded the chimera protein FKBP-GD-CT, including nFKBP-GD-CT and cFKBP-GD-CT (Fig. 6A and Extended Table 1). We first used AlphaFold 3 and PyMOL to predict and visualize the 3D structure of nFKBP-GD-CT and cFKBP-GD-CT (Fig. 6B), followed by Chai Discovery and ChimeraX to analyze the interaction between FKBP-GD-CT and AP20187, including hydrogen (H) bonds and other contacts. We found that AP20187 binding induced distinct conformational changes in both nFKBP-GD-CT and cFKBP-GD-CT (Fig. 6C, D).

A Schematic illustration of the design of the chimera protein FKBP-GD-CTs. B The structure of FKBP-GD-CTs was predicted via AlphaFold3 and then visualized via PyMOL (3D structure) and ChimeraX (PAE plot). N/A: not applicable. C, D The interaction of AP20187 and FKBP-GD-CTs was predicted via AlphaFold3 and then visualized via PyMOL (3D structure) and ChimeraX (PAE plot). N/A: not applicable. Hydrogen bonds (H-bonds, blue) and contacts (purple) were identified between the AP20187 (green) and the free GD-CT (gray or marine).
Biological characterization of the chimera protein FKBP-GD-CT
To assess the subcellular localization of FKBP-GD-CT, we transfected HEK293 cells with nFKBP-GD-CT or cFKBP-GD-CT and performed confocal microscopy and subcellular fractionation analyses. Our results revealed that nFKBP-GD-CT or cFKBP-GD-CT localized to both the cytoplasm and the plasma membrane (Fig. 7A, B), indicating that the Myr motif successfully facilitates their membrane targeting. Next, to assess FKBP-GD-CT stability, HEK293 cells transfected with these constructs were treated with CHX and/or AP20187, followed by immunoblot. We found that AP20187 treatment significantly extended the half-life of nFKBP-GD-CT or cFKBP-GD-CT. (Fig. 7C). Finally, to test whether FKBP-GD-CT inhibits the translocation of GD-NT to the plasma membrane or its release into culture medium, HEK293 cells transfected with these constructs were subjected to subcellular fractionation and immunoblot. The data showed that nFKBP-GD-CT or cFKBP-GD-CT reduced GD-NT release into the culture medium, and AP20187 further enhanced this inhibitory effect (Fig. 7D).

A Distribution of FKBP-GD-CT by fluorescence microscopy. HeLa cells were transfected with Flag-FKBP-GD-CT individually. 18 h later, the cells were fixed with methanol, stained with mouse FITC-Flag antibody, and counterstained with DAPI. The distributions of these proteins were visualized by fluorescence microscopy. B HEK293 cells were transfected with the indicated constructs. 16 h later, the cells were harvested and subjected to sample preparation using the Thermo Fisher Subcellular Fractionation Kit. Different fractions were subjected to IB analysis with the indicated antibodies. GAPDH, NA/K-ATPase α1, and LSD1 served as internal controls. C HEK293 cells were transfected with the indicated constructs. 16 hours later, the cells were treated with CHX (50 µg/mL) and/or AP20187 (100 nM) and then harvested at different time points for IB analysis with the indicated antibodies. D 293-tetO-GD-NT cells were transfected with the indicated constructs. Different concentrations of AP20187 were added at the same time as transfection. 14 h later, Dox (2 µg/mL) was added to induce the expression of GD-NT. The cells were harvested at 8 h post-Dox for subcellular fractionation. The proteins in culture medium, membrane, cytoplasm, as well as WCL were subjected to IB analysis with the indicated antibodies. StrataClean beads were used to precipitate and enrich proteins from the culture medium. Data are representative of at least two independent experiments.
