Introduction
Soybean gray mold, caused by the necrotrophic fungal pathogen Botrytis cinerea, has emerged as a devastating disease threatening global soybean production, with yield losses reaching 30–50%1,2. This pathogen typically invades host tissues through wounds or natural openings, leading to characteristic symptoms including leaf blotches, stem wilting, and fruit rot3,4. Its broad host adaptability enables it to infect over 200 economically important crops, including soybean, resulting in substantial economic losses. Although chemical fungicides remain the primary control strategy, their overuse has not only driven the evolution of multidrug resistance in pathogens but also disrupted soil microbial communities5. Consequently, biocontrol strategies utilizing plant growth-promoting rhizobacteria (PGPR) have gained prominence as sustainable alternatives6,7, highlighting the urgency to develop sustainable alternatives such as PGPR-based biocontrol.
Bacillus velezensis, first isolated from the Vélez River estuary in Málaga, Spain, by Ruiz-García et al. in 1999 and formally described in 20058, has emerged as a model Gram-positive PGPR due to its broad-spectrum antimicrobial activity and plant growth-promoting traits9,10. Its biocontrol mechanisms involve the synthesis of functional metabolites (e.g., cyclic lipopeptides, polyketides, siderophores), coupled with nutrient competition, biofilm formation, and induction of systemic resistance (ISR) in host plants10,11,12.
Notably, accumulating evidence demonstrates the efficacy of B. velezensis against B. cinerea: For instance, cell-free supernatants from strain YTQ3 suppress mycelial growth (> 60% inhibition) and spore germination of B. cinerea in a dose-dependent manner13, while strain BE1 significantly reduces postharvest gray mold incidence in tomato fruits14. Additionally, this bacterium exhibits protective effects against soybean root rot (caused by Fusarium oxysporum), Phytophthora root rot (Phytophthora sojae), and pustule disease (Xanthomonas axonopodis pv. glycines)15,16,17. However, its potential for controlling soybean gray mold remains unexplored.
Recent studies highlight the role of B. velezensis in reshaping plant-microbe interactions through modulation of rhizosphere exudates. These root-secreted metabolites, including sugars, organic acids, and amino acids, serve as key signaling molecules in rhizosphere communication18,19. B. velezensis can induce the secretion of antimicrobial compounds such as cinnamic acid and malic acid, which directly inhibit pathogens (e.g., Ralstonia solanacearum) while recruiting beneficial microbes (e.g., Pseudomonas spp., Streptomyces spp.) to establish pathogen-suppressive microbiomes18,20. Metabolomic analyses reveal that this bacterium reprograms plant metabolic pathways, particularly trehalose biosynthesis and phenylpropanoid metabolism, thereby altering rhizosphere metabolite profiles and optimizing microbial community structure21,22. Furthermore, lipopeptides (e.g., fengycin) secreted by B. velezensis synergize with root exudates to suppress pathogens, while enhanced biofilm formation ensures long-term rhizosphere colonization23,24. Nevertheless, the metabolic reprogramming mechanisms and microbiome-mediated immune regulation in soybean-B. cinerea interactions remain poorly understood.
In this study, integrated metabolomic and metagenomic analyses were employed to elucidate how B. velezensis ES2-4 enhances soybean resistance by specifically inducing the secretion of antimicrobial metabolites (e.g., oxalic acid, eicosane) and reconstructing rhizosphere microbial networks. Our findings innovatively decipher the microbiome engineering-mediated plant immunity mechanisms, offering a theoretical foundation for developing rhizosphere-focused biocontrol strategies against soybean gray mold.
Results
Alterations in rhizosphere exudate profiles following co-inoculation of soybean with strain ES2-4 and B. cinerea
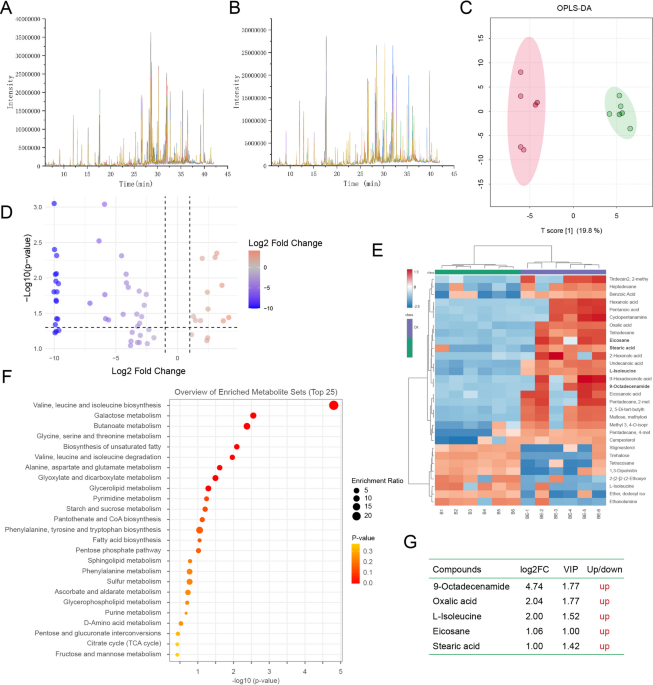
To investigate the dynamic changes in soybean rhizosphere exudates under individual or co-inoculation with B. velezensis ES2-4 and B. cinerea, comprehensive metabolite profiling was conducted using GC-MS. Total ion chromatogram (TIC) analysis from 24 biological replicates (6 per group) showed high inter-sample peak overlap within each treatment group (Fig. 1A,B, S1A-B), with consistent retention times and ion intensities, confirming methodological reproducibility and instrument stability. Orthogonal partial least squares-discriminant analysis (OPLS-DA) revealed clear separation between the BE (ES2-4 + B. cinerea) and B (B. cinerea alone) groups (Fig. 1C), and distinct metabolic divergence between the E (ES2-4 alone) and CK (control) groups (Fig. S1C). Screening with VIP ≥ 1 and |log2FC| ≥1 thresholds identified 33 differentially abundant metabolites in the BE vs. B comparison (11 upregulated, 22 downregulated), including oxalic acid, eicosane, stearic acid, and 9-octadecenamide (Fig. 1D). The E vs. CK comparison revealed 14 differential metabolites (7 upregulated, 7 downregulated), with shared biomarkers (e.g., eicosane, oxalic acid), indicating conserved stress-responsive mechanisms (Fig. S1D). Ward’s hierarchical clustering based on Euclidean distances showed tight intra-group clustering and distinct inter-group expression patterns of differential metabolites (Fig. 1E, S1E). KEGG pathway enrichment analysis indicated that E vs. CK differential metabolites were primarily linked to starch/sucrose metabolism and unsaturated fatty acid biosynthesis (Fig. 1F)25,26. In contrast, BE vs. B comparisons implicated 11 pathways, including branched-chain amino acid metabolism and glyoxylate/dicarboxylate cycling, with unsaturated fatty acid biosynthesis being the most enriched (Fig. S1F). The upregulation of four metabolites—9-octadecenamide, oxalic acid, eicosane, and stearic acid—was observed in the BE group prior to B. cinerea infection (Fig. 1G). These findings demonstrate that B. velezensis ES2-4 significantly induces the upregulation of key rhizosphere exudates, including oxalic acid and stearic acid, during B. cinerea infection in soybean plants.

