
Introduction
Immunotherapy represents one of the most promising therapeutic approaches in oncology, offering significant potential to improve cancer treatment outcomes. This strategy leverages immune system to specifically target cancer cells and gained considerable attention in recent years. Among the emerging immunotherapeutic approaches, antibody-drug conjugates (ADCs) have revolutionized cancer therapy by combining the precision of monoclonal antibodies with the potency of cytotoxic drugs1. ADCs constitute an emerging class of combination therapies designed to selectively target tumor cells by conjugating a monoclonal antibody with a therapeutic payload, such as a cytotoxic agent, immune-stimulatory cytokines, or an antimitotic compound. ADCs enable the selective delivery of cytotoxic agents directly to malignant cells, minimizing off-target toxicity and improving the therapeutic index2,3. By the end of 2021, fourteen ADCs had received U.S. Food and Drug Administration (FDA) approval, and more than 100 ADCs were in various stages of clinical development worldwide4,5,6. While ADCs have shown remarkable efficacy in treating various cancers, in recent years the range of payloads using antibody-mediated delivery to human cells has expanded beyond traditional cytotoxic agents, including steroids7, oligonucleotides8, bifunctional degraders9, epigenetic modulators10, and other molecules11.
The selection of an appropriate target antigen12 is the most crucial determinant of an ADC efficacy, selectivity and tolerability. Ideally, the target antigen should be highly expressed on the surface of cancer cells, ensuring optimal accessibility for ADC binding. Additionally, the internalization rate of the antigen-ADC complex plays a pivotal role, as effective internalization is required for intracellular drug release13. Epidermal growth factor receptor (EGFR) has emerged as a key target in ADC development due to its aberrant overexpression in multiple malignancies, including colorectal carcinoma (CRC), non-small cell lung cancer (NSCLC), and head and neck squamous cell carcinoma (HNSCC)14,15,16. Cetuximab (Cet), an FDA-approved anti-EGFR monoclonal antibody, is commonly used as a first-line therapy in combination with chemotherapy for metastatic CRC17. While Cet primarily functions by inhibiting EGFR signaling and suppressing tumor cell proliferation, it can also exert antitumor activity through antibody-dependent cell-mediated cytotoxicity (ADCC) by engaging Fc receptors on natural killer (NK) cells, γδ T lymphocytes, monocytes, and granulocytes18.
In this context, there has been a growing interest in developing innovative ADCs that can combine tumor cell targeting with immune system stimulation to elicit a localized and specific antitumor response19. This dual action can help achieve more effective and durable treatment responses. One promising approach involves the use of aminobisphosphonates (N-BPs), specifically zoledronic acid (ZA, Fig. 1), which exhibit antitumor effects, being able to selectively enhance the proliferation and antitumor activity of a specific subset of human γδ T cells, known as Vγ9Vδ2 T cells. ZA exerts its anticancer effects by entering monocytes, macrophages, endothelial cells, and tumor cells, where it inhibits farnesyl pyrophosphate synthase (FPPS), a crucial enzyme in the mevalonate pathway responsible for cholesterol biosynthesis. This inhibition leads to the accumulation of phosphate antigens (pAg), such as isopentenyl pyrophosphate (IPP), which in turn activates Vδ2 T cells20,21. Despite its clinical success in bone-related conditions22, ZA has several limitations, including poor bioavailability, limited cellular uptake, and tendency to accumulate in bone tissue, which hinders its effectiveness against non-bone tumors23,24.
In our previous studies we developed an ADC that covalently conjugates Cet with ZA (Cet-ZA), with a defined drug-antibody ratio (DAR) of 4.3. This conjugate retained significant reactivity and effectively modulated signaling pathways involved in CRC25. In addition to its direct antitumor effects, Cet-ZA expanded and activated Vδ2 T cells, a subset of gamma-delta T cells known for their ability to recognize and eliminate tumor cells. This dual mechanism, direct targeting of tumor cells and immune activation, offers a novel approach for enhancing anticancer immunity26. This effect was also observed when tumor-associated fibroblasts (TAFs) derived from CRC mucosa were treated with Cet-ZA27. These findings indicate that Cet-conjugation may serve as an effective strategy for directing N-BPs toward tumor cells, thereby reducing their preferential accumulation in bone tissue. Moreover, the targeted delivery of ZA to the tumor site via Cetuximab may facilitate the local recruitment of γδ T cells, which can then be activated within the tumor microenvironment (TME) to exert antitumor immune responses.
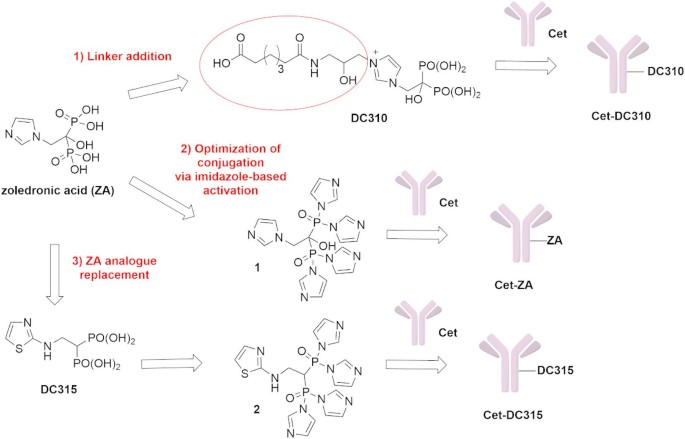
In this study, our aim was to explore the antitumor activity of Cet conjugated with novel N-BPs using two different strategies (Fig. 1). At first, we investigated alternative conjugation strategies involving a linker addition to ZA to generate compound DC310 or a minor modification to the imidazole-based activation of ZA. Our main objective was to improve DAR, to optimize drug release kinetics, and to enhance targeting specificity. Furthermore, a ZA structural modification was assessed by replacing ZA with its aminothiazole derivative, DC315 (Fig. 1). This alternative molecule was designed to improve affinity for tumor cells, reduce accumulation in bone tissue, and optimize pharmacokinetic properties. Indeed, Matsumoto et al. reported28 the development of ZA analogues, with enhanced antitumor activities by introducing minor structural changes, such as replacing the imidazole group with other nitrogen-containing heterocycles. These analogues, particularly the aminothiazole derivative DC315 and its pivoxil prodrug29, demonstrated superior inhibition of tumor cell growth compared to ZA in a series of leukemia/lymphoma and carcinoma cell lines. Indeed, both hematopoietic and solid tumor cell proliferation was inhibited with an approximately 5000-fold less amount using these novel bisphosphonates instead of ZA28. We have recently demonstrated that Cet can be linked to other classical N-BPs such as ibandronate (IBA) or risedronate (RIS) and reacted similarly to native Cet with some CRC cell lines26. More importantly, Cet-ZA, Cet-RIS or Cet-IBA, can inhibit the growth of WT EGFR+ Caco-2 CRC cells better than the native Cet. To achieve this functional effect, the amount of ZA linked to Cet was about 100-fold less than the dose required to inhibit Caco-2 cell growth with soluble ZA. In addition, Cet-ZA and Cet-RIS ADC increased the cytotoxicity exerted by Vδ2 T cells against some CRC cell lines either with a WT EGFR-pathway or KRAS-mutated compared to that triggered with the native Cet26. These findings may suggest that carrying the ZA inside the tumor cells through the engagement of EGFR with an antibody can increase the antitumor effect of N-BPs. Based on all these reports, DC315 incorporation into ADCs may offer additional advantages compared to Cet conjugated with classical N-BPs in targeting CRC cells inhibiting cell growth directly and activating antitumor cytotoxicity of Vδ2 T cells. The new ADCs, Cet-DC310 and Cet-DC315, were evaluated through various assays to assess their reactivity and effects on the growth of some CRC cell lines and their ability to stimulate Vδ2 T cell-mediated antitumor activity. These experiments were conducted in conventional monolayer and 3D culture models to compare the efficacy of these ADCs and to assess their potential application in CRC treatment.
To our knowledge, this is the first study that translates the activity of the novel N-BP DC315 into an antibody–drug conjugate, demonstrating the antitumor efficacy of Cet-DC315 in primary CRC cells and patient-derived organoids, together with its ability to trigger cytotoxic γδ T cells.

Chemical structure of zoledronic acid and its aminothiazole analogue DC315 and novel strategies for Cet conjugation with N-BPs.
Materials and methods
Chemical synthesis
For synthesis of ADCs Cet-DC310, Cet-ZA and Cet-DC315 see Supplementary Information.
Bioanalytical characterization of ADCs by MALDI-MS
To determine DAR, the Matrix Assisted Laser Desorption Ionization Mass Spectrometry (MALDI-MS) was performed as described previously25. Briefly, native antibody Cet and the proper ADC were desalted with the Amicon® Ultra 0.5mL 10KDa MWCO (Merck Millipore). Then, 2µL of desalted Cet or ADC were mixed with 2µL of a saturated solution of s-DHB MALDI matrix (Bruker Daltonics, GmbH) in 0.1% TFA in acetonitrile: water (50:50, v/v). The mixture was then spotted on the MALDI plate and left to dry in the air. Subsequently, 1µL of the saturated matrix solution was added to each spot and left to dry again. The UltrafleXtreme MALDI mass spectrometer (Bruker Daltonics, GmbH) operating in linear mode, over the mass range m/z 30–220 kDa was used for the acquisition of mass spectra.
