
Introduction
Non-coding RNAs (ncRNAs) are key components of epigenetic mechanisms with a pivotal role in gene regulation activities1. Among the various types of ncRNAs, circular RNAs (circRNAs) are a relatively new and understudied form, having been discovered less than three decades ago2,3. Unlike linear RNAs, circRNAs are more stable4 and are formed by undergoing back-spicing events that produce a covalently closed loop structure by the ligation of 5′ and 3′ ends5,6,7. Initially, the circRNAs were believed to represent splicing errors with no known functions8. Although it is now widely accepted that some circRNAs have crucial roles to play in the regulatory mechanisms of genes and their biological processes, an in-depth analysis of the functions of circRNAs is still needed9,10.
Many studies have now demonstrated that circRNAs can function as miRNA sponges11,12,13 and regulate gene expression through interactions via either RNA or proteins4,14,15 Until recently, the majority of the studies on circRNAs were comprehensively carried out in animals, however, once Wang et al. (2014)16 reported the presence of circRNAs in diverse genera, including fungi, protists and the model plant Arabidopsis thaliana, there has been an increase in studies reporting the identification and analysis of circRNAs in many plants, including Arabidopsis, rice17, wheat18, barley19, soybean20, tomato21, potato22, maize23, tea24, cucumber25, cotton26, apple27 and black pepper28. Most of these studies, however, are limited to a few tissues or stress conditions and involve a small number of RNA-seq datasets, resulting in the identification of a few hundred to some thousand circRNAs. In tea (C. sinensis), reports on circRNAs are primarily limited to specific conditions or tissues. A previous study identified a total of 3,174 circRNAs from 6 circular RNA-seq data sets (i.e., three replicates each of leaf buds and young leaves) and analyzed to highlight their potential roles in photosynthesis or metabolite biosynthesis during the leaf development process24. Recently, studies have examined circRNAs in tea during leaf bud to young leaf development29, under chilling stress30, during winter dormancy and spring flushing31, and in response to Helopeltis theivora infestation32.
Tea, an important plantation crop, is consumed as a morning drink around the world. Botanically, there are three types of tea i.e., C. sinensis (L.) O. Kuntze aka China type, C. assamica (Masters) aka Assam type, and C. assamica ssp. lasiocalyx (Planchon ex Watt) referred as Cambod type, and each of these has its own distinct taste or quality33. Being a commercial commodity, the price vis-à-vis profitability of tea depends upon the cup quality of the brewed product. This quality is influenced by several factors, including environmental factors, the altitude of the plantation, the genotypes and ages of the plants, and the quality of the green tissue used in tea production. In the present study, we identified a large number of stress- and tissue-specific circRNAs from public datasets, focusing on the circRNAs specific to TV-1 genotype of C. assamica and its various tissues. Recently, a similar effort was undertaken using a large number of publicly available RNA-seq datasets on tea to determine tissue-specific long non-coding RNAs (lncRNAs) associated with aroma formation pathways, which can regulate the quality trait of the plant34.
With advancements in high-throughput sequencing and the availability of efficient tools and strategies, the research community is well-equipped to identify and report a greater number of circRNAs across diverse species. The strategy defined here in the present study, using publicly available RNA-seq data, may serve as a valuable guide for researchers aiming to identify and analyze circRNAs in significant numbers in other species as well. Moreover, the analysis supports the fact that, rather than focusing on some key genes, a deeper study of circRNAs, their roles and their interactions with protein-coding genes is necessary. Furthermore, this is the first report of a database comprising tissue- and stress-specific circRNAs of tea, and the circRNA data generated here at this large scale could serve as a foundation for comprehensive structural and functional studies of circRNAs in tea.
Plant materials and experimental methods
Data source for prediction of circRNAs
De novo identification of circRNAs required greater sequencing depth, and for tea (C. sinensis), substantial amount of RNA-seq data is available at NCBI. Before processing this large amount of data for circRNA prediction and analysis, RNA-seq data of 5 different tissues of TV-1 genotype of Indian tea (C. assamica) were generated35 and used to standardize the pipeline (Table S1). Total RNA was extracted from 5 different tissues of TV-1 viz. third leaf, active bud, banji bud, flower bud, and young stem using TRIzol reagent followed by isolation of mRNA, synthesis of cDNA, and ligation of adapter. Subsequently, RNA-seq libraries were sequenced on Illumina Hiseq2000 platform for each of the 5 tissues. This dataset was used in the pipeline for the identification and classification of circRNAs in TV-1 genotype (Table S2), followed by differential expression analysis to identify tissue-specific circRNAs and their functional annotation (Fig. 1). Further, at NCBI, there were 13 Bioprojects having 81 Pair-end (PE) RNA-seq data for different stress-related conditions (Table S3) and 15 Bioprojects having 146 PE RNA-seq data belonging to 17 different tissues (Table S4). These PE RNA-seq data were downloaded for the in-silico prediction and analysis of circRNAs following the standardized pipeline.

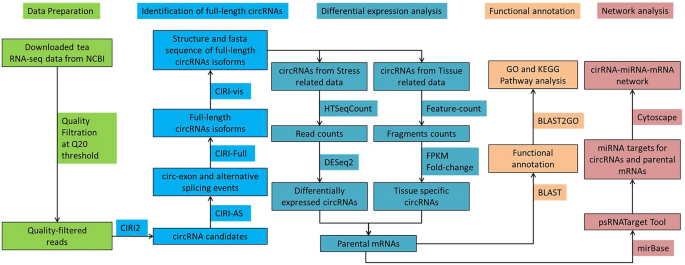
Pipeline for identification of tea circRNA used in the present study.
Identification and characterization of circRNAs in tea
The RNA-seq raw data was quality filtered using NGSQCToolKit Version v2.3.336 at a Q20 threshold. The filtered data were then used for the identification of circRNA with CIRI237 by initially mapping the filtered reads to the reference tea (C. sinensis) genome38 using Burrows-Wheeler Aligner (BWA)39. Further, we have used CIRI-AS40 for the detection of circ-exon and alternative splicing events. Finally, the reconstruction of full-length circRNAs isoforms was performed using CIRI-full41, followed by the generation of FASTA sequences and visualization of the circular structure of full-length circRNAs using CIRI-vis. The predicted full-length circRNA isoforms were classified as exonic, intronic, and intergenic based on their originating position in the reference genome sequence.
Comparison with known circRNAs in other plants
The identified circRNAs were BLAST searched against the PlantcircBase42 using a threshold of E-value < 1e-05 to classify them as conserved or novel circRNAs.