Metabolomic analysis of soybean root exudates under Bacillus velezensis ES2-4 treatment. (A,B) Total ion chromatograms (TIC) of metabolites between B and BE groups, showing distinct intensity profiles across retention times. (C) OPLS-DA score plot demonstrating clear metabolic separation between B and BE groups. (D) Volcano plot highlighting significantly upregulated (red) and downregulated (blue) metabolites (|log2FC| > 1.5, VIP > 1.0). Key antifungal metabolites, including oxalic acid), were markedly elevated. (E) Heatmap of the top 25 enriched metabolite clusters, with red indicating high abundance between B and BE groups. (F) Overview of enriched metabolic pathways mapping was conducted using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. (G) Table summarizes VIP scores, log2FC values, and regulation trends of differential metabolites.
Strain ES2-4 elicits soybean resistance against B. cinerea through rhizosphere exudate-mediated mechanisms
To investigate the regulatory effects of soybean rhizosphere secretions on the chemotactic behavior of B. velezensis ES2-4, we analyzed the impact of key differential metabolites on bacterial growth and chemotaxis using a semi-solid plate assay. The results revealed that oxalic acid, eicosane, and 9-octadecenamide significantly increased the colony area of B. velezensis ES2-4 (p < 0.05) within a 0.05–1 mM concentration range, demonstrating a concentration-dependent pattern that suggests their role in inducing directional chemotaxis (Fig. 2A–E). In contrast, stearic acid exhibited a unique concentration response: while low concentrations (0.05 mM) markedly enhanced chemotactic motility, higher concentrations progressively attenuated this effect, indicating potential metabolic inhibition or receptor saturation (Fig. 2A,E). Further analysis of antifungal activity revealed that all four metabolites significantly suppressed the growth of B. cinerea at 0.05 mM. Notably, eicosane, 9-octadecenamide, and stearic acid exhibited enhanced inhibitory effects at higher concentrations (0.1–1 mM), with stearic acid showing the strongest pathogen inhibition (Fig. 2F–I). However, oxalic acid did not exhibit enhanced antifungal activity beyond 0.05 mM, suggesting distinct molecular targets or regulatory mechanisms compared to the other metabolites (Fig. 2F,I). These findings collectively suggest that B. velezensis ES2-4 may enhance plant systemic resistance by modulating root secretion of oxalic acid, eicosane, and 9-octadecenamide, which synergistically promote bacterial chemotaxis and suppress pathogen growth.

Inhibitory effects of key root exudate metabolites induced by Bacillus velezensis ES2-4 on Botrytis cinerea colony growth. (A) Representative images of strain ES2-4 treated with oxalic acid, eicosane, 9-octadecenamide, or stearic acid at concentrations ranging from 0 to 1 mM. (B–E) Quantitative analysis of colony diameter (cm) of strain ES2-4 treated with oxalic acid, eicosane, 9-octadecenamide, or stearic acid at concentrations ranging from 0 to 1 mM. (F–I) Quantitative analysis of colony diameter (cm) of B. cinerea treated with oxalic acid, eicosane, 9-octadecenamide, or stearic acid at concentrations ranging from 0 to 1 mM. Note: the bars indicate the mean ± SE (standard error) of three replicates, and different letters indicate significant differences from the other treatments (p < 0.05).
Strain ES2-4 modulates rhizosphere microbiome diversity and structure in response to B. cinerea infection
This study evaluated soybean rhizosphere microbiome variations across treatments using α- and β-diversity indices. For bacterial communities, the Sobs, Chao, and Ace indices followed the order E > CK > BE > B (Table S1, Fig. 3A–D), indicating that B. velezensis ES2-4 enhanced bacterial richness in both healthy and B. cinerea-infected rhizospheres, while B. cinerea (Group B) significantly reduced bacterial richness. The BE group (ES2-4 pretreated before infection) partially mitigated this reduction. Shannon index analysis revealed lower diversity in Group B compared to Groups E and CK, while the BE group exhibited higher diversity than Group B, and Group E surpassed CK, suggesting that ES2-4 improved bacterial diversity in both healthy and diseased plants. In contrast, fungal communities displayed opposite trends (Table S2). Groups B and BE exhibited higher Sobs, Chao, and Ace indices than Groups CK and E, indicating that B. cinerea increased fungal richness, while ES2-4 (Groups E and BE) reduced fungal richness in both healthy and infected plants. Simpson index analysis confirmed higher fungal diversity in Group B compared to other treatments, with the BE group showing a significant decline relative to Group B, highlighting ES2-4’s inhibitory effect on pathogen-enhanced fungal diversity. Hierarchical clustering supported these findings, with BE/B groups and E/CK groups forming distinct clusters (Fig. 3E,F). PCoA based on Bray-Curtis distance revealed significant β-diversity differences in bacterial communities among treatments (PERMANOVA: R² = 0.669, p = 0.001), with PC1 and PC2 collectively explaining 24.15% of the variation (Fig. 3G). B. cinerea-treated groups (B and BE) separated from E and CK groups along PC1, while ES2-4 effects differentiated BE from Group B along PC2. Fungal communities showed similar β-diversity differences (R² = 0.5676, p = 0.001), with PC1 and PC2 explaining 24.48% of the variation (Fig. 3H), mirroring bacterial clustering patterns. In conclusion, B. velezensis ES2-4 alleviated B. cinerea-induced dysbiosis in the soybean rhizosphere by differentially regulating bacterial and fungal community diversity and structure.

Microbial diversity and community structure analysis under experimental treatments. (A) Sobs index on species level. (B) Chao index on species level. (C) Ace index on species level. (D) Shannon index on species level. (E) Cluster analysis between samples based on the Bray–Curtis distance of species abundance. (F) Heatmap of Bray-Curtis distances revealing closer microbial similarity within treated groups. (G) PCoA dimensionality reduction analysis based on the Bray–Curtis distance of species abundance of bacteria. (H) PCoA dimensionality reduction analysis based on the Bray–Curtis distance of species abundance of fungi. (I) Beta diversity boxplots highlighting increased inter-group dissimilarity (Bray-Curtis distance: 0.15–0.35).