Determination of the dissociation constant (Kd) by flow cytometry
The binding affinity of antibodies to their target was assessed by flow cytometry as previously described30. Briefly, RKO, HCT-116 and Caco-2 CRC cell lines cells were incubated for 30 min at 4 °C with increasing concentrations of antibody (Cetuximab and Cet-DC315) ranging from 0.002 to 200 µg/mL, followed by the anti-human Alexa Fluor 647 antiserum (Invitrogen, Thermo Fisher Scientific, Waltman, USA) at 4 °C. Negative controls were stained with anti-human Alexa Fluor 647 antiserum alone. After washing with PBS supplemented with 1% Fetal Bovine Serum (FBS, Gibco™ One Shot™ Fetal Bovine Serum Gibco, Life Technologies, Monza, Italy) at 4 °C, the cells were analyzed using Cytoflex S (Beckman Coulter). The mean fluorescence intensity (MFI), corrected for background fluorescence, was used as a measure of bound antibody. The dissociation constant (Kd) was calculated by fitting the experimental binding data using nonlinear regression (competitive binding, one site-specific binding model) in GraphPad Prism (GraphPad Software, San Diego, CA, USA).
The binding curves were fitted to the following equation:
$${text{Y = Bmax }} times [{text{L}}]/({text{Kd}} + [{text{L}}])$$
where Y is the specific binding (MFI), Bmax is the maximal binding, and [L] is the concentration of the antibody.
Cell lines and cell cultures
The Caco-2, HCT-116, LS-180, RKO, and SW-620 CRC human cell lines were obtained from Policlinico San Martino hospital unit of Cell Bank and authenticated by STR analysis. These cell lines were cultured in adherent flask (Sarstedt, Nümbrecht, Germany) in RPMI-1640 medium supplemented with 1% penicillin/streptomycin and l-glutamine and 10% FBS in a humidified incubator at 37 °C with 5% CO2. At the 50–70% of confluence, the CRC cells were detached from the plastic substrate with Trypsin-EDTA 0.5% (Gibco Life Technologies). After extensive washes the cells were used in functional experiments. Cell spheroids were generated in 3D by culturing CRC tumor cells in an AggreWell™ 400 (StemCell™ Vancouver, Canada) 24w plate for 48 h with DMEM-F12 (Gibco) in serum-free medium (SFM), supplemented with EGF (Peprotech Europe, London UK) at 10 ng/ml final concentration (≥ 1 × 106 IU/mg). The culture spheroid cell cultures were checked over time by an IX51 inverted microscope (Olympus, Germany) and used for functional experiments after two days for additional 5 days. Cell spheroids size was monitored by time-lapse imaging by a CellCyte X™ scanner (Cytena GmbH, Germany). Cells vitality was estimated by the uptake of the cell death probe C. Live Tox green (10 nM final concentration, Cytena GmbHinvitrogen). Established CRC patient-derived organoids (PDO) were 3D cultured in 5ul Geltrex domes (Gibco, cat.A14133-02) using DMEM-F12 with B27 (w/o Vit-A, Gibco, cat.12587-010), 25ng/ml animal-free huEGF (Peprotech, cat.AF100-15), 0.5μM A83-01 (SelleckChem) and 0.15μM LDN-193189 (SelleckChem), as previously described25.
Flow cytometry and confocal microscopy
The CRC cell lines were incubated with serial dilutions (200–0.002 µg/mL/106 cells) of Cet or Cet-DC315 or Cet-DC310 for 1 h at 4 °C, followed by the anti-human Alexa Fluor 647 antiserum (Invitrogen). Negative controls were stained with anti-human Alexa Fluor 647 antiserum alone. PDO were cultured for 24 h in medium without EGF, extracted from the extracellular matrix, trypsinized and stained with Cet or Cet-DC315 (2 µg/mL) followed by the anti-human Alexa Fluor 647 antiserum, or with an anti-human CD326 (EpCAM) primary antibody (BD Biosciences, San Jose, CA, USA), followed by a PE-conjugated anti-mouse secondary antibody (Invitrogen, Thermo Fisher Scientific) to confirm the purity of epithelial cells. Samples were analyzed by Cytoflex S (Beckman Coulter) and results were expressed as log of mean fluorescence intensity or percentage of positive cells. For confocal microscopy, PDO were extracted from their matrix and cultured in suspension to speed-up Ab diffusion. Floating PDO were incubated with Cet-DC315 (2 µg/mL) for 1 h. Afterwards, they were fixed with 4% paraformaldehyde (Sigma Aldrich, Merck, Darmstadt, Germany), and permeabilized using 0.1% Triton X-100 (Sigma Aldrich, Merck). Fluorescent labeling was performed using an anti-human Alexa Fluor 488-conjugated antiserum. Finally, nuclei were counterstained with DAPI (Invitrogen, Thermo Fisher) and the sample immediately observed by a Fluoview FV4000 Confocal Laser Scanning Microscope (Evident Scientific, Waltham, MA, USA), capturing 10, 1 μm thick, Z-stacks by a 20x optic. 2D color deconvolution was applied to each stack, to reduce the noise created by the thickness of 3D PDO.
Analysis of CRC cell line proliferation
Caco-2 or HCT-116 CRC cell lines were cultured in adherent 96-well flat-bottomed plates (Sarstedt, Nümbrecht, Germany) in complete medium for different period of time. Cet or Cet-DC310 or Cet-DC315 (all of them at the concentration of 2 µg/mL) was added at the onset of the culture and the cell growth was followed by recording the cultures with a CellCyte X™ scanner for 120 h. Images were taken every 12 h and the percentage of the confluence of the culture were determined with the CellStudio software integrated in the scanner. The control cultures (CTR) were represented by tumor cells cultured in medium without the addition of antibody. Some experiments were performed on spheroids generated after culture of 2.5 × 105 cells/well in an AggreWell™ 400 24-well plate for 48 h, in complete medium. Afterwards, Cet or Cet-DC310 or Cet-DC315 (2 µg/mL) were gently added to the cell cultures which were further incubated for 120 h. Spheroid size was analyzed on images taken with the CellCyte X™ scanner and quantified using the CellStudio software. This culture method generated a very large number of individual spheroids (n = 1200) of a similar size in a short period.
In vitro expansion of Vδ2 T lymphocytes
Blood of healthy donors was obtained after signed informed consent from the blood transfusion centre of Ospedale Policlinico San Martino, Genoa, at the moment of donation while the specimens from CRC patients were obtained after signed informed consent according to the Ligurian Regional Ethic Committee approval, PR163REG2014, renewed in 2017. All methods were performed in accordance with the relevant guidelines and regulations. Peripheral blood mononuclear cells (PBMC) were obtained as described31 from buffy coat of healthy adult donor density gradient centrifugation using lymphocyte-H separation medium (Cedarlane, Burlington, Canada). Highly purified T cells were isolated from PBMC cell suspensions using the RosetteSep HLA T cells enrichment cocktail (Stem Cell Technologies) as previously described25. This cell population was composed of > 95–98% T cells, the remaining 2–5% of leukocytes consisted of B cells, and no CD14+ monocytes were detected, as proved by specific immunofluorescence with anti-CD14 mAb25. The evaluation of Vδ2 T cell expansion by the Cet-DC315 ADC in co-cultures of the irradiated CRC cell line LS-180 and T cells was performed as shown25. Indeed, the LS-180 CRC cell line can trigger the preferential growth of Vδ2 T cells when incubated with soluble ZA. Briefly, purified T cells (105 cells) were co-cultured with irradiated (5000rads) LS-180 CRC cells (104 cells) in 96-well U-bottom plates (Sarstedt) in 200 µL of complete RPMI-1640 medium, with or without Cet-DC315 or Cet (2 µg/mL). After 24 h, 100 µL of medium was discarded and 100 µL of human and animal-free recombinant interleukin 2 solution (IL2, Peprotech, Thermo Fisher Scientific) was added to each well (30 IU/ml final concentration). Cell growth was monitored by microscopy examination and cell cultures were split 1 in 2 with medium plus IL2 when necessary. The Vδ2 expression was analyzed at the indicated time points (usually at day 14 and 21) by indirect immunofluorescence assay using the anti-Vδ2 antibody, clone γδ123R3, IgG1 isotype (home-made supernatant) and the anti-CD3 antibody JT3A (home-made supernatant of 289/11/F10 subclone, IgG2a isotype), followed by isotype specific AF647 anti-IgG1 or PE anti-IgG2a antiserum (Thermo Fisher Scientific). Cells stained with the second reagent alone represented the negative control. Samples were run on a Cytoflex S flow cytometer and results analyzed by the CytExpert 2.4 software.