FKBP-GD-CT blocks GD-NT-mediated pyroptosis in a more efficient way
To evaluate the inhibitory capacity of FKBP-GD-CT against GD-NT-mediated pyroptosis, we employed two cell models: the 293-tetO-GD-NT cells and the murine macrophage cell line iBMDM. In the 293-tetO-GD-NT model, the cells were transfected with the original GD-CT or the chimeric FKBP-GD-CT. After verifying their expression via Immunoblot (Fig. 8A), pyroptosis was assessed using two approaches: First, the cells without fixation were stained with DAPI and subjected to fluorescence microscopy, in which the DAPI-positive cells represent dying cells. The results showed that FKBP-GD-CT significantly reduced dying cells, an effect further enhanced by AP20187 (Fig. 8B). Next, flow cytometry was performed to quantify the dying cells, which revealed less pyroptosis in the cells transfected with FKBP-GD-CT compared to the original GD-CT, with AP20187 amplifying this reduction. Notably, cFKBP-GD-CT showed superior inhibitory efficacy to nFKBP-GD-CT (Fig. 8C, D).

A 293-tetO-GD-NT cells were transfected with the indicated constructs. 24 h later, the cells were treated with Dox (2 µg/mL) and/or AP20187 (100 nM). 16 h after Dox treatment, the cells were subjected to IB analysis with the indicated antibodies. B Similar to (A), except that, 8 h after Dox treatment, the cells were stained with DAPI (1 µg/mL) and imaged under immunofluorescence microscope. DAPI-positive cells were considered pyroptotic cells in this context. 24 h after Dox treatment, the cells floating in culture medium were removed, and the adherent cells (considered as living cells) were subjected to phase-contrast imaging. C,D Similar to (A), except that 14 h after Dox treatment, the cells were analyzed by flow cytometry using the Annexin V/PI staining kit. Shown are the proportion analysis of surviving cells (C)and the dot plot (D). Note: Annexin-V/PI double-negative cells were defined as surviving cells. E iBMDM cells were constructed to stably express EV (iBMDM-EV) or Flag-nFKBP-GD-CT (iBMDM-cFKBP-GD-CT). The expression of cFKBP-GD-CT was confirmed through IB analysis with the indicated antibodies. F iBMDM-EV or iBMDM-cFKBP-GD-CT were treated with LPS (1 µg/mL, 6 h) followed by Nigericin (2 µM, 3 h). AP20187 (1 µM) was added at the same time as LPS. The floated cells were removed, and the adherent cells (considered as living cells) were subjected to phase-contrast imaging. In C, differences among groups were analyzed by two-tailed Student’s t-test (means ± s.e.m). Error bars represent the variation range of duplicated experiments. Data are representative of at least two independent experiments.
In iBMDM cells, pyroptosis can be triggered with LPS + Nigericin (Nig), allowing evaluation of FKBP-GD-CT’s blocking efficacy in a relatively physiological setting. We generated stable cell lines expressing cFKBP-GD-CT or empty vector control via lentiviral transduction (Fig. 8E). These cells were pretreated with LPS and/or AP20187 for 6 h, then treated with Nigericin for 3 h. Consistent with the findings obtained from 293-tetO-GD-NT, AP20187 substantially enhanced cFKBP-GD-CT’s inhibition of pyroptosis in iBMDM cells (Fig. 8F). Thus, the engineered FKBP-GD-CT colocalizes with GD-NT and dimerizes upon AP20187 binding to achieve prolonged stability, effectively overcoming GD-CT’s low efficiency.
Discussion
Gasdermins are predominantly expressed in the epithelium of the gastrointestinal tract and skin [39]. For a long time, gasdermins have been documented as regulators of epithelial cell growth, hair formation [40, 41], hearing loss [42], and carcinogenesis [43, 44]. In 2015, Gao et al. found that the GSDMA3 N-terminal fragment (GA3-NT) induces autophagy and subsequent cell death, which can be blocked by the GSDMA3 C-terminal fragment (GA3-CT) or the C-terminal fragments of other gasdermin members. The inhibitory function of GA3-CT was further emphasized by studying a loss-of-function mutant Y344H of full-length gasdma3 (GA3-FL-Y344H). Y344H, located in the C-terminal domain of GA3-FL, abolishes the inhibitory capability of the GA3 C-terminal domain and makes GA3-FL-Y344H toxic like GA3-NT [45]. Later, gasdermin-mediated cell death was identified as “pyroptosis” [5], which does not depend on autophagy. These findings suggest that gasdermins potentially regulate a variety of biological processes when being maintained in their full-length form [43, 44, 46], but specifically induce pyroptosis upon cleavage to release the cytolytic N-terminal fragments [47]. The intricacies of gasdermin processing remain to be explored for further understanding of the mechanisms underlying cell death.
