
Introduction
Angelman syndrome (AS) is a severe neurodevelopmental disorder caused by loss of maternal (mat)UBE3A in the brain [1,2,3,4,5,6,7]. In neurons, a long non-coding RNA (lncRNA) called Ube3a-ATS silences paternal (pat)UBE3A in cis [8, 9], making matUBE3A the sole source of neuronal UBE3A. Current clinical trials for AS utilize antisense oligonucleotides (ASOs) to target and downregulate Ube3a-ATS and unsilence patUBE3A [10,11,12,13]. These ASOs require repeated intrathecal injections, which could limit their use to only those who live near major medical centers [10,11,12,13,14].
Our group recently developed a CRISPR/Cas9 AAV vector that targets small nucleolar (sno) RNA C/D box cluster 115 (Snord115) repeats to enduringly unsilence patUBE3A (>17 months) [15]. We delivered this vector to an AS mouse model with a single or dual intracerebroventricular (i.c.v.) injection early in life (embryonic day 15.5 [E15.5] and/or postnatal day 1 [P1]), as UBE3A may play an essential role during neurodevelopment [16, 17]. AAV is currently the most well-studied and effective gene therapy delivery vector for central nervous system disorders, particularly because biodistribution in the brain is widespread [18,19,20,21,22,23]. However, AAV has a packaging limit of ~4.6 kilobases (kb) [24]. Despite being a promising therapeutic tool, Cas9 is large (~3–4 kb, depending on Cas9 variant) relative to the ~4.6 kb AAV packaging limit. Another genome-modifying tool, ZFNs, is smaller (~2 kb including the both binding domains and both FokI nuclease domains) and may be better suited for delivery with AAV. Another potential limitation of Cas9 therapies involves its safety profile. Up to 78% of humans possess pre-existing immunity to Cas9 [25, 26], potentially rendering these therapies ineffective.
Genome editors, such as ZFNs, hold promise for introducing permanent changes into the genome. This leads to expression effects that persist for at least several months, demonstrated by our use of active CRISPR-Cas9 nuclease [15]. One group recently utilized a non-nuclease zinc finger artificial transcriptional factor (ATF) containing a Kruppel-associated box (KRAB) domain, targeted to the promoter region of Ube3a-ATS to facilitate its downregulation [27, 28]. This vector unsilenced patUBE3A up to 5 weeks in an adult model of AS with a single injection. However, it also downregulated levels of Snord116, which can lead to Prader-Willi syndrome (PWS). Another group employed a Cas13 strategy, targeted to the RNA of Ube3a-ATS and avoiding cleavage of the DNA. This vector unsilenced patUBE3A up to 4 months in vivo [29]. While these treatments hold promise, the persistence of epigenome and RNA-targeting editing remains largely understudied in neurons and in the brain [30, 31], particularly in a long-lived species like humans.
Multi-targeting AAV vectors knockdown Ube3a-ATS more effectively than single-target vectors [15, 32]. Here, we designed and tested ZFNs that target multiple Snord115 repeats while avoiding pairs that had predicted off-target sites elsewhere in the genome. The small size of ZFNs allowed us to package a ZFN pair into a single bicistronic AAV vector and evaluate efficacy of Ube3a-ATS knockdown and patUBE3A unsilencing in vitro and in vivo.
Results
Multi-target zinc finger nucleases unsilence paternal UBE3A in cultured neurons
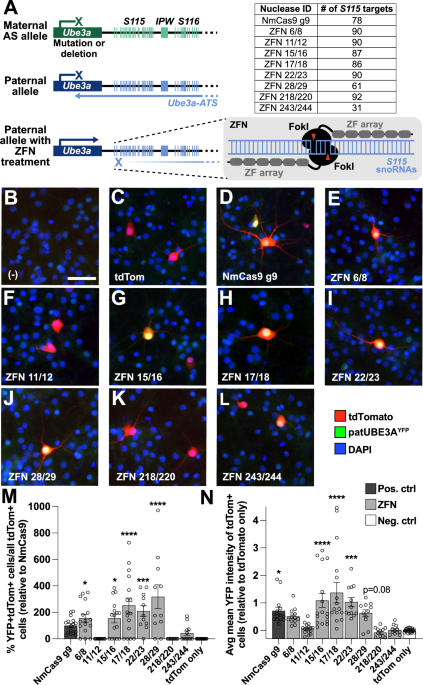