Differential expression of circRNAs
To identify stress-related or tissue-specific circRNAs, differential expression analysis was performed using RNA-seq data mapped to the reference tea genome38. For stress-related circRNAs, read counts were calculated for the identified circRNAs with HTSeqCount and analyzed with the DESeq2 package43 to detect differentially expressed circRNAs (DECs) by applying the Wald test with a significance threshold of P-value ≤ 0.05 and abs(log2 fold-change) ≥ 1 between any two groups or samples analyzed.
For the differential expression analysis of the circRNAs identified in the tissue-related data, featureCounts44 from Subread v1.5.045 was used to count fragments, followed by tissue enrichment classification based on FPKM values by TEnGExA46. The circRNAs with FPKM > 1 in atleast 1 tissue sample were considered as expressed circRNAs and further classified as ‘Tissue Enriched’ (FPKM > 1 and FPKM fold change ≥ 5 compared to all other tissues), ‘Group Enriched’ (FPKM > 1 and FPKM fold change ≥ 5 in a group of 2 or more (but not all) tissues compared to all other tissues), ‘Tissue Enhanced’ (FPKM > 1 and FPKM fold change ≥ 5 compared to the average FPKM in all other tissues), ‘Expressed in all’ (Detected in all tissues with FPKM > 1 but not present in any of the first 3 categories), ‘Mixed 1’ (Detected in fewer than all tissues but all with FPKM > 1 but not present in any of the first 3 categories), ‘Mixed 2’ (Detected in any number of tissue with atleast 1 having FPKM > 1 but not all)47.
Functional annotation and KEGG Pathway analysis
To predict the potential functions of DECs, the respective parental mRNAs were BLAST searched against the Viridiplante database (with E-value ≤ 1e-10), and BLAST2GO v5.2.548 was used to assign Gene ontology (GO) categories annotation followed by pathway analysis using the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database49.
Interaction analyses of circRNAs and/or mRNAs with miRNAs
The psRNAtarget tool50 was used at default parameters with a cut-off score of ≤ 4 for mRNA and ≤ 4.5 for circRNA 51 to predict the miRNA targets by aligning against miRBase release 21.052. The potential connections between different sets of circRNA, miRNA, and mRNA were drawn and visualized using Cytoscape v3.7.153.
Database development
A user-friendly database, Tea CircRNA DataBase (TCDB), was developed using XAMPP server with PHP, Java, MySQL, HTML, and CSS for the back- or front-end functionalities, allowing users to search and extract circRNAs based on the user-selected criteria. The TCDB can be accessed at http://indianteagenome.in:8080/tcdb/.
Results
Identification and characterization of predicted circRNAs of tea
A total of 135.414 million high-quality RNA-seq reads from 5 different tissues of TV-1 genotype (Table S1) were processed using the CIRI2 and CIRI-full pipelines to detect a total of 3,052 full length circRNA isoforms with lengths ranging from 37 to 585 nucleotide bases with a minimum of 1 to a maximum of 5 circ-exons (Table S2). These identified circRNAs were categorized as exonic (74.77%), intronic (6.72%), and intergenic (16.58%) with 1.93% remaining unclassified (Table S2). From the publicly available data sets, a total of 59,575 full-length circRNAs were identified from 178 RNA-seq data sets, which included 61 stress-related (Table S3) and 117 tissue-related data sets (Table S4). Of these circRNAs, 79.34% were characterized as exonic, 14.66% as intergenic, 4.73% as intronic, and the remaining 1.27% as others (Table S5, Fig. 2a). The exonic and intronic circRNAs were further characterized as sense and antisense, with 73.94% and 26.06%, respectively, in the exonic class and 70.71% and 29.29%, respectively, in the intronic class. The lengths of the 59,575 full-length circRNAs varied from 26 to 19,157 nucleotide bases, with 36,352 (i.e., 61.02%) having a length between 101 and 200 bases (Fig. 2b). The predicted circRNAs from the public data sets, contained up to 7 circ-exons, including 77.94% and 18.55% with 1 and 2 circ-exons, respectively (Fig. 2c). There were up to 4201 Back Splicing Junction (BSJ) reads found in the predicted circRNAs.

Comparative analysis of predicted tea circRNAs. (a) Classification of circRNAs as exonic, intronic and intergenic. (b) Length wise distribution of circRNAs. (c) Number of exons per circRNA. (d) Number of circRNAs per scaffold.
In the reference CSS genome38 (Wei C et al., 2018), there were 14,051 scaffolds and 33,932 genes were predicted from 3,116 scaffolds, indicating that no genes were predicted from 10,935 scaffolds. Additionally, these 10,935 scaffolds showed a very low abundance of circRNAs with only 31 detected, which is just 0.05% of the total identified circRNAs (Table S6). Moreover, no circRNAs were found in 12,012 scaffolds, and only 604 of these scaffolds contained more than 1 gene. Ninety scaffolds were identified with over 100 circRNAs, with a maximum of 710 circRNAs in a single scaffold (scaffold304, measuring 1,409,464 bp length) (Fig. 2d). The total predicted 59,575 circRNAs were found to originate from 12,166 parental protein-coding genes. Most of these genes served as the source for multiple circRNAs, while only 4,551 genes were found to have one circRNA each (Table S7).
A total of 25,651 circRNAs were detected from stress-related RNA-seq data sets, while 39,750 circRNAs were identified from tissue-related data sets, with only 5,826 circRNAs being common to both. Therefore, while 19,825 circRNAs were exclusively present in stress-related data, the tissue-related data set contained 33,924 such circRNAs (Fig. S1). In the stress-related data, almost a similar number of circRNAs (~ 516) per RNA-seq file were detected for both treated and control data sets. For tissue-related data, the samples were categorized into 17 different classes, comprising a total of 39,750 non-redundant circRNAs (Table S8). The accumulation of circRNAs was high in some tissues. For example, high numbers of circRNAs were detected in the 2nd leaf, 4th leaf, apical bud, a bud and 2 leaves, flower bud, and a bud and a leaf with over 100 circRNAs per Gb raw RNA-seq bases, while each of the flowers, early-stage lateral bud, roots, and tender shoot had fewer than 50 circRNAs per Gb raw RNA-seq bases (Table S8).