In other experiments co-cultures of PDO (OMCR16-005TK or OMCR19-010TK) and purified T cells isolated from PBMC of healthy donors were performed as described25. Briefly, PDO were harvested from the culture domes and put into 96-U bottomed microplates after washing to eliminate geltrex matrix. Approximately, 104 organoid cells were co-cultured with 105 purified T cells in RPMI complete medium with Cet or Cet-DC315 (2 µg/mL) or in medium alone as control cultures. IL2 was added after 24 h of incubation and after 3–5 days the culture medium was changed until proliferation was detected by microscopy examination. The cell cultures were split when necessary 1 in 2 and the percentage of Vδ2 T cell was determined by indirect immunofluorescence at day 21. Finally, in additional experiments, cellular suspensions derived from CRC patients (OMCR12-040, OMCR12-041, OMCR12-052, OMCR12-065, OMCR18-009) after digestion of tumor specimens were used25. After thawing, the CRC cell suspensions were analyzed for the presence of Vδ2 T cells. Then, 105 cells were cultured with or without Cet-DC315 ADC or Cet (2 µg/mL) as above and IL2 added after 24 h. Cell growth was microscopically analyzed and cell cultures were split with IL2 when necessary. The percentage of Vδ2 T cells was evaluated by immunofluorescence at day 25. In all experiments, the percentage of Vδ2 T cells was compared to the initial input of Vδ2 T cells and to the percentage of Vδ2 T cells obtained at the same day in cultures incubated with native Cet.
In vitro cytotoxicity
To evaluate the in vitro cytotoxicity of Cet-DC315, we performed experiments on CRC cell lines and PDO. Briefly, Vδ2 T lymphocytes (purity: 90–95%) obtained after stimulation with ZA (1 µM) and expansion with IL2 (30IU/ml, 10ng/ml final concentration) as described25,32,33 were incubated with adherent CRC cells at a E: T ratio of 2:1 for 48 h. Cet (2 µg/mL) or Cet-DC315 (2 µg/mL) were added to the co-cultures of Vδ2 T cells and CRC cell line Caco-2 or PDO from the beginning of the assay. The number of target cells plated was defined using Luna II automatic cell counter (Byosystems, Aligned Genetics Inc) of Caco-2 cells or CRC cells of PDO after dissociation from the 3D structure. The addition of Cet or ADCs allowed us to evaluate the contribution of the antibody and/or the bisphosphonate drug to the cytotoxicity of the Vδ2 effector T cells. This assay quantifies the amount of surviving CRC cells at the end of the assay. Living Caco-2 cells or PDO adhered spontaneously to the culture plates and this allowed the staining of CRC cells. To detect cytotoxicity of CRC cells, each well was gently washed with complete culture medium to discard non-adherent cells (e.g. Vδ2 T cells, dying/died CRC cells). Afterwards, culture wells were stained with crystal-violet and colorimetric intensity evaluated at the wavelength of 594 nm with the fluorimeter VICTOR X5 (PerkinElmer Italia, Milan, Italy) as previously described20,25. The optical density (OD) values were compared with the OD values of the target cells (Caco-2 or PDO) cultured alone and referred to as 100% of viability. Cytotoxicity resulted in a lower OD value in co-cultures of CRC cells and lymphocytes than the OD value of culture of CRC cell alone.
Statistical analysis
A two-tailed unpaired Student’s t-test with Welch correction and one-way ANOVA followed by Tukey’s multiple comparisons test to compare more than two group were applied using the GraphPad Prism software 5.0.
Results
Synthesis of novel ADCs: Cet-DC310 and Cet-DC315
In our efforts to improve Cetuximab-conjugate N-BPs efficacy, we focused on different synthetic approaches. The first strategy involved the insertion of a proper linker between ZA and Cet, in order to improve the conjugation system. The synthesis of this new ADC is illustrated in Supplementary Fig. S1. In our second approach, we aimed to optimize the conjugation efficiency in the synthesis of Cet-ZA, as reported in Supplementary Fig. S2.
Finally, to further enhance its affinity for tumor cells and improve cellular uptake, the structure of ZA was modified by replacing it with its aminothiazole phosphonic acid analogue, DC315, using the conjugation approach described in Supplementary Fig. S2.
MALDI-MS characterization of Cet-DC310 and Cet-DC315
Both the MALDI mass spectra of Cet-DC310 or Cet showed peaks of single, double and triple charged forms of (Fig. 2 panel A and C). Considering the peak corresponding to the single charged form of Cet-DC310 at m/z of 155,859 compared to the same charged species observed for native Cet at 151,101, the measured DAR was of approximately 6.8. This indicates an improvement of 1.5 fold compared to Cet-ZA already published that reported a DAR 4.325. As observed for Cet-DC310, the MALDI analysis of Cet-DC315 evidenced the presence of single, double and triple charged species for both Cet linked to DC315 and native Cet (Fig. 2 panel B). In this case the calculated DAR from the difference between the monoprotonated species at m/z 158,910 for Cet-DC315 and 151,101 for Cet, was of approximately 23. This indicates a strong amount of DC315 linked to the antibody.

Matrix Assisted Laser Desorption Ionization (MALDI) mass spectra of Cetuximab conjugated with DC310 (A, Cet-DC310), Cetuximab conjugated with DC315 (B, Cet-DC315) or native antibody (C, Cetuximab). The molecular weight (MW) of the peak of single, double and triple charged molecules are indicated in each panel. The different MW of the ADC compared to the native antibody indicates the covalent conjugation of DC310 or DC315 to the antibody Cet.
Reactivity of Cet-DC310 and Cet-DC315 with CRC cell lines and patient-derived organoids
To determine the reactivity of Cet-DC310 or Cet-DC315 with CRC cell lines expressing the EGFR, we incubated the Caco-2, RKO, LS-180, HCT-116 and SW-620 cells with either Cet-DC310, Cet-DC315 or native Cet for comparison followed by Alexa Fluor 647 labeled anti-human isotype specific Ig antiserum and samples have been analyzed by flow cytometry (Fig. 3). We selected these cell lines because Caco-2 and RKO exhibit wild-type EGFR signaling, HCT-116 is EGFR-positive but carries a KRAS mutation, and SW-620 also harbors a KRAS mutation and displays very low reactivity to the anti-EGFR antibody Cetuximab.
Cet-DC310 exhibited a binding profile comparable to that of Cet across all tested concentrations (0.02–200 µg/mL) in Caco-2 cells. In LS-180 cells, Cet-DC310 showed higher reactivity than Cet within the 0.02–20 µg/mL range. In contrast, in HCT-116 cells, its reactivity overlapped with that of Cet at higher concentrations (20–200 µg/mL) but was reduced at lower concentrations (0.02–2 µg/mL) (Fig. 3 Panel A). Overall, the staining pattern of Cet-DC310 closely mirrored that of Cet, with both antibodies effectively labeling EGFR⁺ CRC cell lines, while showing minimal staining of the EGFRdull SW-620 cell line.
Similarly, Cet-DC315 displayed a reactivity pattern higher and comparable to Cet at 200 and 20 µg/mL, respectively, but its binding was notably reduced between 2 and 0.02 µg/mL (Fig. 3 Panel B). For instance, in HCT-116 cells at 2 µg/mL, the mean fluorescence intensity ratio (MFIR) of Cet-DC315 was approximately half that of Cet (MFIR ≈ 50 vs. ≈ 100). As observed for Cet-DC310, Cet-DC315 also mirrored the staining pattern of Cet, selectively labeling EGFR⁺ CRC cell lines while exhibiting low reactivity with the EGFRdull SW-620 cell line.
The reactivity dose of 2 µg/mL/106 cells was selected for further functional experiments.
To quantitatively assess whether EGFR recognition was preserved after DC315 conjugation, we determined the apparent dissociation constant (Kd) of native Cet and Cet-DC315 by flow cytometry–based binding assays. Binding curves were generated by incubating EGFR-expressing cells with increasing concentrations of antibody and MFI values (background-corrected) were fitted to a single-site binding model. As reported in Supplementary Table 1, Cet demonstrated a Kd of 0.01–0.02 µg/mL, whereas Cet-DC315 exhibited a higher Kd, ranging from 24 to 189 µg/mL across the three cells lines analyzed. These results are consistent with a significative reduction in affinity upon conjugation. Importantly, the maximum binding capacity (Bmax) was higher for the conjugated antibody (Cet-DC315) compared to Cet in all the cell lines tested, indicating that the overall number of accessible EGFR binding sites was not reduced and may even be enhanced upon conjugation (Supplementary Table 1).
These data confirm that although drug conjugation markedly decreases the affinity of Cetuximab for EGFR, the overall binding capacity is maintained, supporting the conclusion that target recognition is largely preserved.
In a set of experiments, the reactivity of Cet-DC315 was assessed in PDO cultured in suspension. PDO were stained with Cet-DC315 followed by an anti-human Alexa Fluor 488-conjugated antiserum, and analyzed by flow cytometry and confocal microscopy. Figure 3 Panel C shows a representative image of floating CRC organoids acquired by phase-contrast microscopy. As illustrated in Fig. 3 Panel D, Cet-DC315 displayed a binding profile comparable to that of Cetuximab in PDO expressing the epithelial marker EpCAM. In line with the flow cytometry results, the confocal microscopy image in Fig. 3 Panel E confirms strong Cet-DC315 binding.