GSDMD was the first characterized and is the most important pyroptosis executioner [5]. Structurally, GSDMD consists of a cytolytic N-terminal domain and an inhibitory C-terminal domain [48]. This intramolecular inhibitory mechanism renders GSDMD non-cytolytic and harmless to host cells [19]. To liberate the cytolytic activity of the N-terminal domain, inflammatory caspases cleave the linker region of GSDMD, separating it into two free fragments: GD-NT and GD-CT [5]. Although the pore-forming activity of GD-NT has been clarified [49], why GD-CT fails to efficiently block GD-NT-mediated pyroptosis remains poorly understood. In this study, we observed that GD-CT interacted with GD-NT and only partially blocked its cytolytic activity. Mechanistically, GD-NT translocates to and forms pores in the plasma membrane, whereas GD-CT is restricted to the cytoplasm. The spatial separation prevents effective inhibition. Additionally, GD-CT is an unstable protein with a short half-life in cells and is rapidly degraded via the proteasome-dependent pathway. Therefore, our results suggest that the low inhibitory efficiency of GD-CT results from its spatial separation and fast degradation.
Pyroptosis can be detrimental in conditions such as cancer, rheumatoid arthritis, inflammatory bowel disease, autoinflammatory syndromes, and neuroinflammation [15, 17, 50,51,52]. Given that GD-CT is not functionless but can regulate the extent of pyroptosis, it could potentially be bioengineered for therapeutic applications in pyroptosis-related diseases. To increase the inhibitory ability of GD-CT, we created the chimera protein FKBP-GD-CT by fusing GD-CT with an N-terminal Myr motif and two FKBP12F36V domains. The Myr sequence targets the protein to the plasma membrane, bringing it to the site of GD-NT pore formation. The FKBP12F36V domains, in the presence of the small molecule dimerizer AP20187, confer GD-CT a prolonged half-life. These two strategies significantly enhance the inhibitory efficiency of GD-CT.
In summary, we demonstrate the molecular basis underlying the limited inhibitory efficacy of GD-CT against GD-NT-mediated pyroptosis. To overcome this limitation, we engineered the chimera protein FKBP-GD-CT. Thus, our work helps to understand the regulation mechanisms of gasdermin-mediated pyroptosis and proposes innovative therapeutic strategies. Notably, our CID-based design of FKBP-GD-CT provides a uniquely powerful tool for dissecting pyroptosis mechanisms in vitro. Currently, in vivo delivery and tissue-specific expression of FKBP-GD-CT are technically challenging, hindering its therapeutic potential for pyroptosis-related diseases. Further studies are warranted to address these limitations.
Methods
Data reporting
No statistical methods were used to predetermine the sample size. The experiments were not randomized. Investigators were not blinded to allocation during experiments and outcome assessment.
Cell lines and cell culture conditions
HEK293 cells (SCSP-502) and HeLa cells (SCSP-504) were obtained from the Cell Bank of the Chinese Academy of Sciences. Immortalized bone marrow-derived macrophages (iBMDM, EDC00071) were obtained from Editgene. These cell lines were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100 U/mL penicillin, and 100 µg/mL streptomycin at 37 °C under 5% CO₂. All cell lines were routinely screened for mycoplasma contamination using PCR and authenticated based on morphological characteristics.