Comparative analysis of circRNA
To identify conserved circRNAs, a BLAST search was conducted against the PlantcircBase database. About 12.75% (389) of the 3,052 predicted circRNAs from the TV-1 genotype exhibited homology with 1,240 circRNA sequences in PlantCircBase 4.0 (Table S9). These 1,240 sequences represent just 1.08% of the total entries in the database, which is consistent with the recently published works on circRNAs in tea29 (Tong et al., 2018). Hence, 12.75% of predicted circRNAs in TV-1 were conserved with 1.08% of previously reported plant circRNAs, while 87.25% were identified as novel. Among the 389 circRNAs in TV-1, 317 (i.e., 81.49%) showed homology with circRNAs of C. sinensis (Table S10).
Of the 59,575 tea circRNAs identified from public data sets, 10.87% (6,475) circRNAs exhibited homology with 6,043 circRNA sequences in PlantCircBase 4.0 (Table S11), representing 5.25% of the total plant circRNAs in the database (Table S9). Thus, only 10.87% of the identified tea circRNAs are conserved with 5.25% of known circRNAs, while 89.13% (53,100) are considered novel in terms of sequence homology. These differences could be due to the fact that very few circRNAs have been reported for tea to date in the standard database like PlantCircBase.
Differentially expressed circRNA in tea
Differential expression analysis of 3,052 circRNAs identified from 5 different tissues of the TV-1 genotype revealed that 2,733 (i.e., 89.55%) were expressed (having FPKM > 1 in at least 1 tissue sample) (Table S12). Among these expressed circRNAs, 6.22%, 10.61%, and 3.55% were further categorized as ‘Tissue Enriched’, ‘Group Enriched’ (expressed in a group of 2 to 4 tissues), and ‘Tissue Enhanced’, respectively (Table 1). In addition to these three main classes, 78.63% were classified as ‘Expressed in all’, while a very small portion, 0.62% and 0.37%, represented the ‘Mixed 1’ and ‘Mixed 2’ classes, respectively. Among 170 ‘Tissue-Enriched’ circRNAs, the majority were expressed in the 3rd leaf tissue (130 circRNAs), only 21 in flower bud, and 1 in young stem. Similar trends were observed for the 97 ‘Tissue Enhanced’ circRNAs, with counts of 66, 11 and 9 in the 3rd leaf, flower bud, and young stem, respectively.
Among the 39,750 circRNAs identified in publicly available tissue-related data sets, 65.84% (i.e., 26,170 circRNAs) were expressed, as indicated by their FPKM-fold values (Table S13). Among these expressed circRNAs, 2.63% were categorized as ‘Tissue Enriched’, 1.16% as ‘Group Enriched’ (expressed in a group of 2 to 7 tissues), 15.20% as ‘Tissue Enhanced’, 58.51% as ‘Expressed in all’, 13.68% as ‘Mixed 1’ and 8.82% as ‘Mixed 2’ (Table 2). Further, among the 687 ‘Tissue Enriched’ circRNAs, the highest expression was observed in tender-shoots (162 circRNAs), followed by flowers (20), lateral-buds (103), roots (8), seeds (65), and the 6th leaf (61) (Table S14). A small number of tissue-enriched circRNAs were found in other tissues, including flower-bud (37), seedling (8), mature-leaf (8), 4th leaf (7), a-bud-a-leaf (4), stem (4), a-bud-2-leaf (2), axillary-bud (2), and apical-bud (1) (Table S14). Notably, no tissue-enriched circRNAs were detected in the 2nd-leaf, despite it having one of the highest (111) number of circRNAs per Gb raw RNA-seq bases (Table S8). Similar trends were observed for ‘Tissue Enhanced’ circRNAs, with high numbers in lateral-bud (631), tender-shoot (605), and the 6th leaf (603), while fewer were found in axillary-bud (41), apical-bud (30), and the 2nd leaf (25) (Table S15).
For stress-related circRNAs, the DESeq2 package was used to detect DECs by comparing control and treated datasets across nine different Bioprojects (Table 3). A total of 1,463 circRNAs were found to be differentially expressed (up- or down-regulated), originating from 902 parental mRNAs. The highest number of DECs (809) was observed in the Bioproject PRJEB11522 (comparing PEG or NaCl treatments with control), followed by 548 DECs in the Bioproject PRJNA288922 (for Methyljasmonate treatment vs control analysis). In PEG vs control comparison, 351 circRNAs were up-regulated and 351 were down-regulated, while in NaCl vs control analysis, 294 circRNAs were up-regulated and 184 were down-regulated. To identify the common circRNAs between the NaCl and PEG treatments, further analysis revealed that 224 were up-regulated in both treatments, and 162 were down-regulated in both, indicating that these circRNAs may have cross-talk between NaCl and PEG treatments. In contrast, 127 circRNAs were found to be up-regulated exclusively under PEG treatment, compared to just 70 that were up-regulated by NaCl treatment, suggesting that these circRNAs are specific to stress. Similarly, 189 circRNAs were down-regulated exclusively in PEG, while only 22 were down-regulated in NaCl. Notably, no circRNAs exhibited up-regulation in PEG while being down-regulated in NaCl, and vice versa. When comparing PEG vs NaCl expression analysis (i.e., NaCl treatment followed by PEG treatment), 60 circRNAs were found to be up-regulated and 63 were down-regulated. Among these 60 up-regulated circRNAs, 43 were also up-regulated in the PEG vs control analysis discussed above. This indicates that 17 circRNAs (as 60–43 = 17) get up-regulated specifically with NaCl treatment followed by PEG treatment, while 308 circRNAs (as 351–43 = 308) get up-regulated after PEG treatment only. Similarly, 21 circRNAs were down-regulated exclusively after NaCl treatment followed by PEG treatment, and 309 achieved down-regulation after PEG treatment only, as there were 42 circRNAs down-regulated in both PEG vs control and PEG vs NaCl comparisons.
The Bioproject PRJNA315669 focuses on identifying the potential candidate genes involved in the regulation of pollen tube elongation by nitrogen oxide under low-temperature stress. However, no DECs were found in the NOxide vs control analysis. In the cold vs control analysis, 9 circRNAs were found to be up-regulated and 5 down-regulated. In the NOxide vs cold analysis, there were 3 up-regulated and 19 down-regulated circRNAs. Although there were no DECs that were either up- or down-regulated in both analysises, interesting patterns were observed when comparing the DECs from cold vs control and NOxide vs cold. All 9 circRNAs that were up-regulated in cold vs control, were down-regulated in NOxide vs cold analysis. Additionally, of the 5 circRNAs that were down-regulated in the cold vs control, 3 were up-regulated in the NOxide vs cold analysis, suggesting that NOxide treatment can reverse the regulation established after cold treatment.