Altogether, these findings indicate that the conjugation of DC310 or DC315 to Cet altered only partially the interaction with the EGFR. This modification, detected only at very high concentration of ADC, can be dependent on the strong DAR of Cet-DC315 that may induce a steric hindrance among the antibody binding portion and the recognized epitopes of EGFR antigen.

Reactivity of Cet-DC310 and Cet-DC315 with CRC cell lines and PDO. The indicated CRC cell lines have been tested for the reactivity with Cet-DC310 (A) and Cet-DC315 (B) ADC compared to that of native Cet. (A) Upper histograms, titration of Cet-DC310 (red) and native Cet (grey) antibodies with the Caco-2, LS-180, HCT-116 and SW-620 CRC cell lines. Cell lines were incubated with the indicated concentration of antibodies followed by the anti-human Alexa Fluor 647 antiserum. Results are expressed as mean fluorescence ratio (ratio between the mean fluorescence intensity (MFI) of Cet-DC310 or Cet and the MFI of cells incubated with the second reagent alone). The results have been plotted with GraphPad Prism software. Lower panel. Overlay histograms of Cet (grey), Cet-DC310 (red) antibodies at 2 µg/mL and negative control (CTRL, black, second reagent alone). Results are expressed as log far red fluorescence intensity vs. cell number. (B) Upper histograms, titration of Cet-DC315 (red) and native Cet (grey) antibodies with the Caco-2, RKO, HCT-116 and SW-620 CRC cell lines. Cell lines were incubated with the indicated concentration of antibodies followed by the anti-human Alexa Fluor 647 antiserum. Results are expressed as mean fluorescence ratio. Lower panel. Overlay histograms of Cet (grey), Cet-DC315 (red) antibodies at 2 µg/mL and negative control (CTRL, black, second reagent alone). (C) Representative image of floating OMCR19-003TK PDO cultured in suspension, acquired using phase-contrast Leica DMIRB microscope. The inset on the right is a magnified view of selected PDO. (D) Representative flow cytometric analysis of OMCR19-003TK PDO stained with Cet or Cet-DC315 followed by anti-human Alexa Fluor 488-conjugated antiserum. Negative control is represented by PDO incubated with the second reagent alone. PDO were also stained with an anti-EpCAM antibody followed by anti-mouse PE-conjugated to identify epithelial cells. Results are expressed as percentage of positive cells. (E) Representative confocal microscopy image showing strong surface binding of Cet-DC315 (Alexa Fluor 488 green) in OMCR19-003TK PDO. Nuclei were counterstained with DAPI. Images: 1: DAPI; 2: Alexa Fluor 488; 3: Merge; 4: voxel projection of the Z stack.
Cet-DC315 and Cet-DC310 effect on CRC cell line proliferation
It has been reported the anti-EGFR antibody Cet can inhibit the cell proliferation of CRC cell lines with a WT-EGFR-mediated signaling34. This is the main reason as this antibody is used for the treatment of some metastatic CRC35. To test whether Cet-DC310 and Cet-DC315 ADC can inhibit the proliferation of CRC cell lines like native Cet: the Caco-2 and HCT-116 cell lines were used both in conventional culture conditions and in a 3D culture system resembling tumor micromasses. The Caco-2 cell line expresses a WT EGFR-mediated signaling and Cet antibody can affect its growth as reported34 while HCT-116, showing the G13D KRAS mutation, should be refractory to Cet-mediated inhibition. First, we cultured the two cell lines in conventional adherent plates. It is evident that the inhibitory effect of Cet-DC315 ADC is greater than that exerted by native Cet (Fig. 4 left Panel A). Indeed, inhibition of about 75% of Caco-2 cell proliferation has been detected in cultures with Cet-DC315 after 120h. In contrast, only 25% inhibition was detectable with native Cet after the same culture period. As expected, no effect of Cet-DC315 or Cet was observed on HCT-116 cell growth. To determine whether the ADC can influence the Caco-2 cell growth in a 3D culture system, spheroids of Caco-2 cell line were obtained using AggreWellTM400 plates, as previously described20. Again, Cet-DC315 exerted a very strong inhibitory effect on the increment of the diameter of Caco-2 spheroids. This effect was markedly greater than that of native Cet (mean of 145 μm of diameter for CTR vs. 128 μm for Cet and 60 μm for Cet-DC315). No effect was observed on the generation of HCT-116 spheroids (Fig. 4 right panel A).
Similar experiments were performed to evaluate whether Cet-DC310 ADC can still inhibit the proliferation of CRC cell lines like native Cet. The Caco-2 and HCT-116 cell lines were cultured in conventional adherent plates and treated with Cet and Cet-DC310 at 2 µg/mL. In contrast to Cet-DC315, treatment with Cet-DC310 did not result in a significant inhibition of cell proliferation in either Caco-2 or HCT-116 CRC cell lines (Fig. 4 panel B).
Altogether, these findings indicate that Cet-DC315 ADC not only inhibits the growth of CRC cells with a WT-EGFR pathway, but also that its activity is increased compared to Cet. This increase is detected also when micromasses of CRC are growing, possibly supporting the idea that this novel ADC could be more active than Cet during the onset of expansion of localized tissue micrometastasis.

Effect of Cet-DC315 and Cet-DC310 ADC on the growth of CRC cell lines. (A) Caco-2 (upper) or HCT-116 (lower) cell lines were seeded in flat-bottomed plates without (CTR, no antibody, black) or with 2 µg/mL of native Cet (grey) or Cet-DC315 (red). Cell growth was represented as the degree of confluence by taking images of n = 5–6 culture well replicates at the indicated time points (0, 24, 48, 72, 96, 120 h). The percentage of confluency was estimated by CellStudio software, integrated with the CellCyte XTMplate scanner. The results are expressed as mean ± SD of percentage confluency. In right panel A spheroids were generated from Caco-2 cells in the AggreWell-400 plates after 5 days of culture without (CTR) or with 2 µg/mL of the indicated antibodies. The spheroid diameter was measured in n = 50 spheroids randomly chosen. The results have been plotted with GraphPad Prism software and are the mean ± SD of 50 spheroids. (B) The Caco-2 (upper) or the HCT-116 (lower) were seeded in flat-bottomed plates without (CTR, no antibody, black) or with 2 µg/mL of native Cet (grey) or Cet-DC310 (red). Cell growth was represented as the degree of confluence by taking images of n = 4–6 culture well replicates at the indicated time points (0, 24, 48, 72 h). The percentage of confluency was estimated by CellStudio software, associatedintegrated with the CellCyte XTMTM plate scanner. The results are expressed as mean ± SD of percentage confluency. ****p < 0.001 comparing the cell confluence of Cet-DC315 vs. Cet or vs. CTR or the area of spheroids of Cet-DC315 vs. Cet or vs. CTR of (A). For both panels statistical analysis was performed using one-way ANOVA with Tukey’s multiple comparisons test; detailed results are reported in Supplementary Table 2.
Expansion of Vδ2 T lymphocytes by ADC Cet-DC315
It is well-established that N-BPs such as ZA can trigger the activation and consequent proliferation of Vδ2 T lymphocytes in the presence of IL236. To determine whether Cet-DC135 ADC can be processed by CRC cells inducing the expansion of Vδ2 T cells, peripheral blood T lymphocytes were co-cultured with the CRC cell line LS-180 with Cet-DC315 (2 µg/mL) and IL2 was added after 24 h. Indeed, it has been shown previously that LS-180 CRC cells can present the small pyrophosphate antigens such as isopentenylpyrophoshate (IPP), derived from the pharmacological effect of ZA, to Vδ2 T cells favoring their proliferation in the presence of IL231. An evident increase of the percentage of Vδ2 T cells was detected by indirect immunofluorescence assay during the culture. In particular, at day 14 about the 60% of cells found in co-cultures were represented by Vδ2 T cells. On the other hand, similar percentages of Vδ2 T cells were found in co-cultures incubated with native Cet compared to the starting Vδ2 T cell population (1–3% vs. 0.5-4% of Vδ2 T cells at time zero) (Fig. 5, left panel). Further, we analyzed whether the observed effect on the expansion of Vδ2 T cells could be detected when purified T cells were incubated under the same experimental conditions with PDO. To this aim, PDO cultured in geltrex domes in appropriate culture conditions were harvested, geltrex matrix washed away and CRC cells cultured with T cells at the effector target (T: PDO) ratio of 10:1. On day 25, a strong increment in the percentage of Vδ2 T cells was found (Fig. 5, middle panel). While no expansion of Vδ2 T cells was detectable with PDO cultured with Cet.
Eventually, we analysed whether Cet-DC315 could stimulate the growth of Vδ2 T cells present in cell suspensions from CRC mucosa specimens. To this aim, cell suspensions from CRC mucosa were obtained by enzymatic digestion, and after thawing were put in culture with either Cet-DC315 or Cet as antibody control. IL2 was added after 24 h and cell cultures followed by microscopic examination. The cell proliferation in the presence of Cet-DC315 and IL2 was evident after about 10 days. However, given the relatively low amount of CD45+ cells (i.e. identified as leukocytes) in the starting populations (5–25%), the cell cultures were assessed by indirect immunofluorescence at day 25 to assess the amount of Vδ2 T cells. While the Vδ2 T cells were almost undetectable (0.01–0.5%) at the starting time point they increased up to 30–35% on day 25 (Fig. 5, right panel). Altogether, these data would suggest that DC315 carried by anti-EGFR antibody Cet can be processed by cells from CRC mucosa and it efficiently stimulates Vδ2 T cell growth.