Active Cas9 nuclease multi-targeted to a repetitive Snord115 snoRNA region in Ube3a-ATS can unsilence patUBE3A in neurons [15]. We first sought to determine if active ZFNs targeted to this repetitive Snord115 region were also capable of knocking down Ube3a-ATS and unsilencing patUBE3A as a potential therapeutic tool for AS. We obtained eight pairs of ZFNs (CompoZr® custom ZFN platform; MilliporeSigma), each targeting various sequences and numbers of Snord115 repeats (Fig. 1A; Supplementary Table 1). We selected ZFN pairs that target numerous Snord115 repeats, while avoiding predicted off-target sites elsewhere in the genome (based on PROGNOS analysis) [33]. We then transiently transfected ZFNs into primary cortical neuron cultures from Ube3am+/patYFP mice. These mice contain a YFP knocked-in to the paternally-inherited Ube3a allele to visualize (Fig. 1B–L) and quantify (Fig. 1M, N) unsilencing of patUBE3A-YFP [15, 34,35,36,37,38]. As a positive control, we also transfected neurons with a Neisseria meningitidis Cas9 (NmCas9) guide RNA (gRNA) plasmid (Fig. 1D) that multi-targets the Snord115 region and unsilences patUBE3A-YFP [32]. In all wells, except for our negative control (Fig. 1B), we co-transfected with a CaMKII:tdTomato (tdTom) expression plasmid to identify transfected neurons (Fig. 1C–L). While two ZFN pairs did not unsilence patUBE3A-YFP (Fig. 1F, K, M, N), several pairs unsilenced as well or better than our positive control NmCas9 gRNA (Fig. 1E, G–J, L–N). The ZFN 243/244 pair did not unsilence patUBE3A-YFP effectively, despite the target region for this pair overlapping with the SaCas9-Sajw33 gRNA target region that robustly knocks down Ube3a-ATS in cultured neurons and in mice [15]. ZFN pair 28/29 yielded the highest percentage of YFP+tdTom+ cells (Fig. 1M), and ZFN pair 17/18 yielded the highest average mean YFP intensity of all tdTom+ cells (Fig. 1N), prompting us to select these pairs for subsequent experiments. These results indicate that ZFNs multi-targeted to Snord115 repeats can unsilence patUBE3A.

A The Ube3a/Ube3a-ATS locus (left), illustrating where multi-target ZFN pairs target Snord115 (S115) repeats within Ube3a-ATS, and how many S115 repeats each pair targets (right). S116, Snord116 repeats. IPW, imprinted Prader-Willi region. Representative images of Ube3am+/patYFP neurons (B) not transfected, transfected with (C) CamkIIa promoter:tdTomato (tdTom) only, or co-transfected with tdTom and (D) NmCas9 as a positive control, or (E–L) with a pair of EF-1-alpha promoter:ZF binding domain:Fok1 nuclease plasmids, where each plasmid carries one half of the ZFN pair. Nuclei are shown in blue (DAPI). Scale bar, 50 μm. M, N Quantification of co-stained images as shown in (C–L). n = 11–24 culture wells per condition, 4 images quantified per well. Error bars, standard error of mean (S.E.M.). One-way ANOVA, Dunnett’s multiple comparison test to tdTom condition; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Given that future applications in vivo would require an efficient delivery system, we next cloned our two best-performing multi-target ZFN pairs into bicistronic AAV vectors, where a T2A peptide separated each zinc finger array (Fig. 2A; pAAV-ZFN17/18 and pAAV-ZFN 28/29) [39]. We also tested NmCas9 g9 in an AAV vector as a positive control (Fig. 2A; pAAV-NmCas9 g9). We used a human synapsin 1 (hSyn1) promoter to express Cas9 and ZFNs selectively in neurons [40, 41]. After packaging each vector into AAV2/1, we transduced Ube3am+/patYFP primary cortical neuron cultures with different viral titers (Fig. 2B). We incubated neurons with virus from 3 days-in-vitro (DIV) to 10 DIV and collected total RNA to evaluate gene expression changes. Ube3a-ATS knockdown and patUbe3a unsilencing in neurons treated with ZFN17/18 were comparable to our NmCas9 g9 positive control, particularly at the highest multiplicity-of-infection (MOI) tested (50000 MOI; Fig. 2C, D). In contrast, ZFN 28/29 marginally knocked down Ube3a-ATS and unsilenced patUbe3a at the highest titer tested (Fig. 2C, D). All vectors expressed their respective nuclease domain (Supplementary Fig. 1A), and both ZFN vectors expressed both FokI domains (Supplementary Fig. 1B, C). ZFN17/18 and NmCas9 g9 also downregulated Snord115 snoRNA expression (Fig. 2E). Neither ZFN nor NmCas9 significantly downregulated expression of upstream Snord116 snoRNAs (Fig. 2F), deletion of which is associated with Prader-Willi syndrome [42,43,44].