GO and KEGG analysis
During the functional annotation of circRNA-parental genes or mRNA targets of tissue-enriched or tissue-enhanced circRNAs in the TV-1 genotype, only 2 genes were identified as involved in the phenylpropanoid biosynthesis pathway. These genes encode a crucial enzyme, i.e., lactoperoxidase which aids in the enhancement of lignin-derived end-components in this pathway. Similarly, the KEGG pathway analysis of parental genes associated with 1,527 circRNAs specific to the TV-1 genome identified 5 genes involved in the phyenylpropanoid biosynthesis pathway. These genes encode ammonia-lyase (which initiates the pathway by converting phenylalanine to cinnamic acid or tyrosine to p-coumaric acid) and lactoperoxidase enzyme (Fig. S2), indicating that these circRNAs may play a significant role in tea aroma formation.
Interestingly, functional annotation of 12,166 circRNA-parental genes from all predicted circRNAs in publicly available data revealed a total of 80 tea genes encoding 8 enzymes in the phenylpropanoid biosynthesis pathway (Fig. S3). These 8 enzymes, including ammonia-lyase (ec:4.3.1.24 and ec:4.3.1.25), O-methyltransferase (ec:2.1.1.68 and ec:2.1.1.104), ligase (ec:6.2.1.12), reductase (ec:1.2.1.44), dehydrogenase (ec:1.1.1.195), gentiobiase (ec:3.2.1.21), and lactoperoxidase (ec:1.11.1.7) are involved in and enhance most steps from the beginning to the end in the phenylpropanoid biosynthesis pathway. Of the 464 circRNAs predicted in these 80 genes, only 12 were tissue-enriched (8 in flowers, 3 in tender-shoot, 1 in root), and 44 were tissue-enhanced (11 in flowers, 9 in 6th leaf, 7 in tender shoot, 3 in each of flower buds, roots, and others, 2 in each of 4th leaf, stem and lateral buds, 1 in each of seed and seedling).
In addition to the phenylpropanoid biosynthesis pathway, these 12,166 genes were found to be involved in a total 136 KEGG pathways, with more than 20 enzymes encoded in 10 of these pathways, including purine metabolism (27 enzymes), cysteine and methionine metabolism (27), starch and sucrose metabolism (26), pyruvate metabolism (24), glycolysis/gluconeogenesis (23), amino sugar and nucleotide sugar metabolism (23), aminoacyl-tRNA biosynthesis (22), porphyrin and chlorophyll metabolism (20), and phenylalanine, tyrosine and tryptophan biosynthesis (20) (Table S16).
circRNA-miRNA-mRNA network
CircRNAs may function as miRNA sponges, however, the identification of circRNAs lacking putative miRNA binding sites suggests that many circRNAs do not act as miRNA sponges. Among the total of 1,463 DECs, 82.29% (i.e., 1,204 circRNAs) were found to contain at least one binding site, with a total of 3,552 miRNAs at a score ≤ 4.5, and 259 circRNAs (17.70%) were predicted to have no miRNA binding sites. Similarly, among the 687 ‘Tissue Enriched’ circRNAs, 590 (85.88%) were identified as having at least one binding site, encompassing a total of 2,380 miRNAs, while 97 circRNAs (14.12%) were predicted to lack putative miRNA binding sites. These findings indicate that circRNAs may have functions other than binding to miRNAs. In both of these sets, some well-known miRNA families, such as miR156, miR164, miR166, and miR395 were predicted to be targeted by more than 100 circRNAs each. However, only 11 circRNAs were found to be common between these two sets, associated with nine parental genes. For this subset, 22 miRNA targets were identified from 8 circRNAs (at a score ≤ 4.5), and 720 miRNA targets were identified from the 9 parental genes (at a score ≤ 4). A circRNA-miRNA network and a circRNA-miRNA-mRNA network were created using Cytoscape for this dataset of 11 circRNAs, their 9 parental genes, and the miRNA targets of these circRNAs and parental genes (Fig. 3a and b). In the circRNA-miRNA network, it was observed that circRNAs could have anywhere from zero to multiple (4 in this case) miRNA targets (Fig. 3a) without any inter-connections. In contrast, the circRNA-miRNA-mRNA network interconnects these circRNA-miRNA sets by incorporating mRNAs and their miRNA targets in the network. Even within this small dataset, an independent circRNA-miRNA network was identified that lacked any mRNA connections or inter-connections with other networks or sets (e.g., Tea_circ_053028 has 4 miRNA targets but no mRNA targets and no inter-connections with miRNA targets from other circRNAs or mRNAs) (Fig. 3b).

(a) circRNA-miRNA network. (b) circRNA-miRNA-mRNA network of DECs.
CircRNA database
The sequence and all important features of circRNAs predicted in the present analysis can be accessed through our user-friendly in-house database through the link http://indianteagenome.in:8080/tcdb/. This database allows users to easily search and extract circRNA sequences filtered by circRNAs length, circ-exons counts, BSJ counts, parental gene or scaffold, classification type, and expression (Fig. 4). Additionally, the BLAST search option allows users to input their query sequences and match them against the circRNAs in the database, providing detailed information on the matches.

Snapshots of TCDB database showing Home page (center) with different tabs including “circRNAs” tab for displaying tabular view, “Search” tab for searching circRNAs based on various features, “Blast” tab for BLAST searching user specified sequence against predicted circRNAs and “Help” tab for navigating throughout the database.
Discussion
Predicting non-coding RNAs and their role in controlling gene expression in eukaryotic cells is an emerging field. Among the many non-coding RNAs, circRNA is comparatively less known for its role in controlling gene expression. With the availability of a large amount of publicly accessible RNA-seq data, researchers can easily mine circRNAs, provided the sequencing depth is sufficient54. The analysis with RNA-seq data sets from tea can help initiate similar analysis for other organisms as well. This study provides the largest profiling of tissue- and stress-specific circRNAs in tea, or any single plant species, derived from public RNA-seq data, with an attempt to investigate their possible roles in transcriptional and post-transcriptional regulation. While opting for the type of RNA-seq data (single or paired-end), it was observed that, although there are circRNA prediction algorithms specifically designed for single-end (SE)10,55 or paired-end (PE) data56,57,58,59,60,61, the majority of the algorithms provide options to use either of the two. More importantly, developers agree that PE data should be preferred to increase the sensitivity and reliability of the results7. While CIRI62 has been used unbiasedly and accurately for detecting circRNAs from RNA-seq data with high sensitivity7,63,64, the advanced version CIRI237 was found to be even more trustworthy for de novo prediction of circRNAs59,65. Hence, only PE data was used for circRNA prediction with CIRI2. Although CIRCexplorer2 is also capable of identifying full-length circRNA transcripts, CIRI-Full was chosen as the primary tool as it is specifically optimized for processing large datasets, ensuring accurate detection of circRNAs across multiple samples and efficiently manages large volumes of data, maintaining high accuracy in identifying circRNA events41,66.