In contrast to Cet-DC315, peripheral blood T lymphocytes co-cultured with the CRC cell line LS-180 treated with Cet-DC310 (2 µg/mL) + IL2 did not show any γδ T cell expansion at any of the time points tested (data not shown).

Cet-DC315 ADC can trigger the expansion of Vδ2 T cells. Left panel: peripheral blood T cells purified by negative selection kit (> 98% CD3 + TCR+) were cultured with irradiated LS-180 CRC cells at the ratio of 10:1 either without (CTR, black circles) or with the indicated antibodies (Cet, grey circles, or Cet-DC315 red circles) at 2 µg/mL in complete medium. IL2 was added after 24 h of incubation. Cell cultures were evaluated on day 14 for the expression of Vδ2 TCR by indirect immunofluorescence and flow cytometry analysis. Results are the mean ± SD of T cells from n = 6 different healthy donors. Middle panel: highly purified T cells from n = 2 healthy donors were co-cultured with 2 PDO (OMCR16-005TK or OMCR19-010TK) in complete RPMI 1640 medium in the same experimental conditions described for the left panel. The percentage of Vδ2T cells was evaluated on day 25 of culture and the results are the mean ± SD of n = 4 independent experiments performed. Right panel: CRC cell suspension from 4 patients (see materials and methods section) were incubated in the same experimental conditions described in the previous panels and the percentage of Vδ2T cells was estimated on day 25 of culture. The results are the mean ± SD from n = 4 independent experiments. The evaluation of Vδ2 TCR expression was performed by polychromatic indirect immunofluorescence flow cytometry, using the anti-Vδ2 and the anti-CD3 antibodies followed by anti-isotype specific second reagents labelled with AF647 and PE respectively. The results have been plotted with GraphPad Prism software. Statistical significance was assessed using a one-way ANOVA with Tukey’s multiple comparisons test. ****p < 0.0001).
Triggering of Vδ2 T cell cytotoxicity on CRC cells by ADC Cet-DC315
It is well established that the ADCs composed of Cet linked to N-BPs such as ZA or ibandronate (Cet-IBA) or risedronate (Cet-Ris) can induce potent antitumor cytotoxicity in 2D- and 3D-cultured CRC models25,26. To evaluate whether the novel conjugate Cet-DC315 could elicit activation of Vδ2 T cells against CRC cell lines or PDO, we conducted cytotoxicity assays using Caco-2 cell line (Fig. 6A) and CRC PDO (Fig. 6B) pre-incubated with either Cet or Cet-DC315 at a concentration of 2 µg/mL. Vδ2 T cells were expanded from PBMCs of three healthy donors with ZA as described31 and co-cultured with Caco-2 cell line or PDO at an effector-to-target (E: T) ratio of 2:1 in the presence of the ADCs. After 48 h of incubation, surviving adherent CRC cells were then quantified by crystal violet staining. As shown in Fig. 6, Cet-DC315 induced the strongest cytotoxic effect against both Caco-2 cell line and PDO (Fig. 6A,B). It is of note that at the E: T ratio used, Vδ2 T cell-mediated cytotoxicity of CRC cells was negligible. On the contrary, the addition of native Cet could increase the cytotoxic effect indicating that a detectable degree of ADCC was elicited. More importantly, the incubation of Cet-DC315 further enhanced the lytic effect exerted by Vδ2 T cells (0.7 OD of Cet-DC315 vs. 1.0 of only Cet, about 30% of increase of cytotoxicity). This finding supports that Cet-DC315, compared to the native Cet, increase the anti-tumor effect of Vδ2 T lymphocytes. In contrast, Cet-DC310 did not trigger any detectable cytotoxic activity under the same experimental conditions (data not shown). Taken together, these findings would indicate that Cet-DC315 metabolized by the target cells triggers the stimulation of Vδ2 T cells while the uncleavable Cet-DC310 N-BPs linked anti-EGFR antibody does not.

Cet-DC315 ADC triggers Vδ2T cell cytotoxicity against CRC cell lines and PDO. Caco-2 cell line (Tumor: T) (A) and the PDO OMR19-003TK (T) (B) were incubated with Cet (2 µg/mL) or Cet-DC315 (2 µg/mL) and 24 h later cocultured with Vδ2 T cell at the effector: target ratio of 2:1. After 48 h, the cell cultures were stained with crystal violet to quantify surviving/adherent cells. The optical density (OD) of the eluted stain was evaluated by a VICTOR X5 multilabel plate reader at 594 nm. CTR: OD of cultured tumor cells only. Data represent mean ± SD of n = 3–6 cell replicates (all individual points are shown). Cet = T+Vδ2+Cet; Cet-DC315 = T+Vδ2+Cet-DC315. Statistical significance was assessed using a one-way ANOVA with Tukey’s multiple comparisons test. *p < 0.05; **p < 0.01.
Discussion
Immunotherapy represents one of the most promising therapeutic approaches in oncology. Recently, there has been growing interest in developing innovative strategies that allow tumor targeting and simultaneous activation of a local and specific antineoplastic immune response. In this regard, ADCs have been developed and are widely used for the treatment of various cancers. These agents exert a powerful and specific antitumor effect by targeting tumor cells expressing the antigen recognized by the antibody. The cytotoxic drug, along with the antibody, enters the target cells and disrupts essential metabolic or structural cellular functions, leading to apoptosis and/or proliferative blockade1.
We focused on developing antibodies conjugated with aminobisphosphonates and evaluating their potential to expand and enhance the antitumor activities of Vδ2 T cells. Recently, we generated ADCs composed of Cet covalently linked to different N-BPs such as ZA, ibandronate or risedronate26. These novel ADCs exert a potent antitumor effect and expand Vδ2 T cells against CRC cells, suggesting that they could effectively direct ZA mainly to tumor cells while reducing the tendency of soluble N-BPs to accumulate in bone tissue. Altogether, these findings suggest that these ADCs can exert a stronger antitumor effect by two mechanisms: (1) directly affecting tumor cell proliferation and (2) indirectly activating an immune response.
In our previous studies, we developed a novel ADC by conjugating Cet to ZA (Cet-ZA), achieving a DAR of 4.3 while retaining reactivity comparable to the unconjugated antibody, effectively modulating multiple pathways involved in CRC25,26,27.
Building upon these promising results, we aimed to optimize our system of ADC conjugation by exploring alternative strategies, including the improvement of DAR. Recent advancements in ADC technologies have emphasized the potential of high DAR platforms to significantly enhance therapeutic outcomes37,38. Traditionally, ADCs with DARs of 2–4 were favored due to concerns over aggregation, fast clearance, and reduced efficacy associated with higher DAR ADCs39,40. However, novel systems have demonstrated that well-designed high-DAR ADCs—up to DAR 18 and even DAR 44—can achieve superior efficacy while maintaining stability and tolerability in vivo41,42.
Advantages of high-DAR ADCs include improved payload delivery efficiency, particularly important for targeting low-abundance antigens43. This is beneficial in overcoming limitations such as low antigen density and internalization rates in poorly vascularized tumor regions. On the disadvantage side, increasing DAR inherently increases hydrophobicity, which can lead to aggregation, reduced solubility, and faster plasma clearance and could compromise pharmacokinetics and tissue penetration43. To mitigate this, the use of highly polar payloads or specific linkers have been proposed in literature39,44.
Based on these considerations, our first approach involved the introduction of an appropriate spacer to improve both the DAR and conjugation efficacy. The resulting ADC Cet-DC310, although endowed with an increased DAR of 6.8, failed in functional cell-based assays. This strategy was unsuccessful possibly due to the use of an uncleavable linker, which likely prevented the release of DC310. Indeed, Cet-DC310 well reacted with EGFR+ CRC cell lines, but it neither inhibited tumor cell proliferation nor triggered the Vδ2 T cell proliferation and/or activation of CRC cell killing. This finding would suggest the inability of cells to lead to the accumulation of small pyrophosphate antigens , due to the effect of DC310 on mevalonate pathway. This lack would impair the consequent presentation of small phosphate antigens through butyrophilins and Vδ2 T cell activation45,46.
The second approach focused on the optimization of our conjugation system combined with structural modifications in ZA structure. For this purpose, we employed an improved conjugation method involving the direct formation of the activated phosphonic acid as a imidazole phosphonoamidate, similar to the approach that we already reported for Cet-ZA, but with significant modifications. This conjugation specifically targeted lysine residues by using a direct phosphonoamidation reaction between the phosphonic acids and the amine groups of lysine residues. This reaction is commonly applied in peptide conjugation and the linkage of free phosphoric acids to DNA47. The formation of a phosphonoamidate bond has been well-established in the field of targeted therapy, with similar reactions used in the creation of oligonucleotide-peptide conjugates, such as Fludarabine-(5’phosphoramidate)-[anti-IGF-1R]48, Gemcitabine-(5’phosphoramidate)-[anti-IGF-1R]49, and various diagnostic agents.