Strain ES2-4 alters taxonomic composition and enriches beneficial microbes in soybean rhizosphere under pathogen stress
Taxonomic annotation based on the NR database revealed that bacteria dominated the soybean rhizosphere microbiota (mean relative abundance: 99.61%), followed by viruses (0.16%), eukaryotes (0.13%), and archaea (0.09%). Bacterial communities consisted of 162 phyla, 269 classes, 482 orders, 998 families, 3,906 genera, and 27,342 species, while fungal communities included 10 phyla, 39 classes, 100 orders, 258 families, 436 genera, and 739 species (Fig. 4A). Genus-level Venn analysis identified 3,497 bacterial genera shared across all treatments (Fig. 4B). The dominant bacterial genera included Rugosimonospora, Reticulibacter, Actinomadura, Pseudonocardia, Alphaproteobacteria, Bradyrhizobium, Streptomyces, and Nitrobacteraceae (Fig. 4C). Treatment with strain ES2-4 significantly increased the abundance of Actinomadura and Ktedonobacter in healthy plants, while reducing the abundance of Edaphobacter. In infected plants, ES2-4 pretreatment decreased the abundance of Rugosimonospora, Reticulibacter, Actinomadura, and Pseudonocardia (Fig. 4C). Differential analysis further revealed higher abundances of Alphaproteobacteria, Proteobacteria, Acidobacteria, Pseudolabrys, Gemmatimonadetes, Betaproteobacteria, and Opitutus in the BE group compared to the B group (Fig. 4D,E). For fungal communities, 324 genera were shared across treatments (Fig. 4F), with dominant genera including Tulasnella, Pseudogymnoascus, Aspergillus, Tulasnellaceae, Rhizopus, Mycoblastus, and Amanita (Fig. 4G). Strain ES2-4 treatment significantly increased the abundance of Aspergillus and Rhizopus in infected plants, while the BE group exhibited higher abundances of Mycoblastus, Amanita, Suillus, and Ceratobasidium compared to other groups (Fig. 4H). Differential analysis confirmed that the BE group had significantly higher abundances of Aspergillus, Daldinia, Mycoblastus, Anaeromyces, Rhizopus, Amanita, and Trichoderma compared to the B group (Fig. 4H,I). In conclusion, strain ES2-4 reshapes microbial community composition by selectively enriching beneficial taxa (e.g., Alphaproteobacteria, Aspergillus) and suppressing pathogen-associated genera under biotic stress.

Microbial community composition and functional diversity across experimental groups. (A) Domain-level taxonomic distribution: Bacteria dominated in all groups, while Eukaryota and Archaea were minor components. (B) Venn diagrams identified 3,497 shared microbial taxa across groups. (C) Relative abundances of major genus present in the bacterial communities under different treatments. (D) Comparative analysis of rhizosphere bacteria in group E and CK at genus level (Top15). (E) Comparative analysis of rhizosphere bacteria in group B and BE at genus level (Top15). (F) Venn diagrams identified 324 shared microbial taxa across groups. (G) Relative abundances of major genus present in the fungal communities under different treatments. (H) Comparative analysis of rhizosphere fungi in group E and CK at genus level (Top15). (I) Comparative analysis of rhizosphere fungi in group B and BE at genus level (Top15).
Strain ES2-4 coordinates metabolic rewiring and defense activation in the rhizosphere to suppress B. cinerea
KEGG functional annotation revealed distinct metabolic potentials across the treatment groups. The metabolic category accounted for the highest proportion of functional annotations in all groups: 51.84% in the B group, 51.41% in the BE group, 50.71% in the CK group, and 50.72% in the E group (Fig. 5A). Environmental information processing represented 16.90% (B), 16.79% (BE), 16.45% (CK), and 16.50% (E), while cellular processes made up 11.55% (B), 11.76% (BE), 11.75% (CK), and 11.69% (E). Genetic information processing accounted for 10.74% (B), 10.96% (BE), 11.51% (CK), and 11.59% (E), with organismal systems showing the lowest values: 3.66% (B), 3.71% (BE), 3.86% (CK), and 3.83% (E). Comparative analysis of KEGG pathways demonstrated significant treatment-specific effects. In ES2-4-treated healthy plants (E vs. CK), starch/sucrose metabolism (p = 0.01219) and the PI3K-Akt signaling pathway (p = 0.02157) showed increased gene abundance (Fig. 5B). For pathogen-infected plants pretreated with ES2-4 (BE vs. B), several metabolic pathways were significantly downregulated, including metabolic pathways (p = 0.01219), microbial metabolism in diverse environments (p = 0.01219), cofactor biosynthesis (p = 0.01219), nucleotide sugar biosynthesis, fatty acid metabolism (p = 0.01219), and butanoate metabolism (p = 0.03671). In contrast, pathways such as two-component systems (p = 0.01219), purine metabolism (p = 0.01219), nucleotide metabolism, and glycan biosynthesis/metabolism (p = 0.01219) were upregulated in the BE group (Fig. 5C).

Functional and statistical analyses of metabolic pathways and microbial interactions. (A) Circular plot of metabolic subsystem distribution.Color gradients represent functional categories. (B) Bar plot of Wilcoxon rank-sum test results for pathway mapping was conducted using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. Significant differences (p < 0.05) included upregulated starch/sucrose metabolism (p = 0.0121) and downregulated phosphatidylinositol signaling (p = 0.0121). Green/red markers denote 95% confidence intervals. (C) Bar plot of differential metabolic pathway mapping was conducted using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. Error bars indicate inter-group variance. (D) Circular plot of cellular process distribution. (E) COG functional comparison analysis of rhizosphere microorganisms in E and CK group. (F) COG functional comparison analysis of rhizosphere microorganisms in B and BE group.
COG functional classification grouped microbial proteins into three major categories. Metabolic functions dominated across treatments: 44.29% (B), 44.29% (BE), 44.04% (CK), and 43.88% (E), followed by cellular processes/signaling: 23.93% (B), 24.30% (BE), 25.75% (CK), and 25.54% (E). Information storage/processing had the lowest proportions: 18.16% (B), 17.94% (BE), 17.43% (CK), and 17.69% (E) (Fig. 5D). Differential analysis revealed significant enrichment in the BE group compared to the B group in pathways such as signal transduction mechanisms (p = 0.03671), translation/ribosome biogenesis (p = 0.01219), DNA replication/recombination/repair (p = 0.02157), defense mechanisms (p = 0.03671), cell cycle control/division (p = 0.03671), cellular motility (p = 0.03671), and extracellular structure formation (p = 0.03671) (Fig. 5E).
In conclusion, strain ES2-4 reprograms rhizosphere functional processes by suppressing pathogen-promoting metabolic pathways and enhancing stress-responsive and defense-associated functions, thereby establishing a microbially-driven defense network against B. cinerea.
Strain ES2-4 modulates microbial interaction networks to enhance pathogen resistance in soybean rhizosphere
Spearman correlation networks (|r| ≥ 0.7, p < 0.05) revealed distinct interaction patterns between bacterial and fungal communities at the genus level. For bacterial networks, the CK group exhibited the highest complexity (nodes = 49, edges = 388, average degree = 15.837, clustering coefficient = 0.753), with a balanced distribution of positive and negative edges (50.77% vs. 49.23%). In contrast, strain ES2-4 treatment in healthy plants (E group) slightly reduced network complexity (nodes = 48, edges = 379, average degree = 15.792, clustering coefficient = 0.736), but increased the proportion of positive edges to 54.35% (Fig. 6A-B). Pathogen infection (B group) drastically simplified bacterial networks (nodes = 47, edges = 139, average degree = 5.915), while ES2-4 pretreatment (BE group) partially restored network complexity (nodes = 50, edges = 154, average degree = 6.16) (Fig. 6C-D). Keystone genus analysis (Table S3, S5) demonstrated that ES2-4 enhanced the synergism between beneficial bacteria (e.g., Bradyrhizobium-Streptomyces) while suppressing pathogen-associated genera (e.g., Ralstonia) in network centrality. Fungal networks exhibited contrasting dynamics. ES2-4 treatment in healthy plants (E group) reduced the number of edges from 180 (CK) to 108, with a lower average degree (4.408 vs. 7.347), but a higher clustering coefficient (0.645) (Fig. 6E-F). Pathogen challenge (B group) increased fungal connectivity (edges = 159, average degree = 6.49), whereas ES2-4 pretreatment (BE group) weakened the interaction intensity (edges = 104, average degree = 4.426) and elevated the proportion of positive edges to 68.27% (Fig. 6G-H). Fungal keystone analysis (Table S4, S6) indicated that ES2-4 reduced the network centrality of pathogenic fungi (e.g., Fusarium) while strengthening antagonistic genera (e.g., Trichoderma), thereby optimizing community stability. In summary, strain ES2-4 enhances soybean’s ecological defense against B. cinerea by differentially restructuring bacterial synergy networks and fungal antagonism networks, establishing a host-beneficial microbiome equilibrium.