Plasmids
pDB-His-MBP-mGSDMD (Addgene, 123365) was a gift from Hao Wu and was used as a template to generate Flag-GD-CT, HA-GD-CT, Flag-GD-FL, Flag-GD-NT via the standard PCR cloning strategy. The cDNA sequences for nmFlag-GD-FL and FKBP-GD-CT constructs were generated through custom gene synthesis (Gencefe Biotech, China) and were subcloned into pcDNA3.1 or pTRIPZ-TetOn-based viral vectors via the standard PCR cloning strategy. An illustrative diagram of the structural composition of nmFlag-GD-FL and FKBP-GD-CT is shown in Fig. 4C and Fig. 6A, respectively. The amino acid sequences of all GSDMD constructs are provided in Extended Table 1. pcDNA3.1-based vectors were used for transient transfection. The pLenti-CMV-based viral vector and pTRIPZ-TetOn-based viral vector were used to create 293-tetO-GD-NT cells and iBMDM-cFKBP-GD-CT cells, respectively. All plasmids were verified by DNA sequencing and immunoblot analysis.
Reagents and antibodies
Doxycycline (D3447) was obtained from Sigma-Aldrich. Subcellular Protein Fractionation Kit (78840) was obtained from Thermo Fisher. StrataClean resin (400714) and chemical competent cells (200315) were obtained from Agilent. For immunoblotting, anti-Flag (F1804), anti-Flag (F7425), and anti-HA (H6908) were obtained from Sigma-Aldrich. Anti-GSDMD (ab219800) was obtained from Abcam. pS166-RIP (65746), pS227-RIP3 (93654), pS358-MLKL (91689), LC3B (83506) and p62 (88588) were obtained from CST. Anti-β-Actin (sc-47778), anti-GST (sc-138), and anti-Na/K-ATPase α1 (sc-21712) were purchased from Santa Cruz Biotechnology. For immunofluorescence, anti-Rabbit conjugated with FITC (GB22303) and anti-Mouse conjugated with AF594 (GB28303) were obtained from Servicebio.
Stable cell lines
Lentivirus was produced in HEK293 cells by transfection of the lentiviral vector with psPAX2 (Addgene) and pMD2.G (Addgene). Lentiviral supernatants were collected, filtered through 0.45 µm filters, and used to transduce HEK293 cells. Polybrene infection/transfection reagent (Millipore, 10 µg/mL) was added to increase the efficiency of lentiviral infection. After 2 days of transduction, puromycin (Sigma, 2 µg/mL) was added to select the transduced cells. Empty lentiviral vectors were used to generate control cells. The expression of relevant genes in stable cell lines was verified by immunoblot.
Cytotoxicity assay and cell viability assay
To quantify pyroptotic cell death, cell death and cell viability were performed using the Non-Radioactive Cytotoxicity Assay kit (G1780, Promega) and the CellTiter-Glo Luminescent Cell Viability Assay kit (G7571, Promega), respectively, according to the manufacturer’s instructions. Briefly, 5 × 103 cells were cultured in 96-well plates with opaque walls. At the desired time points, cell death was determined by titrating the amount of lactate dehydrogenase (LDH) released into the culture medium. The maximum LDH control was obtained by lysing cells with the kit-provided LDH Release Reagent. Absorbance was measured at 490 nm using a multifunctional microplate reader (PerkinElmer EnSight). Cell viability was determined based on the ATP levels within cells, and the corresponding luminescence signals were quantified using the same PerkinElmer EnSight platform.
Immunoblots and immunoprecipitation
Cells were lysed in EBC buffer (50 mM Tris pH 7.5, 120 mM NaCl, 0.5% NP-40) supplemented with protease inhibitors (A32953, Thermo Fisher) and phosphatase inhibitors (B15002, Bimake). Protein concentrations of cell lysates were measured using the Beckman Coulter DU-800 spectrophotometer and the Bio-Rad protein assay reagent. Equal amounts of total proteins were resolved by SDS-PAGE and immunoblotted with the indicated antibodies. For immunoprecipitation, cell lysates containing 1 mg of total proteins were incubated with anti-Flag agarose (A2220, Sigma) or anti-HA agarose (A2095, Sigma) for 4 hours at 4 °C. Precipitates were washed three times with EBC buffer and resolved by SDS-PAGE, followed by immunoblot analysis with the indicated antibodies.