In our previous work25, we already applied this conjugation strategy to the synthesis of Cet-ZA. In that case, the reaction was performed in an imidazole buffer 0.1 M pH = 6 with EDC as the activating agent, leading to the in situ formation of the imidazole intermediate of ZA, which was not isolated25. In our new approach, the antibody-drug conjugation was still performed via phosphonoamidation but using N,N’-carbonyl diimidazole (CDI) as the activating agent for the phosphonic groups. CDI is one of the most widely used activating reagents, frequently employed for N-acetylation reactions, such as amide and amine synthesis via the acylimidazole intermediate50. Additionally, CDI is well-known for its ability to activate phosphate groups, converting them into highly reactive intermediates, thus facilitating conjugation between phosphates and primary amines51.
This new conjugation system was at first adopted using ZA as payload and then modifying ZA structure to give DC315, where the imidazole heterocycle was replaced by an aminothiazole moiety and the hydroxyl group in the α-position relative to the two phosphonic acids was replaced by an hydrogen atom. DC315 was part of a series of ZA analogues developed by Matsumoto et al.28 through minor structural changes, such as replacing the imidazole group with nitrogen-containing heterocycles like pyridine, pyrimidine, or thiazoles. The lack of the hydroxyl group in alpha to phosphonic acids could contribute to reduce bone binding in vivo and have a positive effect on the bioavailability of the payload. These analogues were evaluated on several tumor cell lines using cell viability assay, where DC315 demonstrated an antitumor activity comparable or even greater than ZA28.
The crucial step of this new conjugation system is the formation and isolation of the phosphonoimidazole analogues of ZA or DC315, designated as compound 1 or 2, respectively (Fig. 1 and Supplementary Fig. S2). 1 or 2 were obtained through a CDI-mediated activation, and a purification step. The purified derivatives 1 or 2 were then reacted with abundant solvent-accessible Lys residues of Cet in imidazole buffer 0.1 M at pH 6, without any other interferences to prevent any side reactions. This novel technique applied to Cet-ZA synthesis afforded a good conjugation with a DAR of 4.5, comparable to that previously reported25. Surprisingly, the analogous reaction between Cet and 2 resulted in a markedly improved conjugation efficiency under the same conditions, producing a DAR of 23 for Cet-DC315. The enhanced DAR signified a substantial improvement in the conjugation process, which we hypothesized could lead to increased therapeutic efficacy. The improvement in conjugation efficiency could be explained by the higher chemical stability of DC315 with respect to ZA. In fact, as already reported by other authors52, the hydroxyl group in the α-position relative to the two phosphonic acids in ZA is a key structural element that affects its reactivity, determining chemical instability and increasing bone binding.
The analysis of Cet-DC315 and Cet-DC310 reactivity with a panel of CRC cell lines and PDO indicated that the change in conjugation procedure and the presence of different N-BPs did not alter the interaction between the anti-EGFR antibody Cet and the EGFR. This is a prerequisite to suggest the use of the ADCs for therapeutic purposes53. It is to be noted that Cet-DC310 reacted better than Cet with EGFR, except for HCT-116 cell line. Whereas Cet-DC315 showed a lesser reactivity compared to Cet.
The Kd of Cet-DC315 was higher than that of Cet, consistent with a lower binding affinity (see Supplementary Table 1). This clearly indicates that DC315 linked to the anti-EGFR antibody affects the interaction with the EGFR. However, in vitro functional experiments indicated that Cet-DC315 retained similar effects to those ADC conjugated with other N-BPs characterized by higher affinity. This suggests that a partial loss of affinity does not necessarily compromise functional activity, which can be compensated by additive mechanisms, such as the combination of Cet antiproliferative effect and the cytotoxic activity of DC315. Thus, Cet-DC315 may be an useful experimental tool to further study the in vivo functionality of anti-EGFR antibody linked to N-BPs.
Similar examples have been reported in the literature, where ADCs with reduced affinity compared to the unconjugated antibody nevertheless exhibit enhanced functional efficacy through the additive or synergistic actions of the targeting antibody and the cytotoxic payload. This approach underscores the potential of ADCs to maintain or even enhance therapeutic efficacy despite modifications that may lower binding affinity54.
A notable aspect is that Bmax values showed a relevant increase for the conjugated antibody in comparison to native Cet, suggesting that receptor-accessible sites were not reduced by steric hindrance. On the contrary, this increase may reflect structural changes induced by conjugation that render certain epitopes more accessible, potentially further enhancing the functional performance of Cet-DC315.
Roughly, the optimal reactivity of both the ADCs was 2 µg/mL. More relevant, the functional behavior of the two ADCs on CRC cell lines is different. Cet-DC315 can markedly increase the inhibitory effect on the growth of the WT-EGFR cell line Caco-2 exerted by the unconjugated antibody Cet. This would imply that the interaction of this ADC with the EGFR may influence the EGFR-mediated signal transduction better than Cet. This effect was evident also on the growth of 3D cell spheroids of Caco-2. No effect was detected on the growth of the KRAS-mutated cell line HCT-116. The proliferation of HCT-116 is independent on the signal mediated through the interaction between the EGF and EGFR because of the activating KRAS downstream mutation34. Altogether, these findings are in line with what observed using Cet conjugated with classical N-BPs such as zoledronate, risedronate or ibandronate26. On the other hand, Cet-DC310 did not increment the inhibition of cell growth observed with Cet and this was not in line with the increment of Cet-DC310 reactivity with EGFR. Altogether, these observations may indicate that the conjugation of the Cet antibody with different N-BPs modify the ligand binding capacity of Cet. However, this modification does not lead to an impaired inhibition of CRC cell growth at an ADC concentration 100–200 fold lower than that usually found in the plasma of patients treated with ADC55. Furthermore, Cet-DC315 can trigger the expansion of Vδ2+ T cells in co-cultures with the CRC cell line LS-180. This implies that DC315 linked to Cet may be processed in LS-180, leading to the production of small pyrophosphate antigens, which in turn are presented at the cell surface to trigger the Vδ2 T cell proliferation. This effect is similar to that shown with Cet conjugated with other classical N-BPs25,26. The finding that Cet-DC315 can stimulate Vδ2+ T cell growth in co-cultures of CRC PDO may further support the functional relevance of the generation of this ADC and its possible application in a clinical setting, although relevant limitations to this purpose will be listed below. Finally, the expansion of Vδ2 T cells using cell suspensions from CRC specimens indicates that Cet-DC315 antibody is efficient in an experimental condition mimicking strictly the in vivo situation. Indeed, CRC-derived cell suspensions contain several types of cells typically present in the TME including CRC cells, different leukocyte subsets and stromal cells31,56,57,58.
Cet-DC315 triggers the cytolytic activity of Vδ2 + T cells against the CRC cell line Caco-2 and CRC organoid OMCR19-003TK, supporting the hypothesis of its superior anti-CRC activity compared to native Cet, at least in these in vitro experimental models.
Our present data using PDO and CRC mucosa cell suspension are more representative of the human context than models artificially reconstructed in mice. Nevertheless, the present findings should be further analyzed and confirmed in more complex 3D models such as assembloids or heterotypic spheroids under dynamic experimental conditions. Indeed, these in vitro culture systems may better represent the cell heterogeneity of the TME present in CRC59,60. The presence of tumor-associated fibroblasts as well as of other immune cells, besides the epithelial-derived components of organoids, is crucial to properly evaluate the immune response to a novel ADC53,59,61,62. Furthermore, it is important to analyze in the depth the biochemical mechanisms underlying the greater inhibition of cell growth of Caco-2 cell line found with Cet-DC315 compared to native Cet. The definition of these mechanisms could help to design similar N-BPs and conjugated Cet-based ADC with a more evident direct antitumor effect.
The most relevant limitation of the present study is the absence of in vivo validation experiments to assess the toxicity and efficacy of Cet-DC315 ADC against CRC. Such evaluation is essential to establish the translational potential of this ADC, since the demonstration of antitumor activity in murine models is generally considered the proof-of-concept step required before moving to clinical applications of therapeutic antibodies63.
Although it is technically feasible to test the efficacy of antibody and of Vδ2 + T cells in mice, several factors limit the biological significance of these models63. The first one is that the γδT cell are different in mice compared to humans and this determines that murine tumor and γδ T cell response are not representative of the human response64,65,66,67,68. Further, only adoptive treatment with human Vδ2 T cells could be assessed in immunosuppressed or immune-deficient mice carrying a human tumor, a condition far from what occurs in humans65. Nevertheless, the direct therapeutic activity of Cet-DC315 could be tested in CRC orthotopic models, where the use of PDO-derived xenografts represents one of the most feasible approaches to mimic human tumor biology69,70,71,72.
In addition, in vivo testing of Cet-DC315 should not be limited to CRC, but extended to other carcinomas in which EGFR plays a pivotal role in tumor growth and therapeutic targeting, including head and neck cancers73 and non-small cell lung cancer74. The absence of data on Cet-DC315 activity in these tumor settings represents another limit of our study.