In the present study, while 3,052 circRNAs were predicted from in-house generated data comprising 135.414 million reads of a popular Indian tea, i.e., the TV-1 genotype, 59,575 circRNAs were identified from 178 publicly available RNA-seq data sets of tea from NCBI. Over 80% of circRNAs in both sets originated from exonic or intronic regions, which is quite similar to those predicted in other plant species including rice, Arabidopsis, wheat, tomato, soybean, and apple17, 27,67,68,69, indicating the effectiveness of this approach in predicting circRNAs in tea. Most parental protein-coding genes (7615 out of 12,166) produced more than one circRNA, while 4,551 genes produced only one. This could be due to the fact that circRNAs are formed by alternative back-splicing or from different pairs of exons/introns within the same parental gene17.
Out of the total 59,575 circRNAs from public data sets, only 9.77% (5,826 circRNAs) were shared between stress- and tissue-related data, while 19,825 circRNAs (77.29%) out of 25,651 were specifically present in stress-related data, and 33,924 circRNAs (85.34%) out of 39,750 were specifically found in tissue-related data. Moreover, 89.55% of the 3,052 predicted circRNAs in Indian tea and 65.84% of the 39,750 circRNAs identified in tissue-related public data sets were found to be differentially expressed in different tissues or groups of tissues. These results support the idea that circRNAs are not only often expressed in a tissue-specific manner but could also be stress-specific17,18,20,25,29,59, 70,71,72 in controlling gene expression.
The total number of circRNAs reported here in tea is the highest for any single plant species, with PlantCircBase 4.0 having 40,311 and 38,938 circRNAs in O. sativa and A. thaliana, respectively. Out of the 59,575 tea circRNAs generated from public data sets, only 10.87% share homology with known plant circRNAs, while a substantial 89.13% are novel plant circRNAs. Among 3,052 circRNAs from Indian tea, 389 were found to be homologous with 1,240 known plant circRNAs in PlantCircBase, and 317 (i.e., 81.49%) of those shared homology with circRNAs of C. sinensis. Moreover, 1,393 out of the 3,052 circRNAs in Indian tea were found to be common among 59,575 circRNAs from public data sets, leaving 1,659 circRNAs that are exclusively present in Indian tea. Furthermore, it was observed that 1,495 and 1,527 of these 1,659 circRNAs shared no homology with any plant circRNAs or circRNAs of C. sinensis from PlantCircBase, respectively. Clearly, 1,527 are strictly specific to Indian tea (TV-1) and 94% (i.e. 1436) of those included 1,122, 166, and 14 circRNAs that showed expression for ‘Expressed in all’, ‘Group-enriched’, and ‘Mixed’, respectively. The remaining 134 circRNAs were found to be expressed (either enhanced or enriched) in different tissues, with the maximum number observed in the 3rd leaf (82 circRNAs), followed by 27, 10, 9, and 6 circRNAs expressed in the flower bud, banji-bud, young stem, and active bud, respectively. The GO-KEGG pathway analysis of 948 parental protein-coding genes associated with these 1,527 Indian tea specific circRNAs revealed their involvement in various biological pathways. Notably, 5 genes were found to encode ammonia-lyase and lactoperoxidase enzymes in the phyenylpropanoid biosynthesis pathway, guiding the initial and final steps, respectively. Furthermore, as expected, the KEGG pathway analysis of all parental protein-coding genes of circRNAs from public data sets identified 80 genes encoding 9 important enzymes in the phenylpropanoid biosynthesis pathway, with a total of 464 circRNAs predicted in these 80 genes. The significant number of circRNAs from the genes involved in phenylpropanoid biosynthesis confirms their possible role in the secondary metabolite (catechins etc.) biosynthesis24,73,74 and underscores the need to study circRNAs, their roles, and their interactions with protein-coding genes, rather than only focusing on the key genes.
A total of 1,463 DECs were detected, which were found to be either up- or down-regulated under different stress conditions, including drought, salt, methyljasmonate, and NOxide. The in-depth comparative analysis of these DECs confirms several key observations. Some circRNAs were either up- or down-regulated in more than one stress condition, while others were specifically regulated in only one type of stress condition. Although no circRNAs exhibited up-regulation in PEG treatment and down-regulation in NaCl treatment (or vice-versa), 38 circRNAs were found to be up- or down-regulated with NaCl treatment followed by PEG treatment. The differential expression analysis of circRNAs obtained from the data with NOxide treatment under low-temperature stress indicated that NOxide treatment can reverse the regulation established due to cold treatment, as all 9 circRNAs that were up-regulated and 3 out of 5 circRNAs that were down-regulated in cold treatment were found to be down- and up-regulated, respectively, in the NOxide vs cold study.
The circRNAs may have important regulatory roles, as the circRNA-miRNA interaction analysis revealed that more than 82% of DECs and tissue-enriched circRNAs contained at least 1 miRNA binding site with high affinities for important miRNA families, including miR156, miR164, miR166 and miR395, which are known to play regulatory roles in development, growth and responses to stress in various plants75,76,77,78,79,80,81,82,83,84,85,86,87. However, since 14 to 17% of the circRNAs in these sets were found to have no putative miRNA binding sites, this confirms reports by earlier researchers that, although circRNAs mainly function as miRNA sponges9,20,88, this may not be the only defined function of plant circRNAs89.
Although this study provides valuable insights into circRNA prediction using available RNA-seq datasets, we acknowledge limitations including the lack of biological replicates in some datasets, with some Bioprojects having as few as two to three replicates, while others had up to 12 or 18. This variation may affect the robustness and consistency of the predictions. Additionally, the absence of experimental validation is noted as a limitation. Nevertheless, the bioinformatics framework presented here is expected to serve as a foundation for future experimental validation efforts by other researchers.