Pyroptosis detection by flow cytometry
Cell pyroptosis was quantified using an Annexin V-FITC/PI detection kit (KeyGEN BioTECH, KGA1102) according to manufacturer guidelines. Briefly, 1–5 × 10⁵ cells were collected using EDTA-free trypsin with strict limitation of digestion time to minimize false positives. After PBS washes (300 × g, 5 min), the cells were resuspended in 500 μL binding Buffer. Dual staining was performed by adding 5 μL Annexin V-FITC and 5 μL propidium iodide (PI), followed by gentle mixing and incubation at room temperature for 15 min, protected from light. Samples were acquired using the BD Accuri™ C6 Flow Cytometer (BD Biosciences) and analyzed using FlowJo software (Tree Star).
Immunofluorescence staining and microscopy
For immunofluorescence staining, the dishes with a special glass bottom were used. Cells were fixed with 100% methanol (chilled at −20 °C) at room temperature for 5 min. To minimize nonspecific binding of the antibodies, the cells were incubated with blocking solution containing 1% BSA, 22.52 mg/mL glycine in PBST (PBS + 0.1% Tween 20) for 30 min. For immunostaining, cells were incubated with the primary antibody in blocking solution (1% BSA in PBST) overnight at 4 °C in a humidified chamber and subsequently incubated with the secondary antibody in blocking solution for 1 h at room temperature in the dark. The cells were counterstained with DAPI (1 µg/mL), mounted, and subjected to imaging under the LSM 880 confocal microscope (Carl Zeiss AG, Germany) or the Axio Observer 3 widefield fluorescence microscope (Carl Zeiss AG, Germany).
For fluorescence microscopy of living cells, PI (10 µg/mL) or DAPI (1 µg/mL) was added to the culture medium. 10 minutes later, the cells were imaged under the same widefield fluorescence microscope.
Statistical analysis
Student’s t-test was used to determine the differences between the two groups. The results are presented as the mean and standard error of the mean (s.e.m). Statistical significance was assigned to P < 0.05.
Data availability
Original data are available upon request. The full length uncropped original western blots are shown in the ‘Supplementary Material’.
References
-
Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci. 2017;42:245–54.
-
Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111–6.
-
Zou J, Zheng Y, Huang Y, Tang D, Kang R, Chen R. The versatile gasdermin family: their function and roles in diseases. Front Immunol. 2021;12:751533.
-
Privitera G, Rana N, Armuzzi A, Pizarro TT. The gasdermin protein family: emerging roles in gastrointestinal health and disease. Nat Rev. Gastroenterol. Hepatol. 2023;20:366–87.
-
Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–5.
-
Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547:99–103.
-
Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu X, et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature. 2020;579:415–20.
-
Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y. et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science. 2020;368:eaaz7548.
-
Hou J, Zhao R, Xia W, Chang CW, You Y, Hsu JM, et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat Cell Biol. 2020;22:1264–75.
-
Kepp O, Galluzzi L, Zitvogel L, Kroemer G. Pyroptosis – a cell death modality of its kind? Eur J. Immunol. 2010;40:627–30.
-
Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. 2010;11:1136–42.
-
Silveira TN, Zamboni DS. Pore formation triggered by Legionella spp. is an Nlrc4 inflammasome-dependent host cell response that precedes pyroptosis. Infect Immun. 2010;78:1403–13.
-
Doitsh G, Galloway NL, Geng X, Yang Z, Monroe KM, Zepeda O, et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature. 2014;505:509–14.
-
Zuo Y, Chen L, Gu H, He X, Ye Z, Wang Z, et al. GSDMD-mediated pyroptosis: a critical mechanism of diabetic nephropathy. Expert Rev. Mol. Med. 2021;23:e23.
-
Yu P, Zhang X, Liu N, Tang L, Peng C, Chen X. Pyroptosis: mechanisms and diseases. Signal Transduct. Target Ther. 2021;6:128.
-
Wei X, Xie F, Zhou X, Wu Y, Yan H, Liu T, et al. Role of pyroptosis in inflammation and cancer. Cell Mol. Immunol. 2022;19:971–92.
-
Rao Z, Zhu Y, Yang P, Chen Z, Xia Y, Qiao C, et al. Pyroptosis in inflammatory diseases and cancer. Theranostics. 2022;12:4310–29.