A further limitation is the lack of precise definition of the mechanistic action of Cet-DC315. The production of IPP and/or dimethylallyl pyrophopshates (DMAPP) in triggering the Vδ2T cells expansion and anti-tumor cytotoxic activation should be directly demonstrated to support the already known mechanism of N-BPs in the stimulation of immune response46,75.
Another important limitation concerns the high DAR of the Cet-DC315 ADC. Indeed, the reported DAR of ~ 23 for Cet-DC315 is exceptionally high and atypical, compared to clinically developed ADCs76. This high DAR can affect the antibody stability, generate antibody aggregates possibly leading to altered pharmacokinetics and favoring immunogenicity of Cet-DC315 in vivo. Overall, future work should therefore aim to optimize the conjugation process and reduce the DAR of Cet-DC315. In this context, the conjugation of DC315 to the fully human antibody panitumumab could in part reduce the immunogenicity linked to the murine components of the cetuximab77. A lower Cet-DC315 DAR could preserve the functional features of this ADC, as suggested by other ADC preparations composed of Cet and different N-BPs, which displayed DAR values between 3.6 and 7.1 yet retained in vitro activity26. According to that report, the type of N-BP conjugated, rather than its amount, was the main determinant of ADC functionality. Whether soluble DC315 provides superior activity compared to other N-BPs in CRC cell lines such as Caco-2 remains to be fully determined. Indeed, a side-by-side comparison of all the features of Cet-DC315 and Cet-ZA should be performed to define whether is more useful for clinical applications.
In summary, while our results indicate that DC315 represents a promising candidate for the generation of Cet-based ADCs, extensive biochemical, biophysical, and in vivo studies directly comparing different N-BP conjugates will be required to identify the best option to plan a potential therapeutic application of these ADCs.
In conclusion, optimizing the conjugation strategy allowed higher drug loading of Cet-DC315 possibly maintaining antibody reactivity and functionality. The improved conjugation efficiency over Cet-ZA likely reflects the easier synthesis and greater stability of DC315. Given the significantly enhanced DAR, further studies will focus on DC315’s potential in ADC applications. The increased activity of Cet-DC315 supports its evaluation in 3D CRC models to assess EGFR targeting and tumor microenvironment modulation.
Data availability
All raw flow cytometry, proliferation, and cytotoxicity datasets generated during this study have been deposited in Zenodo and are publicly available at [DOI https://doi.org/10.5281/zenodo.17107670].
References
-
Beck, A., Goetsch, L., Dumontet, C. & Corvaïa, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 16, 315–337 (2017).
-
Dumontet, C., Reichert, J. M., Senter, P. D., Lambert, J. M. & Beck, A. Antibody-drug conjugates come of age in oncology. Nat. Rev. Drug Discov. 22, 641–661 (2023).
-
Fuentes-Antrás, J., Genta, S., Vijenthira, A. & Siu, L. L. Antibody-drug conjugates: in search of partners of choice. Trends Cancer. 9, 339–354 (2023).
-
Fu, Z., Li, S., Han, S., Shi, C. & Zhang, Y. Antibody drug conjugate: the ‘biological missile’ for targeted cancer therapy. Signal. Transduct. Target. Ther. 7, 93 (2022).
-
Chari, R. V. J., Miller, M. L. & Widdison, W. C. Antibody-drug conjugates: an emerging concept in cancer therapy. Angew. Chem. Int. Ed. Engl. 53, 3796–3827 (2014).
-
Mullard, A. 2021 FDA approvals. Nat. Rev. Drug Discov. 21, 83–88 (2022).
-
Kern, J. C. et al. Discovery of pyrophosphate diesters as tunable, soluble, and bioorthogonal linkers for site-specific antibody-drug conjugates. J. Am. Chem. Soc. 138, 1430–1445 (2016).
-
Dugal-Tessier, J., Thirumalairajan, S. & Jain, N. Antibody-oligonucleotide conjugates: A twist to antibody-drug conjugates. J. Clin. Med. 10, 838 (2021).
-
Pillow, T. H. et al. Antibody conjugation of a chimeric BET degrader enables in vivo activity. ChemMedChem 15, 17–25 (2020).
-
Cini, E. et al. Antibody drug conjugates (ADCs) charged with HDAC inhibitor for targeted epigenetic modulation. Chem. Sci. 9, 6490–6496 (2018).
-
Hobson, A. D. Antibody drug conjugates beyond cytotoxic payloads. Prog. Med. Chem. 62, 1–59 (2023).
-
Esapa, B. et al. Target antigen attributes and their contributions to clinically approved antibody-drug conjugates (ADCs) in haematopoietic and solid cancers. Cancers 15, 1845 (2023).
-
Xu, S. Internalization, trafficking, intracellular processing and actions of antibody-drug conjugates. Pharm. Res. 32, 3577–3583 (2015).
-
Jones, C., Taylor, M. A. & McWilliams, B. The role of cetuximab as first-line treatment of colorectal liver metastases. HPB (Oxford). 15, 11–17 (2013).
-
Gerber, D. E. & Choy, H. Cetuximab in combination therapy: from bench to clinic. Cancer Metastasis Rev. 29, 171–180 (2010).
-
Yonesaka, K. HER2-/HER3-targeting antibody-drug conjugates for treating lung and colorectal cancers resistant to EGFR inhibitors. Cancers (Basel). 13, 1047 (2021).
-
Saoudi González, N. et al. Cetuximab as a key partner in personalized targeted therapy for metastatic colorectal cancer. Cancers 16, 412 (2024).
-
Correale, P. et al. Cytotoxic drugs up-regulate epidermal growth factor receptor (EGFR) expression in colon cancer cells and enhance their susceptibility to EGFR-targeted antibody-dependent cell-mediated-cytotoxicity (ADCC). Eur. J. Cancer. 46, 1703–1711 (2010).
-
He, L. et al. Immune modulating antibody-drug conjugate (IM-ADC) for cancer immunotherapy. J. Med. Chem. 64, 15716–15726 (2021).
-
Varesano, S., Zocchi, M. R. & Poggi, A. Zoledronate triggers Vδ2 T cells to destroy and kill spheroids of colon carcinoma: quantitative image analysis of three-dimensional cultures. Front. Immunol. 9, 998 (2018).
-
Veeraballi, S., Bandaru, S. S., Kiwan, C., Chan, K. H. & Shaaban, H. S. A multifaceted role of bisphosphonates from palliative care to anti-cancer therapy in solid tumors. J. Oncol. Pharm. Pract. 31, 107–118 (2025).
-
Barbosa, J. S., Paz, A. & Braga, S. S. Bisphosphonates, old friends of bones and new trends in clinics. J. Med. Chem. 64, 1260–1282 (2021).
-
Bhatwadekar, S. et al. Efficacy and safety of Zoledronic acid in sickle cell disease associated bone disorders:single centre experience. Blood 144, 3686 (2024).
-
Van Poznak, C. H. et al. Association of osteonecrosis of the jaw with Zoledronic acid treatment for bone metastases in patients with cancer. JAMA Oncol. 7, 246–254 (2021).
-
Benelli, R. et al. Targeting of colorectal cancer organoids with Zoledronic acid conjugated to the anti-EGFR antibody cetuximab. J. Immunother. Cancer. 10, e005660 (2022).
-
Pisheh, L. et al. EGFR-targeted antibody-drug conjugate to different aminobisphosphonates: direct and indirect antitumor effects on colorectal carcinoma cells. Cancers (Basel). 16, 1256 (2024).
-
Fernandez, J. L. C. et al. Priming of colorectal Tumor-Associated fibroblasts with Zoledronic acid conjugated to the Anti-Epidermal growth factor receptor antibody cetuximab elicits Anti-Tumor Vδ2 T lymphocytes. Cancers (Basel). 15, 610 (2023).
-
Matsumoto, K. et al. Targeting cancer cells with a bisphosphonate prodrug. ChemMedChem 11, 2656–2663 (2016).
-
Tanaka, Y. et al. Anti-Tumor activity and immunotherapeutic potential of a bisphosphonate prodrug. Sci. Rep. 7, 5987 (2017).
-
Hulspas, R. et al. Evaluation of a microfluidic chip-based cell sorter in clinical manufacturing processes to deplete or isolate CD4 regulatory T cells. Transfusion 65, 1502–1508 (2025).
-
Zocchi, M. R. et al. Zoledronate can induce colorectal cancer microenvironment expressing BTN3A1 to stimulate effector γδ T cells with antitumor activity. Oncoimmunology 6, e1278099 (2017).
-
Di Mascolo, D. et al. Nanoformulated Zoledronic acid boosts the Vδ2 T cell immunotherapeutic potential in colorectal cancer. Cancers (Basel). 12, 104 (2019).
-
Wiedemann, G. M. et al. Cancer cell-derived IL-1α induces CCL22 and the recruitment of regulatory T cells. Oncoimmunology 5, e1175794 (2016).
-
Medico, E. et al. The molecular landscape of colorectal cancer cell lines unveils clinically actionable kinase targets. Nat. Commun. 6, 7002 (2015).
-
Li, Q. H. et al. Anti-EGFR therapy in metastatic colorectal cancer: mechanisms and potential regimens of drug resistance. Gastroenterol. Rep. (Oxf). 8, 179–191 (2020).