Correlation network analysis of soybean rhizosphere soil microbial community. (A) Soil microbial community of soybean rhizosphere at the genus level in CK group. Soil microbial community of soybean rhizosphere bacteria at the genus level in groups: (A) CK, (B) E, (C) B. (D) BE. Soil microbial community of soybean rhizosphere fungi at the genus level in groups: (E) CK, (F) E, (G) B. (H) BE. The node size is proportional to the relative abundance of each OTU; Links betwee’s correlation > 0.8 or ≤ 0.8); Different node colors indicate different genus to which the OTU belongs; Line color indicates direction, as shown in the legend.
Discussion
This study integrates metabolomic and metagenomic approaches to elucidate the dual defense mechanisms employed by B. velezensis ES2-4 against B. cinerea-induced soybean gray mold. We demonstrate that ES2-4 reprograms soybean root exudates to enrich antifungal metabolites such as oxalic acid and eicosane, which directly inhibit pathogen growth and enhance the recruitment of beneficial bacteria. Concurrently, ES2-4 restructures the rhizosphere microbiome by enriching taxa including Alphaproteobacteria and Streptomyces, while suppressing pathogenic fungi. Furthermore, network analysis revealed that ES2-4 fosters microbial cooperation and enhances stress-responsive pathways, underscoring its role in establishing a disease-suppressive rhizosphere. These findings provide novel insights into PGPR-mediated metabolic and microbiome engineering, advancing our understanding of sustainable biocontrol strategies.
Plants recruit beneficial microbes via root exudates to enhance stress adaptation, a process potentiated by PGPR27. Similarly, Bacillus velezensis can enhance plant resistance to biotic stress by inducing changes in plant rhizosphere exudates. For example, B. velezensis SAAS-63 promotes the participation of phenylalanine in the synthesis of lignin precursors and coumarins, thereby enhancing plant resistance28. B. velezensis can also induce plants to produce other resistance substances; for instance, B. velezensis YC89 increases the amino acid content in sugarcane root exudates29. Therefore, the recruitment of B. velezensis by plant rhizosphere exudates, and its reciprocal effect on the plant itself to induce changes in root exudate composition, represent core biological processes in plant–B. velezensis interactions. Our metabolomic analyses reveal that ES2-4 induces the accumulation of key metabolites—including oxalic acid, eicosane, stearic acid, and 9-octadecenamide—in both healthy and infected plants. In plate experiments, these substances significantly promoted the physicochemical functions of strain ES2-4, consistent with the findings of Jin et al.20. Notably, eicosane—a recognized antimicrobial effector in plant–microbe interactions30,31,32, previously reported mainly as a bacterial antibiotic—was for the first time shown to be plant-derived under Bacillus induction, highlighting a previously unexplored mechanism of microbiome-mediated immunity. This plant-originated eicosane not only directly inhibits B. cinerea but also facilitates the recruitment of beneficial bacteria.
Pathway enrichment indicated that ES2-4 modulates starch/sucrose metabolism and unsaturated fatty acid biosynthesis, which are critical for membrane integrity and defense signaling33,34,35. The enrichment of fatty acid metabolism pathways in both metabolomic and metagenomic analyses underscores its pivotal role. This aligns with the understanding that unsaturated fatty acids are not only fundamental for membrane integrity but also serve as precursors for jasmonate biosynthesis, a key phytohormone in defending against necrotrophic pathogens like B. cinerea33. Additionally, activation of branched-chain amino acid metabolism likely potentiates jasmonate signaling, enhancing resistance against B. cinerea36,37. Such metabolic restructuring parallels observations in Trichoderma–plant interactions38, suggesting conserved mechanisms for microbiome-enhanced immunity.
Chemotaxis and biofilm formation assays confirmed that ES2-4 selectively responds to root-secreted metabolites. Low concentrations of oxalic acid, eicosane, and stearic acid significantly promoted bacterial motility and biofilm formation, corroborating earlier reports on fatty acid-mediated rhizosphere colonization39,40,41,42 Notably, stearic acid exhibited biphasic activity—enhancing biofilm formation at high concentrations while suppressing chemotaxis, suggesting complex concentration-dependent regulation. The antifungal activity of these metabolites further underscores their dual role in pathogen suppression and microbiome recruitment, consistent with studies on Bacillus-induced systemic resistance43.
Metagenomic profiling revealed that ES2-4 counteracts B. cinerea-induced dysbiosis by enhancing bacterial diversity and reducing fungal richness. Enrichment of beneficial genera such as Alphaproteobacteria and Acidobacteriaceae—known for their roles in nutrient cycling and pathogen suppression44,45,46, aligns with prior work on B. subtilis-mediated microbiome remodeling. Additionally, consistent with the study by Sun et al.47., in which B. velezensis stimulated resident Pseudomonas, our metagenomic analysis revealed a significant enrichment of plant-beneficial taxa such as Alphaproteobacteria and Streptomyces in the BE group. This convergence across independent studies suggests that the recruitment of specific bacterial lineages (e.g., Acidobacteria and Proteobacteria) might be a conserved strategy employed by Bacillus species to establish a disease-suppressive rhizosphere.
KEGG and COG analyses indicated upregulation of two-component systems and defense-related pathways, reinforcing microbial stress adaptation and host defense coordination48,49 The upregulation of two-component systems in the BE group indicates enhanced environmental sensing and signal transduction capabilities within the rhizosphere microbiome, a trait crucial for rapid adaptation to biotic stress and previously linked to improved biofilm formation and biocontrol efficacy in pseudomonads48. These functional shifts are consistent with those induced by other biocontrol agents, supporting the broader potential of microbiome-based disease management.