-
Fang Y, Tian S, Pan Y, Li W, Wang Q, Tang Y, et al. Pyroptosis: a new frontier in cancer. Biomed Pharmacother. 2020;121:109595.
-
Dai Z, Liu WC, Chen XY, Wang X, Li JL, Zhang X. Gasdermin D-mediated pyroptosis: mechanisms, diseases, and inhibitors. Front Immunol. 2023;14:1178662.
-
Wang Z, Wang Y, Yang H, Guo J, Wang Z. Doxycycline induces apoptosis of Brucella suis S2 strain-infected HMC3 microglial cells by activating calreticulin-dependent JNK/p53 signaling pathway. Front Cell Infect. Microbiol. 2021;11:640847.
-
Sagar J, Sales K, Seifalian A, Winslet M. Doxycycline in mitochondrial mediated pathway of apoptosis: a systematic review. Anticancer Agents Med Chem. 2010;10:556–63.
-
Mouratidis PX, Colston KW, Dalgleish AG. Doxycycline induces caspase-dependent apoptosis in human pancreatic cancer cells. Int J. Cancer. 2007;120:743–52.
-
He J, Zheng P, Chen Y, Qi J, Ye C, Li D, et al. A new personalized vaccine strategy based on inducing the pyroptosis of tumor cells in vivo by transgenic expression of a truncated GSDMD N-terminus. Front Immunol. 2022;13:991857.
-
Abramson J, Adler J, Dunger J, Evans R, Green T, Pritzel A, et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature. 2024;630:493–500.
-
McIlhinney RA. Membrane targeting via protein N-myristoylation. Methods Mol. Biol. 1998;88:211–25.
-
Utsumi T, Nakano K, Funakoshi T, Kayano Y, Nakao S, Sakurai N, et al. Vertical-scanning mutagenesis of amino acids in a model N-myristoylation motif reveals the major amino-terminal sequence requirements for protein N-myristoylation. Eur J. Biochem. 2004;271:863–74.
-
Kim SJ, Sun EG, Bae JA, Park S, Hong CS, Park ZY, et al. A peptide interfering with the dimerization of oncogenic KITENIN protein and its stability suppresses colorectal tumour progression. Clin Transl. Med. 2022;12:e871.
-
Xu Y, Li S, Yan Z, Ge B, Huang F, Yue T. Revealing cooperation between knotted conformation and dimerization in protein stabilization by molecular dynamics simulations. J Phys. Chem. Lett. 2019;10:5815–22.
-
Zhao Y, Cieplak M. Stability of structurally entangled protein dimers. Proteins. 2018;86:945–55.
-
Rumfeldt JA, Galvagnion C, Vassall KA, Meiering EM. Conformational stability and folding mechanisms of dimeric proteins. Prog Biophys. Mol. Biol. 2008;98:61–84.
-
Voss M, Kleinau G, Gimber N, Janek K, Bredow C, Thery F, et al. A cytosolic disulfide bridge-supported dimerization is crucial for stability and cellular distribution of Coxsackievirus B3 protein 3A. FEBS J. 2022;289:3826–38.
-
Kolos JM, Voll AM, Bauder M, Hausch F. FKBP Ligands-where we are and where to go? Front Pharm. 2018;9:1425.
-
Fegan A, White B, Carlson JC, Wagner CR. Chemically controlled protein assembly: techniques and applications. Chem Rev. 2010;110:3315–36.
-
Spencer DM, Belshaw PJ, Chen L, Ho SN, Randazzo F, Crabtree GR, et al. Functional analysis of Fas signaling in vivo using synthetic inducers of dimerization. Curr Biol. 1996;6:839–47.
-
Amara JF, Clackson T, Rivera VM, Guo T, Keenan T, Natesan S, et al. A versatile synthetic dimerizer for the regulation of protein-protein interactions. Proc Natl Acad Sci USA. 1997;94:10618–23.
-
Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–6.