The database, TCDB, developed in this study, serves as a comprehensive resource for accessing predicted circRNA sequences and their associated features. It enables users to search and extract data based on a variety of filters, including circRNA length, exon counts, BSJ counts, parental gene or scaffold, classification type, and expression levels. The integrated BLAST search functionality further enhances the database by allowing users to compare their query sequences against the circRNAs in the database, providing detailed match information. These features aim to increase the accessibility and utility of the database for advancing circRNA research. Future updates will include the integration of additional reference genomes, expanding its applicability across the diverse tea genomes currently available. Moreover, structural details for each circRNA will be incorporated to improve the database’s comprehensiveness and support further circRNA-related studies.
Overall, a substantial number of circRNAs were predicted in tea from public datasets and presented in a database format for further exploration. The differential expression analysis and interaction analysis with miRNAs and mRNAs corroborate many previously reported findings. This may be attributed to the fact that only full-length circRNAs were predicted and utilized in the study. This underscores that the strategy employed in the present study can be effectively applied to similar analyses in other species.
Conclusion
In this study, the first database comprising tissue- and stress-specific circRNAs in tea was generated and made available for researchers to investigate the possible function of circRNAs and their interactions with parental protein-coding genes. Moreover, there are circRNAs (intronic and intergenic) without any specified parental protein-coding genes, and their study can also open the door to new findings or possible roles for circRNAs. Researchers can follow a similar strategy of using available public RNA-seq data to detect circRNAs for other organisms to generate a good reservoir of circRNAs and aid in unraveling the role of circRNAs.
Data availability
The generated data under this study are available in the supplementary files and in the database named TCDB which can be accessed at “http://indianteagenome.in:8080/tcdb/”.
References
-
Wei, J., Huang, K., Yang, C. & Kang, C. Non-coding RNAs as regulators in epigenetics (Review). Oncol. Rep. 37, 3–9. https://doi.org/10.3892/or.2016.5236 (2017).
-
Nigro, J. M. et al. Scrambled exons. Cell 64(3), 607–613 (1991).
-
Capel, B. et al. Circular transcripts of the testis-determining gene Sry in adult mouse testis. Cell 73(5), 1019–1030 (1993).
-
Li, Z. et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat Struct Mol Biol. 22, 256 (2015).
-
Lasda, E. & Parker, R. Circular RNAs: diversity of form and function. RNA 20, 1829–1842 (2014).
-
Chen, L. L. & Yang, L. Regulation of circRNA biogenesis. RNA Biol. 12, 381–388 (2015).
-
Szabo, L. & Salzman, J. Detecting circular RNAs: bioinformatic and experimental challenges. Nat Rev Genet. 17(11), 679–692. https://doi.org/10.1038/nrg.2016.114 (2016).
-
Jeck, W. R. et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 19(2), 141–157. https://doi.org/10.1261/rna.035667.112 (2013).
-
Hansen, T. B. et al. Natural RNA circles function as efficientmicroRNA sponges. Nature 495(7441), 384–388. https://doi.org/10.1038/nature11993 (2013).
-
Memczak, S. et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495(7441), 333–338. https://doi.org/10.1038/nature11928 (2013).
-
Zheng, Q. et al. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat Commun. 7, 11215 (2016).
-
Wang, K. et al. A circular RNA protects the heart from pathological hypertrophy and heart, failure by targeting miR-223. Eur Heart J. 37(33), 2602–2611 (2016).
-
Xiao, J. et al. Circular RNAs Acting as miRNAs’ Sponges and Their Roles in Stem Cells. J Clin Med. 11(10), 2909. https://doi.org/10.3390/jcm11102909 (2022).
-
Du, W. W. et al. Foxo3 circular RNA retards cell cycle progression via forming ternary complexes with p21 and CDK2. Nucleic Acids Res. 44(6), 2846–2858 (2016).
-
Singh, S., Shyamal, S., Das, A. & Panda, A. C. Global identification of mRNA-interacting circular RNAs by CLiPPR-Seq. Nucleic Acids Res. 52(6), e29. https://doi.org/10.1093/nar/gkae058 (2024).
-
Wang, P. L. et al. Circular RNA is expressed across the eukaryotic tree of life. PLoS ONE 9(6), e90859 (2014).
-
Ye, C. Y., Chen, L., Liu, C., Zhu, Q. H. & Fan, L. J. Widespread noncoding circular RNAs in plants. New Phytol. 208(1), 88–95. https://doi.org/10.1111/nph.13585 (2015).
-
Wang, Y. et al. Identification of circular RNAs and their targets in leaves of Triticum aestivum L. under dehydration stress. Front Plant Sci. 7, 2024 (2016).
-
Darbani, B., Noeparvar, S. & Borg, S. Identification of circular RNAs from the parental genes involved in multiple aspects of cellular metabolism in barley. Front Plant Sci. 7, 776 (2016).
-
Zhao, W. et al. Genome-wide identification and characterization of circular RNAs by high throughput sequencing in soybean. Sci Rep. 7(1), 5636 (2017).
-
Tan, J., Zhou, Z., Niu, Y., Sun, X. & Deng, Z. Identification and functional characterization of tomato circRNAs derived from genes involved in fruit pigment accumulation. Sci Rep. 7, 8594 (2017).
-
Zhou, R. et al. Transcriptome-wide identification and characterization of potato circular RNAs in response to Pectobacterium carotovorum subspecies brasiliense infection. Int J Mol Sci. 19(1), 71 (2018).
-
Chen, L. et al. Circular RNAs mediated by transposons are associated with transcriptomic and phenotypic variation in maize. New Phytol. 217(3), 3 (2018).
-
Wei, T. et al. Circular RNA architecture and differentiation during leaf bud to young leaf development in tea (Camellia sinensis). Planta 248(10), 1–13 (2018).
-
Zhu, Y. X. et al. Identification of cucumber circular RNAs responsive to salt stress. BMC Plant Biol. 19(1), 164. https://doi.org/10.1186/s12870-019-1712-3 (2019).
-
Wang, X. et al. Genome-wide profiling of circular RNAs in the hybridization of two elite inbred lines of Gossypium hirsutum. Industrial Crops and Prod. 170, 113754 (2021).
-
Wang, D., Gao, Y., Sun, S., Li, L. & Wang, K. Expression Characteristics in Roots, Phloem, Leaves, Flowers and Fruits of Apple circRNA. Genes (Basel). 13(4), 712. https://doi.org/10.3390/genes13040712 (2022).
-
Kumar, B. et al. Genome-wide identification and characterization of tissue-specific non-coding RNAs in black pepper (Piper nigrum L.). Front. Plant Sci. 14, 1079221 (2023).
-
Tong, W. et al. Circular RNA architecture and differentiation during leaf bud to young leaf development in tea (Camellia sinensis). Planta 248(6), 1417–1429. https://doi.org/10.1007/s00425-018-2983-x (2018).