Network analysis further illuminated how ES2-4 modulates microbial interactions. Pathogen challenge reduced network complexity, while ES2-4 pretreatment restored bacterial connectivity and increased positive interactions, particularly between Acidobacteria and Proteobacteria—a structure associated with pathogen suppression50. The restoration of bacterial network complexity and increased positive interactions in the BE group resonate with the concept of “network stability” enhancing disease suppression, as demonstrated in the common bean rhizosphere against soil-borne pathogens. Fungal networks were simplified under ES2-4, with reduced centrality of pathogenic taxa like Tulasnella and enhanced roles of antagonists such as Trichoderma, echoing observations in maize rotation systems51. The simplified fungal network with reduced centrality of potential pathogens (e.g., Tulasnella) and strengthened role of antagonists (e.g., Trichoderma) in the BE group mirrors the microbiome restructuring observed in suppressive soils, suggesting that ES2-4 treatment engineers a functional microbial assembly reminiscent of natural disease-suppressive environments.
In conclusion, B.velezensis ES2-4 confers resistance against soybean gray mold through a dual coordinated strategy: first, by reprogramming plant metabolism to enhance the secretion of antifungal root exudates, and second, by restructuring the rhizosphere microbiome to enrich beneficial taxa and functional pathways. This study establishes a new paradigm in PGPR research by linking induced metabolic changes to microbiome-mediated immunity, thereby providing a foundation for developing targeted rhizosphere engineering strategies against soil-borne diseases.
Materials and methods
Bacterial strain, and fungal inoculum
Bacillus velezensis ES2-4, originally isolated from soil and preserved at the Laboratory of Applied Botany, Sichuan Normal University, was maintained in Luria-Bertani (LB) broth supplemented with 25% glycerol and stored at − 80 °C. Prior to experimental use, the strain was reactivated by culturing in LB broth at 37 °C for 24 h. Botrytis cinerea was cultured on potato dextrose agar (PDA) plates at 28 °C for 7 days. Spore suspensions were prepared by flooding the culture surface with sterile distilled water, followed by stirring and filtration through four layers of sterile degreased gauze. Spore concentrations were determined using a hemocytometer and adjusted to the required density with sterile water before use in experiments.
Preparation of soybean root exudates
Soybean seeds were surface-sterilized with 0.1% (w/v) sodium hypochlorite for 5 min, rinsed 5–6 times with sterile distilled water, and placed on moist filter paper in Petri dishes. Germination was induced in darkness at 25 °C using a germination chamber. Uniformly germinated seedlings were transferred to culture bottles containing one-quarter-strength Hoagland nutrient solution for hydroponic cultivation, with roots shielded from light. After two weeks of cultivation, root irrigation treatments were initiated.
The experiment included four treatment groups: (1) B group (B. cinerea-infected soybean plants); (2) E group (B. velezensis ES2-4-treated soybean plants); (3) BE group (soybean plants pre-treated with B. velezensis ES2-4 followed by B. cinerea infection); and (4) CK group (control plants treated with sterile distilled water). In the BE and E groups, 20 mL of B. velezensis ES2-4 suspension (1 × 10⁷ CFU·mL⁻¹) was applied to the rhizosphere 20 days after transplanting. Five days later, the BE and B groups were inoculated with 20 mL of B. cinerea spore suspension (1 × 10⁷ CFU·mL⁻¹). Five days post-inoculation, plant roots were thoroughly rinsed with ultrapure water to remove residual metabolites of B. velezensis ES2-4 and B. cinerea, followed by hydroponic cultivation in ultrapure water for two days. The culture solution was then collected, filtered, and freeze-dried52. Each treatment included five biological replicates.
Root exudates metabolome assay
Sample pretreatment was performed following the method described by Chen et al.53, with minor modifications. Briefly, 3 mg of freeze-dried root exudate powder was dissolved in 1 mL of methanol, and 50 µL of L-2-chloro-phenylalanine (0.3 mg/mL) was added as an internal standard. The mixture was vortexed for 30 s and sonicated on ice for 15 min. The solution was then filtered through a 0.22 μm membrane and centrifuged at 16,000 r/min for 15 min at 4 °C. The supernatant was completely dried under nitrogen gas, derivatized, and homogenized prior to instrumental analysis. Gas chromatography-mass spectrometry (GC-MS) parameters were set according to reference54.
Mass spectrometric data were matched against the National Institute of Standards and Technology (NIST) Mass Spectral Library. Metabolites were identified by comparing their electron ionization (EI) mass spectra, including fragment patterns, with reference standards in the database. Final metabolite confirmation was achieved using the Kyoto Encyclopedia of Genes and Genomes (KEGG) and Human Metabolome Database (HMDB), with reliability further validated against published literature. Semi-quantitative metabolite analysis was performed using the internal standard method, followed by chromatographic peak area normalization, data transformation, and scaling. Normalized datasets were subjected to multivariate statistical analyses, including Principal Component Analysis (PCA), Orthogonal Partial Least Squares Discriminant Analysis (OPLS-DA), volcano plot analysis, and hierarchical clustering. Functional annotation and enrichment analysis of metabolites were conducted using KEGG pathway mapping. All statistical analyses were performed using MetaboAnalyst 5.0.
Effect of soybean rhizosphere exudates on the chemotactic response of B. velezensis ES2-4 and B. cinerea
The qualitative assessment of microbial inhibition was performed following the method described by Soo-Young et al.55, with modifications. Briefly, sterile filter paper discs (6 mm in diameter) were placed at the center of semi-solid agar plates containing gradient concentrations of differential metabolites. A 10 µL aliquot of bacterial suspension was applied to each disc, with three biological replicates per treatment. For fungal inhibition assays, Potato Dextrose Agar (PDA) medium supplemented with varying concentrations of differential metabolites was prepared and poured into 90 mm sterile Petri dishes. Mycelial plugs (6 mm in diameter) were excised from the periphery of 7-day-old B. cinerea colonies and transferred to the center of each PDA plate. Plates were incubated at 25 °C, and colony diameters were measured at 12-hour intervals using a digital caliper. Three biological replicates were included for each treatment.
Biocontrol agent and pathogen treatments and experimental design
Soybean seeds were initially germinated in seedling trays and then transferred into pots for further cultivation. After 20 days of growth under controlled conditions, the experimental treatments were initiated as follows: standard watering without inoculation (CK), inoculation with the biocontrol bacterium ES2-4 (E), application of ES2-4 followed by B. cinerea inoculation (BE), and inoculation with B. cinerea alone (B). In the E and BE groups, a 20 mL suspension of ES2-4 (1 × 10⁷ CFU·mL⁻¹) was applied to the rhizosphere of soybean seedlings 20 days after transplantation. Five days later, the BE and B groups received a 20 mL suspension of B. cinerea spores (1 × 10⁷ CFU·mL⁻¹). Rhizosphere soil samples were collected five days post-fungal inoculation for metagenomic sequencing47,55. Each experimental condition was replicated five times biologically.