-
Kondo, M. et al. Expansion of human peripheral blood γδ T cells using zoledronate. J. Vis. Exp. 3182 https://doi.org/10.3791/3182 (2011).
-
Vogl, A. M. et al. TUB-040, a homogeneous and hydrophilic NaPi2b-targeting ADC with stably linked exatecan, exhibits long-lasting antitumor activity and a well-tolerated safety profile. Mol. Cancer Ther. https://doi.org/10.1158/1535-7163.MCT-25-0254 (2025).
-
Stamati, I. et al. Anti-HER2, high-DAR antibody fragment–drug conjugates with a glucuronide-based MMAE linker–payload demonstrate superior efficacy over IgG-based ADCs. Mol. Cancer Ther. 24, 1295–1307 (2025).
-
Lyon, R. P. et al. Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat. Biotechnol. 33, 733–735 (2015).
-
Sun, X. et al. Effects of drug-antibody ratio on pharmacokinetics, biodistribution, efficacy, and tolerability of antibody-maytansinoid conjugates. Bioconjug. Chem. 28, 1371–1381 (2017).
-
Zacharias, N. et al. A homogeneous high-DAR antibody–drug conjugate platform combining THIOMAB antibodies and XTEN polypeptides. Chem. Sci. 13, 3147–3160
-
Zhang, H. et al. Preparation of an ultrahigh-DAR PDL1 monoclonal antibody-polymeric-SN38 conjugate for precise colon cancer therapy. Biomaterials. 301, 122285 (2023).
-
Jin, S. et al. DNA self-assembly-mediated high drug-antibody ratio ADC platform for targeted tumor therapy and imaging. Nano Today. 58, 102459 (2024).
-
Hamblett, K. J. et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 10, 7063–7070 (2004).
-
Yuan, L. et al. Phosphoantigens glue Butyrophilin 3A1 and 2A1 to activate Vγ9Vδ2 T cells. Nature 621, 840–848 (2023).
-
Vavassori, S. et al. Butyrophilin 3A1 binds phosphorylated antigens and stimulates human γδ T cells. Nat. Immunol. 14, 908–916 (2013).
-
Hu, Q., Deng, X., Yu, X., Kong, J. & Zhang, X. One-step conjugation of aminoferrocene to phosphate groups as electroactive probes for electrochemical detection of sequence-specific DNA. Biosens. Bioelectron. 65, 71–77 (2015).
-
Coyne, C. P. & Narayanan, L. Fludarabine- (C2-methylhydroxyphosphoramide)- [anti-IGF-1R]: synthesis and selectively ‘Targeted’Anti-Neoplastic cytotoxicity against pulmonary adenocarcinoma (A549). J. Pharm. Drug Deliv. Res. 4, 129 (2015).
-
Coyne, C. P. & Narayanan, L. Gemcitabine-(5’-phosphoramidate)-[anti-IGF-1R]: molecular design, synthetic organic chemistry reactions, and antineoplastic cytotoxic potency in populations of pulmonary adenocarcinoma (A549). Chem. Biol. Drug Des. 89, 379–399 (2017).
-
Rannard, S. P. & Davis, N. J. The selective reaction of primary amines with carbonyl imidazole containing compounds: selective amide and carbamate synthesis. Org. Lett. 2, 2117–2120 (2000).
-
Verma, S. K., Ghorpade, R., Pratap, A. & Kaushik, M. P. Solvent free, N,N′-carbonyldiimidazole (CDI) mediated amidation. Tetrahedron Lett. 53, 2373–2376 (2012).
-
Mizuta, S. et al. Synthesis and immunomodulatory activity of fluorine-containing bisphosphonates. ChemMedChem 14, 462–468 (2019).
-
Hafeez, U., Parakh, S., Gan, H. K. & Scott, A. M. Antibody–drug conjugates for cancer therapy. Molecules. 25, 4764 (2020).
-
Datta-Mannan, A. et al. Reducing target binding affinity improves the therapeutic index of anti-MET antibody-drug conjugate in tumor bearing animals. PLoS One. 19, e0293703 (2024).
-
Jiang, D. M. et al. Plasma cetuximab concentrations correlate with survival in patients with advanced KRAS wild type colorectal cancer. Clin. Colorectal Cancer. 22, 457–463 (2023).
-
Li, J. et al. Remodeling of the immune and stromal cell compartment by PD-1 Blockade in mismatch repair-deficient colorectal cancer. Cancer Cell. 41, 1152–1169e7 (2023).
-
Roelands, J. et al. An integrated tumor, immune and microbiome atlas of colon cancer. Nat. Med. 29, 1273–1286 (2023).
-
Kamali Zonouzi, S., Pezeshki, P. S., Razi, S. & Rezaei, N. Cancer-associated fibroblasts in colorectal cancer. Clin. Transl. Oncol. 24, 757–769 (2022).
-
Taglieri, M. et al. Colorectal organoids: Models, imaging, omics, therapy, immunology, and ethics. Cells 14, 457 (2025).
-
Mei, J. et al. Tumour organoids and assembloids: Patient-derived cancer avatars for immunotherapy. Clin. Transl Med. 14, e1656 (2024).
-
Crescioli, S. et al. Antibodies to watch in 2025. MAbs 17, 2443538 (2025).
-
Ruan, D. Y., Wu, H. X., Meng, Q. & Xu, R. H. Development of antibody-drug conjugates in cancer: overview and prospects. Cancer Commun. (Lond). 44, 3–22 (2024).
-
Hodgins, N. O. et al. In vitro potency, in vitro and in vivo efficacy of liposomal alendronate in combination with γδ T cell immunotherapy in mice. J. Control Release. 241, 229–241 (2016).
-
Qu, G. et al. Comparing mouse and human Tissue-Resident γδ T cells. Front. Immunol. 13, 891687 (2022).
-
Pang, D. J., Neves, J. F., Sumaria, N. & Pennington, D. J. Understanding the complexity of γδ T-cell subsets in mouse and human. Immunology 136, 283–290 (2012).
-
Ribot, J. C., Serre, K. & Silva-Santos, B. Developmental and functional assays to study murine and human γδ T cells. Methods Mol. Biol. 1514, 257–267 (2017).
-
Papadopoulou, M., Sanchez Sanchez, G. & Vermijlen, D. Innate and adaptive γδ T cells: How, when, and why. Immunol. Rev. 298, 99–116 (2020).
-
Reis, B. S. et al. TCR-Vγδ usage distinguishes protumor from antitumor intestinal γδ T cell subsets. Science 377, 276–284 (2022).
-
High, P. P.C. et al. Cetuximab increases LGR5 expression and augments LGR5-targeting antibody-drug conjugate efficacy in patient-derived colorectal cancer models. BioRxiv 2025.06.18.660406. https://doi.org/10.1101/2025.06.18.660406 (2025).
-
Blanco-Domínguez, R. et al. Dual modulation of cytotoxic and checkpoint receptors tunes the efficacy of adoptive delta one T cell therapy against colorectal cancer. Nat. Cancer. 6, 1056–1072 (2025).
-
Chen, C., Fu, Q., Wang, L., Tanaka, S. & Imajo, M. Establishment of a novel mouse model of colorectal cancer by orthotopic transplantation. BMC Cancer. 25, 405 (2025).
-
Bürtin, F., Mullins, C. S. & Linnebacher, M. Mouse models of colorectal cancer: Past, present and future perspectives. World J. Gastroenterol. 26, 1394–1426 (2020).
-
Van den bossche, V. et al. PPARα-mediated lipid metabolism reprogramming supports anti-EGFR therapy resistance in head and neck squamous cell carcinoma. Nat. Commun. 16, 1237 (2025).
-
Shi, Y. et al. Cetuximab-Immunoliposomes loaded with TGF-β1 SiRNA for the targeting therapy of NSCLC: Design, and in vitro and in vivo evaluation. Int. J. Mol. Sci. 26, 1196 (2025).
-
Gober, H. J. et al. Human T cell receptor gammadelta cells recognize endogenous mevalonate metabolites in tumor cells. J. Exp. Med. 197, 163–168 (2003).
-
Wang, R. et al. Antibody-drug conjugates (ADCs): current and future biopharmaceuticals. J. Hematol. Oncol. 18, 51 (2025).
-
Mosch, R. & Guchelaar, H. J. Immunogenicity of monoclonal antibodies and the potential use of HLA haplotypes to predict vulnerable patients. Front. Immunol. 13, 885672 (2022).
Acknowledgements
This research was funded by the Fondazione AIRC (IG-21648 to A.P.) and Ricerca Corrente from the Italian Ministry of Health 2025 to L.R. The authors thank Villa Montallegro Spa for the generous donation that supported this work.
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cuffaro, D., Di Gregorio, L., Mangini, C. et al. Anti-tumor efficacy and Vδ2 T-cell activation via EGFR antibody-drug conjugates featuring novel aminobisphosphonates. Sci Rep 15, 38774 (2025). https://doi.org/10.1038/s41598-025-22669-w
-
Received:
-
Accepted:
-
Published:
-
Version of record:
-
DOI: https://doi.org/10.1038/s41598-025-22669-w