-
Huang, J. et al. Evolutionary landscape of tea circular RNAs and its contribution to chilling tolerance of tea plant. Int J Mol Sci. 24(2), 1478. https://doi.org/10.3390/ijms24021478 (2023).
-
Baruah, P. M., Bordoloi, K. S., Gill, S. S. & Agarwala, N. CircRNAs responsive to winter dormancy and spring flushing conditions of tea leaf buds. Plant Sci. 336, 111828. https://doi.org/10.1016/j.plantsci.2023.111828 (2023).
-
Bordoloi, K. S., Baruah, P. M. & Agarwala, N. Identification of circular RNAs in tea plant during Helopeltis theivora infestation. Plant Stress. 8, 100150. https://doi.org/10.1016/j.stress.2023.100150 (2023).
-
Mondal, T. K., Bhattacharya, A., Laxmikumaran, M. & Ahuja, P. S. Recent advance in tea Biotechnology. Plant Cell Tissue Org Cult. 75, 795–856 (2004).
-
Varshney, D. et al. Tissue specific long non-coding RNAs are involved in aroma formation of black tea. Industrial Crops and Prod. 133, 79–89. https://doi.org/10.1016/j.indcrop.2019.03.020 (2019).
-
Rawal, H. C., et al. First chromosome-scale genome of Indian tea (Camellia assamica Masters; syn C. sinensis var assamica) cultivar TV 1 reveals its evolution and domestication of caffeine synthesis. Ind. Crops Prod. 222, 119992 (2024).
-
Patel, R. K. & Jain, M. NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE 7(2), e30619 (2012).
-
Gao, Y., Zhang, J. & Zhao, F. Circular RNA identification based on multiple seed matching. Brief Bioinform. 19(5), 803–810. https://doi.org/10.1093/bib/bbx014 (2018).
-
Wei, C. et al. Draft genome sequence of Camellia sinensis var. sinensis provides insights into the evolution of the tea genome and tea quality. Proc Natl Acad Sci USA 115(18), 4151–4158 (2018).
-
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. https://doi.org/10.1093/bioinformatics/btp324 (2009).
-
Gao, Y. et al. Comprehensive identification of internal structure and alternative splicing events in circular RNAs. Nat Commun. 7, 12060 (2016).
-
Zheng, Y., Ji, P., Chen, S., Hou, L. & Zhao, F. Reconstruction of full-length circular RNAs enables isoform-level quantification. Genome Med. 11(1), 2. https://doi.org/10.1186/s13073-019-0614-1 (2019).
-
Chu, Q. et al. PlantcircBase: A Database for Plant Circular RNAs. Mol Plant. 10(8), 1126–1128 (2017).
-
Anders, S. & Huber, W. Differential expression analysis for sequence count data. Genome Biol. 11(10), R106. https://doi.org/10.1186/gb-2010-11-10-r106 (2010).
-
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general-purpose program for assigning sequence reads to genomic features. Bioinformatics 30(7), 923–930 (2014).
-
Liao, Y., Smyth, G. K. & Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. https://doi.org/10.1093/nar/gkz114 (2019).
-
Rawal, H. C., Angadi, U. & Mondal, T. K. TEnGExA: an R package based tool for tissue enrichment and gene expression analysis. Brief Bioinform. 22(3), bbaa221. https://doi.org/10.1093/bib/bbaa221 (2021).
-
Uhlén, M. et al. Proteomics. Tissue-based map of the human proteome. Science 347(6220), 1260419. https://doi.org/10.1126/science.1260419 (2015).
-
Conesa, A. & Götz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int J Plant Genomics. https://doi.org/10.1155/2008/619832 (2008).
-
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53(D1), D672–D677. https://doi.org/10.1093/nar/gkae909 (2025).
-
Dai, X. & Zhao, P. X. psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res. 39, W155–W159. https://doi.org/10.1093/nar/gkr319 (2011).
-
Liu, T., Zhang, L., Chen, G. & Shi, T. Identifying and Characterizing the Circular RNAs during the Lifespan of Arabidopsis Leaves. Front Plant Sci. 8, 1278. https://doi.org/10.3389/fpls.2017.01278 (2017).
-
Kozomara, A. & Griffiths-Jones, S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. https://doi.org/10.1093/nar/gkt1181 (2014).
-
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13(11), 2498–2504. https://doi.org/10.1101/gr.1239303 (2003).
-
Nguyen, M. H., Nguyen, H. N. & Vu, T. N. Evaluation of methods to detect circular RNAs from single-end RNA-sequencing data. BMC Genomics 23(1), 106. https://doi.org/10.1186/s12864-022-08329-7 (2022).
-
Hoffmann, S. et al. A multi-split mapping algorithm for circular RNA, splicing. Trans-splicing and fusion detection. Genome Biol. 15(2), 34 (2014).
-
Salzman, J., Gawad, C., Wang, P. L., Lacayo, N. & Brown, P. O. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS ONE 7(2), e30733 (2012).
-
Salzman, J., Chen, R. E., Olsen, M. N., Wang, P. L. & Brown, P. O. Cell-type specific features of circular RNA expression. PLoS Genet. 9(9), e1003777 (2013).
-
Westholm, J. O. et al. Genome-wide analysis of Drosophila circular RNAs reveals their structural and sequence properties and age-dependent neural accumulation. Cell Rep. 9, 1966–1980 (2014).
-
Zhang, P., Li, S. & Chen, M. Characterization and Function of Circular RNAs in Plants. Front. Mol. Biosci. 7, 91. https://doi.org/10.3389/fmolb.2020.00091 (2020).
-
Nguyen, D. T. et al. Circall: fast and accurate methodology for discovery of circular RNAs from paired-end RNA-sequencing data. BMC Bioinformatics 22, 495. https://doi.org/10.1186/s12859-021-04418-8 (2021).
-
Liao, X. et al. Mitochondrion-encoded circular RNAs are widespread and translatable in plants. Plant Physiol. 189, 1482–1500. https://doi.org/10.1093/plphys/kiac143 (2022).
-
Gao, Y., Wang, J. & Zhao, F. CIRI: an efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 16(1), 4. https://doi.org/10.1186/s13059-014-0571-3 (2015).
-
Hansen, T. B., Veno, M. T., Damgaard, C. K. & Kjems, J. Comparison of circular RNA prediction tools. Nucleic Acids Res. 44(6), e58 (2016).