DNA extraction, PCR amplification, and high-throughput sequencing
Genomic DNA was extracted from six soil samples using the TIANamp Soil DNA Kit (TIANGEN, Beijing, China) following the manufacturer’s protocol. The quality and concentration of the extracted DNA were evaluated via 1% agarose gel electrophoresis and NanoDrop® ND-2000 spectrophotometry (Thermo Scientific, Wilmington, NC, USA) before storage at −80 °C. To amplify bacterial 16 S rDNA and fungal ITS rDNA, the following specific primer pairs were used: 338 F (5’-ACTCCTACGGGAGGCAGCAG-3’) and 806R (5’-GGACTACHVGGGTWTCTAAT-3’) for 16 S rDNA, and ITS1-F (CTTGGTCATTTAGAGGAAGTAA) and ITS4-R (TCCTCCGCTTATTGATATGC) for the ITS rDNA gene. PCR amplification was performed using a T100 Thermal Cycler (Bio-Rad, California, USA) with 2× TransStart® Fast Pfu PCR SuperMix, according to the manufacturer’s instructions56. The thermal cycling conditions were as follows: initial denaturation at 95 °C for 3 min, followed by 27 cycles of 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 45 s, with a final extension at 72 °C for 10 min and a hold at 4 °C. Each sample was amplified in triplicate. PCR products were separated on a 2% agarose gel, purified using the TIANgel Purification Kit (TIANGEN, Beijing, China), and quantified using a Quantus™ Fluorometer (Promega, Beijing, China). The purified amplicons were sequenced on the Illumina NovaSeq PE250 platform by Majorbio Bio-Pharm Technology Co., Ltd. (Shanghai, China) following standard protocols.
Data processing and bioinformatics analysis
Raw FASTQ files were demultiplexed using a custom Perl script developed in-house. Quality filtering and merging of paired-end reads were performed using fastp v0.19.6 and FLASH v1.2.7, respectively, with the following criteria: (1) reads with an average quality score below 20 over a 50 bp sliding window were trimmed, and reads shorter than 50 bp or containing ambiguous bases were discarded; (2) overlapping sequences of at least 10 bp with a maximum mismatch ratio of 0.2 in the overlap region were merged; and (3) samples were demultiplexed based on barcode and primer sequences, allowing exact barcode matches and up to two nucleotide mismatches in the primers.
Operational taxonomic units (OTUs) were identified from the processed sequences using UPARSE v7.157,58, with a 97% sequence similarity threshold. The most abundant sequence within each OTU was selected as its representative. To standardize sequencing depth, 16 S rRNA gene sequences were rarefied to 44,980 reads, while ITS rDNA sequences were rarefied to 84,995 reads, yielding an average Good’s coverage of 99.09%.
Taxonomic classification of OTU representative sequences was conducted using the RDP Classifier v2.2, referencing the SILVA database (v138.1) for bacterial identification and the UNITE database (v138.1) for fungal classification, with a confidence threshold of 0.7. The functional potential of the metagenome was inferred using PICRUSt2 (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States)59, utilizing its integrated pipeline, which includes HMMER for sequence alignment, EPA-NG and Gappa for phylogenetic placement, castor for 16 S gene copy normalization, and MinPath for gene family and pathway predictions, all following the standard PICRUSt2 protocol.
Bioinformatic analyses of the soil microbiota were performed on the Majorbio Cloud platform (https://cloud.majorbio.com). Based on OTU data, rarefaction curves and alpha diversity indices—including observed OTUs, Chao1 richness, Shannon index, and Good’s coverage—were calculated using Mothur v1.30.160. Principal coordinate analysis (PCoA) based on Bray-Curtis dissimilarity was performed using the Vegan v2.5-3 package to assess microbial community similarities among samples. Significant differences between treatments were determined using the Welch t-test in STAMP, while variations in OTU relative abundance across treatments were analyzed using likelihood ratio tests in the “EdgeR” package. A Manhattan plot was generated for visualization using the “ggplot2” package.
Co-occurrence network analysis
Co-occurrence networks were constructed to examine shifts in microbial community interactions for both bacterial and fungal populations. These networks were generated in R (v4.3.1) using Spearman correlation coefficients, incorporating only significant correlations (p < 0.05) with an absolute correlation coefficient (|R|) greater than 0.661. The resulting networks were visualized using the Fruchterman-Reingold layout in Gephi.
Statistical analysis
All quantitative data are presented as the mean ± standard error (SE). Statistical analyses were performed using GraphPad Prism 9.5.0. Analysis of variance (ANOVA) was conducted to assess significant differences, followed by Duncan’s multiple range test for mean separation. A p-value of < 0.05 was considered statistically significant.
Data availability
Sequence data that support the findings of this study have been deposited in the NCBI with the primary accession code PRJNA1303063.
References
-
Savary, S. et al. The global burden of pathogens and pests on major food crops. Nat. Ecol. Evol. 3, 430–439. https://doi.org/10.1038/s41559-018-0793-y (2019).
-
Zhao, Y. et al. Comparison of nutritional diversity in five fresh legumes using flavonoids metabolomics and postharvest Botrytis cinerea defense analysis of peas mediated by Sakuranetin. J. Agric. Food Chem. 72, 6053–6063. https://doi.org/10.1021/acs.jafc.3c08968 (2024).
-
Zhao, Y. et al. Root exudates modulate rhizosphere microbial communities during the interaction of Pseudomonas chlororaphis, β-Aminobutyric Acid, and Botrytis cinerea in tomato plants. J. Plant Growth Regul. 43, 701–714. https://doi.org/10.1007/s00344-023-11128-3 (2024).
-
Hua, L. et al. Pathogenic mechanisms and control strategies of Botrytis cinerea causing post-harvest decay in fruits and vegetables. Food Qual. Saf. 2, 111–119. https://doi.org/10.1093/fqsafe/fyy016 (2018).
-
Shao, W., Zhao, Y. & Ma, Z. Advances in Understanding fungicide resistance in Botrytis cinerea in China. Phytopathology 111, 455–463. https://doi.org/10.1094/phyto-07-20-0313-ia (2021).
-
Nadeem, S. M., Ahmad, M., Zahir, Z. A., Javaid, A. & Ashraf, M. The role of mycorrhizae and plant growth promoting rhizobacteria (PGPR) in improving crop productivity under stressful environments. Biotechnol. Adv. 32, 429–448. https://doi.org/10.1016/j.biotechadv.2013.12.005 (2014).
-
Kumari, R., Pandey, E., Bushra, S., Faizan, S. & Pandey, S. Plant growth promoting rhizobacteria (PGPR) induced protection: A plant immunity perspective. Physiol. Plant. 176, e14495. https://doi.org/10.1111/ppl.14495 (2024).
-
Ruiz-García, C., Béjar, V., Martínez-Checa, F., Llamas, I. & Quesada, E. Bacillus velezensis sp. nov., a surfactant-producing bacterium isolated from the river Vélez in Málaga, Southern Spain. Int. J. Syst. Evol. Microbiol. 55, 191–195. https://doi.org/10.1099/ijs.0.63310-0 (2005).
-
Rabbee, M. F. et al. Bacillus velezensis: A valuable member of bioactive molecules within plant microbiomes. Molecules https://doi.org/10.3390/molecules24061046 (2019).
-
Ye, M. et al. Characteristics and application of a novel species of Bacillus: Bacillus velezensis. ACS Chem. Biol. 13, 500–505. https://doi.org/10.1021/acschembio.7b00874 (2018).
-
Ongena, M. & Jacques Bacillus lipopeptides: versatile weapons for plant disease biocontrol. Trends Microbiol. 16, 115–125. https://doi.org/10.1016/j.tim.2007.12.009 (2008).