-
Pajvani UB, Trujillo ME, Combs TP, Iyengar P, Jelicks L, Roth KA, et al. Fat apoptosis through targeted activation of caspase 8: a new mouse model of inducible and reversible lipoatrophy. Nat Med. 2005;11:797–803.
-
Mallet VO, Mitchell C, Guidotti JE, Jaffray P, Fabre M, Spencer D, et al. Conditional cell ablation by tight control of caspase-3 dimerization in transgenic mice. Nat Biotechnol. 2002;20:1234–9.
-
Fujii T, Tamura M, Tanaka S, Kato Y, Yamamoto H, Mizushina Y, et al. Gasdermin D (Gsdmd) is dispensable for mouse intestinal epithelium development. Genesis. 2008;46:418–23.
-
Runkel F, Marquardt A, Stoeger C, Kochmann E, Simon D, Kohnke B, et al. The dominant alopecia phenotypes bare skin, rex-denuded, and reduced coat 2 are caused by mutations in gasdermin 3. Genomics. 2004;84:824–35.
-
Lunny DP, Weed E, Nolan PM, Marquardt A, Augustin M, Porter RM. Mutations in gasdermin 3 cause aberrant differentiation of the hair follicle and sebaceous gland. J Invest Dermatol. 2005;124:615–21.
-
Mujtaba G, Bukhari I, Fatima A, Naz S. A p.C343S missense mutation in PJVK causes progressive hearing loss. Gene. 2012;504:98–101.
-
Saeki N, Kim DH, Usui T, Aoyagi K, Tatsuta T, Aoki K, et al. GASDERMIN, suppressed frequently in gastric cancer, is a target of LMO1 in TGF-beta-dependent apoptotic signalling. Oncogene. 2007;26:6488–98.
-
Saeki N, Kuwahara Y, Sasaki H, Satoh H, Shiroishi T. Gasdermin (Gsdm) localizing to mouse Chromosome 11 is predominantly expressed in upper gastrointestinal tract but significantly suppressed in human gastric cancer cells. Mamm Genome. 2000;11:718–24.
-
Shi P, Tang A, Xian L, Hou S, Zou D, Lv Y, et al. Loss of conserved Gsdma3 self-regulation causes autophagy and cell death. Biochem J. 2015;468:325–36.
-
Hergueta-Redondo M, Sarrio D, Molina-Crespo A, Megias D, Mota A, Rojo-Sebastian A, et al. Gasdermin-B promotes invasion and metastasis in breast cancer cells. PLoS One. 2014;9:e90099.
-
Liu Z, Busscher BM, Storl-Desmond M, Xiao TS. Mechanisms of gasdermin recognition by proteases. J Mol Biol. 2022;434:167274.
-
Burdette BE, Esparza AN, Zhu H, Wang S. Gasdermin D in pyroptosis. Acta Pharm. Sin. B. 2021;11:2768–82.
-
Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–8.
-
Imre G. Pyroptosis in health and disease. Am J Physiol Cell Physiol. 2024;326:C784–C94.
-
Vasudevan SO, Behl B, Rathinam VA. Pyroptosis-induced inflammation and tissue damage. Semin Immunol. 2023;69:101781.
-
Hu Y, Wang B, Li S, Yang S. Pyroptosis, and its role in central nervous system disease. J Mol. Biol. 2022;434:167379.
Funding
This project was supported by Research Fund for the Doctoral Program of the Fifth Affiliated Hospital of Zhengzhou University (pb2024kyqdj03), Supporting Program for Young Talent Innovation Teams of Zhengzhou University (32320688), Henan Medical Key Technologies R & D Program (LHGJ20220570).
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
We did not include vertebrate or human samples. All methods were performed in accordance with the relevant guidelines and regulations.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, J., Fu, M., Xing, Y. et al. Chemically induced dimerization of GSDMD C-terminal domain blocks GSDMD N-terminal domain-mediated pyroptosis. Cell Death Discov. 11, 456 (2025). https://doi.org/10.1038/s41420-025-02733-0
-
Received:
-
Revised:
-
Accepted:
-
Published:
-
DOI: https://doi.org/10.1038/s41420-025-02733-0