-
Zeng, X., Lin, W., Guo, M. & Zou, Q. A comprehensive overview and evaluation of circular RNA detection tools. PLoS Comput. Biol. https://doi.org/10.1371/journal.pcbi.1005420 (2017).
-
Hansen, T. B. Improved circRNA Identification by Combining Prediction Algorithms. Front. Cell Dev. Biol. 6, 20. https://doi.org/10.3389/fcell.2018.00020 (2018).
-
Zhang, X. O. et al. Diverse alternative back-splicing and alternative splicing landscape of circular RNAs. Genome Res. 26(9), 1277–1287. https://doi.org/10.1101/gr.202895.115 (2016).
-
Jeck, W. R. & Sharpless, N. E. Detecting and characterizing circular RNAs. Nat Biotechnol. 32, 453–461 (2014).
-
Ren, Y. et al. Identification and characterization of circRNAs involved in the regulation of low nitrogen-promoted root growth in hexaploid wheat. Biol Res. 51(1), 43 (2018).
-
Wang, X. S. et al. Identification and functional prediction of soybean Circ RNAs involved in low-temperature responses. J. Plant Physiol. https://doi.org/10.1016/j.jplph.2020.153188 (2020).
-
Misir, S., Wu, N. & Yang, B. B. Specific expression and functions of circular RNAs. Cell Death Differ. 29(3), 481–491. https://doi.org/10.1038/s41418-022-00948-7 (2022).
-
Wang, Z. et al. Identification of circular RNAs in kiwifruit and their species-specific response to bacterial canker pathogen invasion. Front Plant Sci. 8, 413 (2017).
-
Li, N. et al. Identification of Wheat Circular RNAs Responsive to Drought Stress. Sci. Agric. Sicina. 55, 583–4599. https://doi.org/10.3864/j.issn.0578-1752.2022.23.002 (2022).
-
Chen, C. et al. Expression of key structural genes of the phenylpropanoid pathway associated with catechin epimerization in tea cultivars. Front Plant Sci. 8, 702. https://doi.org/10.3389/fpls.2017.00702 (2017).
-
Xia, J. et al. Characterization and expression profiling of Camellia sinensis cinnamate 4-hydroxylase genes in phenylpropanoid pathways. Genes (Basel). 8(8), 193. https://doi.org/10.3390/genes8080193 (2017).
-
Mica, E., Gianfranceschi, L. & Pè, M. E. Characterization of five microRNA families in maize. J Exp Bot. 57(11), 2601–2612 (2006).
-
Curaba, J., Spriggs, A., Taylor, J., Li, Z. & Helliwell, C. miRNA regulation in the early development of barley seed. BMC Plant Biol. 12, 120. https://doi.org/10.1186/1471-2229-12-120 (2012).
-
Xie, K. B. et al. Gradual increase of miR156 regulates temporal expression changes of numerous genes during leaf development in rice. Plant Physiol. 158, 1382–1394. https://doi.org/10.1104/pp.111.190488 (2012).
-
Gu, Y. et al. Identification and characterization of microRNAs in the developing maize endosperm. Genomics 102, 472–478. https://doi.org/10.1016/j.ygeno.2013.08.007 (2013).
-
Ding, H. et al. Identification and functional analysis of miRNAs in developing kernels of a viviparous mutant in maize kernel. Crop J. 1, 115–126. https://doi.org/10.1016/j.cj.2013.07.013 (2013).
-
Deng, P. et al. Global identification of microRNAs and their targets in barley under salinity stress. PLoS ONE 10, e0137990. https://doi.org/10.1371/journal.pone.0137990 (2015).
-
Song, Q. X. et al. Identification of miRNAs and their target genes in developing soybean seeds by deep sequencing. BMC Plant Biol. 11, 5. https://doi.org/10.1186/1471-2229-11-5 (2011).
-
Guo, H., Kan, Y. & Liu, W. Differential expression of miRNAs in response to topping in flue-cured tobacco (Nicotiana tabacum) roots. PLoS ONE 6, e28565. https://doi.org/10.1371/journal.pone.0028565 (2011).
-
Varkonyi-Gasicet, E., Gould, N., Sandanayaka, M., Sutherland, P. & MacDiarmid, R. M. Characterisation of microRNAs from apple (Malus domestica ‘Royal Gala’) vascular tissue and phloem sap. BMC Plant Biol. 10, 159. https://doi.org/10.1186/1471-2229-10-159 (2010).
-
Han, J. et al. Identification of miRNAs and their targets in wheat (Triticum aestivum L.) by EST analysis. Genet. Mol. Res. 12, 3793–3805. https://doi.org/10.4238/2013.September.19.11 (2013).
-
Chen, X. et al. Potential functions of microRNAs in starch metabolism and development revealed by miRNA transcriptome profiling of cassava cultivars and their wild progenitor. BMC Plant Biol. 15, 33. https://doi.org/10.1186/s12870-014-0355-7 (2015).
-
Dong, Q., Hu, B. & Zhang, C. microRNAs and Their Roles in Plant Development. Front Plant Sci. https://doi.org/10.3389/fpls.2022.824240 (2022).
-
Shen, E., Zhao, T. & Zhu, Q. H. Are miRNAs applicable for balancing crop growth and defense trade-off?. New Phytol. 243(5), 1670–1680. https://doi.org/10.1111/nph.19939 (2024).
-
Zuo, J., Wang, Q., Zhu, B., Luo, Y. & Gao, L. Deciphering the roles of circRNAs on chilling injury in tomato. Biochem. Biophys. Res. Commun. 479(2), 132–138. https://doi.org/10.1016/j.bbrc.2016.07.032 (2016).
-
Li, Q. F., Zhang, Y. C., Chen, Y. Q. & Yu, Y. Circular RNAs roll into the regulatory network of plants. Biochem Biophys Res Commun. 488(2), 382–386 (2017).
Acknowledgements
The authors are grateful to the Director of ICAR-National Institute for Plant Biotechnology, New Delhi, 110012 for providing the research facility.
Funding
The authors are grateful to National Tea Research Foundation (Project code: NIPB-526), Tea Board, Ministry of Commerce, Kolkata, India for financial assistance.
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Rawal, H.C., Pal, R.K. & Mondal, T.K. Identification and characterization of tissue- and stress-specific circular RNAs (circRNAs) of tea to generate the largest tea circRNAs data repository. Sci Rep 15, 36999 (2025). https://doi.org/10.1038/s41598-025-94480-6
-
Received:
-
Accepted:
-
Published:
-
DOI: https://doi.org/10.1038/s41598-025-94480-6