-
Vignesh, M., Shankar, S. R. M., MubarakAli, D. & Hari, B. N. V. A novel rhizospheric bacterium: Bacillus velezensis NKMV-3 as a biocontrol agent against Alternaria leaf blight in tomato. Appl. Biochem. Biotechnol. 194, 1–17. https://doi.org/10.1007/s12010-021-03684-9 (2022).
-
Yang, X., Zhang, F., Wang, J., Tian, C. & Meng, X. Characterization of Bacillus velezensis YTQ3 as a potential biocontrol agent against Botrytis cinerea. Postharvest Biol. Technol. 223, 113443. https://doi.org/10.1016/j.postharvbio.2025.113443 (2025).
-
Aboelez, E. M. et al. Biocontrol efficacy of Botrytis cinerea on postharvest tomato fruit by the endophytic bacterium Bacillus velezensis BE1. Physiol. Mol. Plant Pathol. 134, 102427. https://doi.org/10.1016/j.pmpp.2024.102427 (2024). https://doi.org:.
-
Sun, L. et al. Bacillus velezensis BVE7 as a promising agent for biocontrol of soybean root rot caused by Fusarium oxysporum. Front. Microbiol. https://doi.org/10.3389/fmicb.2023.1275986 (2023).
-
Han, X. et al. The Plant-Beneficial rhizobacterium Bacillus velezensis FZB42 controls the soybean pathogen phytophthora Sojae due to Bacilysin production. Appl. Environ. Microbiol. 87, e0160121. https://doi.org/10.1128/aem.01601-21 (2021).
-
Nurcahyanti, S. D., Wahyuni, W. S., Masnilah, R. & Nurdika, A. A. H. Phenol content and peroxidase enzyme activity in soybean infected with Xanthomonas axonopodis pv Glycines with the application of Bacillus subtilis JB12 and Bacillus velezensis ST32. Baghdad Sci. J. https://doi.org/10.21123/bsj.2023.7406 (2023).
-
Gu, Y. et al. The biocontrol agent Bacillus velezensis T-5 changes the soil bacterial community composition by affecting the tomato root exudate profile. Plant. Soil. 490, 669–680. https://doi.org/10.1007/s11104-023-06114-3 (2023).
-
Wang, Y. et al. Analysis of Ginkgo biloba Root Exudates and Inhibition of Soil Fungi by Flavonoids and Terpene Lactones. Plants (Basel) https://doi.org/10.3390/plants13152122 (2024).
-
Jin, Y. et al. Role of maize root exudates in promotion of colonization of Bacillus velezensis strain S3-1 in rhizosphere soil and root tissue. Curr. Microbiol. 76, 855–862. https://doi.org/10.1007/s00284-019-01699-4 (2019).
-
Chen, Q. et al. Chitooligosaccharide enhances plant resistance to P. nicotianae via sugar homeostasis and microorganism assembly. Int. J. Biol. Macromol. 307, 142127. https://doi.org/10.1016/j.ijbiomac.2025.142127 (2025).
-
Sharma, M., Saleh, D., Charron, J. B. & Jabaji, S. A. Crosstalk between Brachypodium root exudates, organic acids, and Bacillus velezensis B26, a growth promoting bacterium. Front. Microbiol. 11, 575578. https://doi.org/10.3389/fmicb.2020.575578 (2020).
-
Wang, B. et al. Secretion and volatile components contribute to the antagonism of Bacillus velezensis 1–10 against fungal pathogens. Biol. Control. 187, 105379. https://doi.org/10.1016/j.biocontrol.2023.105379 (2023). https://doi.org:.
-
Al-Ali, A. et al. Biofilm formation is determinant in tomato rhizosphere colonization by Bacillus velezensis FZB42. Environ. Sci. Pollut Res. Int. 25, 29910–29920. https://doi.org/10.1007/s11356-017-0469-1 (2018).
-
Kanehisa, M. Toward Understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
-
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53, D672–d677. https://doi.org/10.1093/nar/gkae909 (2025).
-
Mashabela, M. D., Piater, L. A., Dubery, I. A., Tugizimana, F. & Mhlongo, M. I. Rhizosphere tripartite interactions and PGPR-mediated metabolic reprogramming towards ISR and plant priming: A metabolomics review. Biology (Basel) https://doi.org/10.3390/biology11030346 (2022).
-
Bai, Y. et al. Using multi-omics to explore the effect of Bacillus velezensis SAAS-63 on resisting nutrient stress in lettuce. Appl. Microbiol. Biotechnol. 108, 313. https://doi.org/10.1007/s00253-024-13153-y (2024).
-
Xie, L. et al. Bacillus velezensis YC89-mediated recruitment of rhizosphere bacteria improves resistance against sugarcane red rot. Chem. Biol. Technol. Agric. 11, 114. https://doi.org/10.1186/s40538-024-00627-4 (2024).
-
Ahsan, T., Chen, J., Zhao, X., Irfan, M. & Wu, Y. Extraction and identification of bioactive compounds (eicosane and dibutyl phthalate) produced by Streptomyces strain KX852460 for the biological control of rhizoctonia Solani AG-3 strain KX852461 to control target spot disease in tobacco leaf. AMB Express. 7, 54. https://doi.org/10.1186/s13568-017-0351-z (2017).
-
Rasulov, B. A. & Pattaeva, M. A. Abiotic/Biotic stress and substrate dictated metabolic diversity of Azotobacter Chroococcum: synthesis of Alginate, antifungal n-Alkanes, Lactones, and Indoles. Indian J. Microbiol. 64, 635–649. https://doi.org/10.1007/s12088-024-01212-x (2024).
-
El-Gendi, H. et al. Foliar applications of Bacillus subtilis HA1 culture filtrate enhance tomato growth and induce systemic resistance against tobacco mosaic virus infection. Horticulturae 8, 301 (2022).
-
He, M. & Ding, N. Z. Plant unsaturated fatty acids: multiple roles in stress response. Front. Plant. Sci. 11, 562785. https://doi.org/10.3389/fpls.2020.562785 (2020).
-
Rezaei-Chiyaneh, E. et al. Intercropping fennel (Foeniculum vulgare L.) with common bean (Phaseolus vulgaris L.) as affected by PGPR inoculation: A strategy for improving yield, essential oil and fatty acid composition. Sci. Hort. 261, 108951. https://doi.org/10.1016/j.scienta.2019.108951 (2020).
-
Shakeri, E., Mohammad, M. S. S. A., Majid, A. D., Ali, T. S., Moradi-Ghahderijani, M. & and Improvement of yield, yield components and oil quality in Sesame (Sesamum indicum L.) by N-fixing bacteria fertilizers and Urea. Arch. Agron. Soil. Sci. 62, 547–560. https://doi.org/10.1080/03650340.2015.1064901 (2016).
-
Li, Y. et al. Isoleucine enhances plant resistance against Botrytis cinerea via jasmonate signaling pathway. Front. Plant. Sci. 12, 628328. https://doi.org/10.3389/fpls.2021.628328 (2021).
-
Pretali, L., Bernardo, L., Butterfield, T. S., Trevisan, M. & Lucini, L. Botanical and biological pesticides elicit a similar induced systemic response in tomato (Solanum lycopersicum) secondary metabolism. Phytochemistry 130, 56–63. https://doi.org/10.1016/j.phytochem.2016.04.002 (2016).
-
Abdelrahman, M. et al. Dissection of trichoderma longibrachiatum-induced defense in onion (Allium Cepa L.) against Fusarium oxysporum f. sp. Cepa by target metabolite profiling. Plant. Sci. 246, 128–138. https://doi.org/10.1016/j.plantsci.2016.02.008 (2016).
-
Ankati, S., Rani, T. S. & Podile, A. R. Changes in root exudates and root proteins in Groundnut–Pseudomonas sp. Interaction contribute to root colonization by bacteria and defense response of the host. J. Plant Growth Regul. 38, 523–538. https://doi.org/10.1007/s00344-018-9868-x (2019).
-
Liu, Y. et al. Identification of Root-Secreted compounds involved in the communication between Cucumber, the beneficial Bacillus amyloliquefaciens, and the Soil-Borne pathogen Fusarium oxysporum. Mol. Plant. Microbe Interact. 30, 53–62. https://doi.org/10.1094/mpmi-07-16-0131-r (2017).
-
Jiang, B., Long, C., Xu, Y. & Han, L. Molecular mechanism of Tsukamurella tyrosinosolvens strain P9 in response to root exudates of peanut. Arch. Microbiol. 205, 48. https://doi.org/10.1007/s00203-022-03387-7 (2023).
-
Yuan, J. et al. Organic acids from root exudates of banana help root colonization of PGPR strain Bacillus amyloliquefaciens NJN-6. Sci. Rep. 5, 13438. https://doi.org/10.1038/srep13438 (2015).
-
Yu, Y. Y. et al. Bacillus-Secreted oxalic acid induces tomato resistance against Gray mold disease caused by Botrytis cinerea by activating the JA/ET pathway. Mol. Plant. Microbe Interact. 35, 659–671. https://doi.org/10.1094/mpmi-11-21-0289-r (2022).
-
Rampelotto, P. H., de Siqueira Ferreira, A., Barboza, A. D. M. & Roesch, L. F. W. Changes in Diversity, Abundance, and structure of soil bacterial communities in Brazilian savanna under different land use systems. Microb. Ecol. 66, 593–607. https://doi.org/10.1007/s00248-013-0235-y (2013).
-
Campos, S. B. et al. Soil suppressiveness and its relations with the microbial community in a Brazilian subtropical agroecosystem under different management systems. Soil Biol. Biochem. 96, 191–197. https://doi.org/10.1016/j.soilbio.2016.02.010 (2016).
-
You, C., Zhang, C., Kong, F., Feng, C. & Wang, J. Comparison of the effects of biocontrol agent Bacillus subtilis and fungicide metalaxyl–mancozeb on bacterial communities in tobacco rhizospheric soil. Ecol. Eng. 91, 119–125. https://doi.org/10.1016/j.ecoleng.2016.02.011 (2016).
-
Sun, X. et al. Bacillus velezensis stimulates resident rhizosphere Pseudomonas stutzeri for plant health through metabolic interactions. ISME J. 16, 774–787. https://doi.org/10.1038/s41396-021-01125-3 (2022).
-
Mikkelsen, H., Sivaneson, M. & Filloux, A. Key two-component regulatory systems that control biofilm formation in Pseudomonas aeruginosa. Environ. Microbiol. 13, 1666–1681. https://doi.org/10.1111/j.1462-2920.2011.02495.x (2011).
-
Pham, J., Liu, J., Bennett, M. H., Mansfield, J. W. & Desikan, R. Arabidopsis histidine kinase 5 regulates salt sensitivity and resistance against bacterial and fungal infection. New Phytol. 194, 168–180. https://doi.org/10.1111/j.1469-8137.2011.04033.x (2012).
-
Mendes, L. W., Mendes, R., Raaijmakers, J. M. & Tsai, S. M. Breeding for soil-borne pathogen resistance impacts active rhizosphere Microbiome of common bean. ISME J. 12, 3038–3042. https://doi.org/10.1038/s41396-018-0234-6 (2018).
-
Niu, J. et al. The succession pattern of soil microbial communities and its relationship with tobacco bacterial wilt. BMC Microbiol. 16, 233. https://doi.org/10.1186/s12866-016-0845-x (2016).
-
Dutta, S., Rani, T. S. & Podile, A. R. Root exudate-induced alterations in Bacillus cereus cell wall contribute to root colonization and plant growth promotion. PLoS One. 8, e78369. https://doi.org/10.1371/journal.pone.0078369 (2013).
-
Tian, L. et al. Foliar application of SiO2 nanoparticles alters soil metabolite profiles and microbial community composition in the Pakchoi (Brassica chinensis L.) rhizosphere grown in contaminated mine soil. Environ. Sci. Technol. 54, 13137–13146. https://doi.org/10.1021/acs.est.0c03767 (2020).
-
Liu, W. et al. Enantioselective effects of Imazethapyr on Arabidopsis Thaliana root exudates and rhizosphere microbes. Sci. Total Environ. 716, 137121. https://doi.org/10.1016/j.scitotenv.2020.137121 (2020).
-
Nam, M. H., Park, M. S., Kim, H. G. & Yoo, S. J. Biological control of strawberry fusarium wilt caused by fusarium oxysporum f. sp. fragariae using Bacillus velezensis BS87 and RK1 formulation. J. Microbiol. Biotechnol. 19, 520–524. https://doi.org/10.4014/jmb.0805.333 (2009).
-
Xie, C. et al. Bacillus velezensis TCS001 Enhances the Resistance of Hickory to Phytophthora cinnamomi and Reshapes the Rhizosphere Microbial Community. Agriculture 15, 86 (2025).
-
Edgar, R. C. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods. 10, 996–998. https://doi.org/10.1038/nmeth.2604 (2013).
-
STACKEBRANDT, E., GOEBEL, B. M. & Taxonomic Note A place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int. J. Syst. Evol. MicroBiol. 44, 846–849. https://doi.org/10.1099/00207713-44-4-846 (1994).
-
Douglas, G. M. et al. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 38, 685–688. https://doi.org/10.1038/s41587-020-0548-6 (2020).
-
Schloss, P. D. et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. https://doi.org/10.1128/aem.01541-09 (2009).
-
Barberán, A., Bates, S. T., Casamayor, E. O. & Fierer, N. Using network analysis to explore co-occurrence patterns in soil microbial communities. Isme j. 6, 343–351. https://doi.org/10.1038/ismej.2011.119 (2012).
Acknowledgements
Not applicable.
Funding
Project supported by the National Natural Science Foundation of China (NO. 32100240).
Ethics declarations
Competing interests
The authors declare no competing interests.
Consent for publication
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, R., Guo, X., Wu, M. et al. Bacillus velezensis ES2-4 modulates root exudation and microbiome remodeling to enhance soybean resistance against gray mold. Sci Rep 15, 37098 (2025). https://doi.org/10.1038/s41598-025-21135-x
-
Received:
-
Accepted:
-
Published:
-
DOI: https://doi.org/10.1038/s41598-025-21135-x