A Design of AAV hSyn1 promoter:NmCas9 (positive control) and AAV hSyn1 promoter:ZFN pairs, where each binding domain-FokI domain are separated by a T2A peptide. Vectors are drawn to scale. B Schematic of mouse cortical neuron cultures untreated or treated at different MOIs, with numbers representing VP/cell. Vectors tested: AAV2/1-NmCas9 with a multi-target gRNA (g9), multi-target ZFN17/18, or multi-target ZFN28/29. RT-qPCR to evaluate expression of (C) Ube3a-ATS, (D) patUbe3a, (E) Snord115 snoRNAs, and (F) upstream Snord116 snoRNAs 7 days following treatment. Normalized to Eif4a2 expression, n = 2–3 culture wells per condition. Error bars, S.E.M. Two-way ANOVA with Dunnett’s multiple comparisons test to g9 treated for main treatment effect; n.s. non-significant, **p < 0.01, ****p < 0.0001. Two-way ANOVA with Dunnett’s multiple comparisons test to 0 MOI treated for simple effects within treatment (across MOI); *p < 0.05, ****p < 0.0001. [inhibitor] or [agonist] vs normalized response non-linear regression analysis for curve fitting. Dashed line indicates normalized values of no virus control, for each dataset.
Active zinc finger nuclease outperforms nickase and inactive ZFNs to knockdown Ube3a-ATS and unsilence paternal Ube3a
Previous work demonstrates the potential use of nickase ZFNs in gene therapies [45]. Moreover, using CRISPR/Cas9 therapeutics, the D10A/H840A-mutated Streptococcus pyogenes Cas9 (SpCas9) mutant can create a transcriptional blockage when bound to DNA [46], and the catalytically inactive NmCas9 (D15A/H587A/N610A) can block Ube3a-ATS transcription to unsilence patUbe3a (data unpublished; H.O.B.). Therefore, we sought to evaluate the extent to which nickase and inactive ZFNs unsilenced patUbe3a relative to active ZFNs. To test this, we mutated our bicistronic pAAV-ZFN17/18 plasmid (Fig. 3A) to contain an amino acid change (Asp450 to Ala; D450A) within the FokI domain at position 450 (relative to the native FokI enzyme) that resulted in catalytically inactive FokI domains (Fig. 3B) [47,48,49]. It is important to note that this ZFN system contains modified FokI domains to promote obligate heterodimerization, as described [50]. We also constructed nickase plasmids where we mutated either the zinc finger (ZF)17 FokI domain (Fig. 3C) or the ZF18 FokI domain (Fig. 3D); the FokI domains would still heterodimerize but would only nick the strand bound by the active array [47]. We then tested whether these mutated constructs cleaved (linearized plasmid) or nicked (relaxed plasmid) DNA in a cleavage/nickase assay (see “Methods”; Fig. 3E) [51], using Nt.BbvCI endonuclease as a positive control for nicking, and SphI as a positive control for double-strand cleavage. As expected, the active ZFN (Fig. 3A) linearized the substrate similarly to SphI (Fig. 3F, G). The nickase versions (Fig. 3C, D) relaxed the substrate similarly to Nt.BbvCI (Fig. 3F, G). The inactive ZFN version (Fig. 3B) did not appear to nick or cleave the plasmid DNA (Fig. 3F, G). Together, these results demonstrate that the zinc finger FokI mutants function as expected.

Schematic of A active ZFN17/18 that cleaves genomic DNA, or various mutated ZF arrays to abolish FokI nuclease activity (D450A) from B both ZF arrays to inactivate the domain, or C, D on one ZF array to nick the genomic DNA on the indicated strand. Amino acid changes are noted in each panel for each ZF array. Arrows indicate FokI ability to nick the indicated strand. E Schematic of in vitro cleavage/nickase assay (IVCA/IVNA). F IVCA/IVNA using plasmid DNA substrate incubated with active (+/+), nickase (+/− or −/+), or inactive (−/−) zinc finger lysates produced in transfected HEK293-T cells. Nt.BbvCI, nicking endonuclease; SphI, cleaving restriction endonuclease. Samples were run on the same gel and imaged together, but were in noncontiguous lanes. G ImageJ quantification of relative band intensities of IVCA/IVNA shown in (F). H Schematic of mouse cortical neuron cultures untreated or treated with 1.0×105 VP/cell of AAV2/1-ZFN vectors shown in (A). I qPCR strategy to detect intact target site (top panel) or AAV integration in either orientation (middle and bottom panels) at the ZFN17/18 target sites following viral transduction. Quantification of J intact target site and K AAV integration. RT-qPCR to evaluate expression of L Ube3a-ATS, M patUbe3a, and N FokI 7 days following treatment. All qPCR data normalized to Eif4a2 expression, n = 3 culture wells per condition. Error bars, S.E.M. One-way ANOVA with Dunnett’s multiple comparisons test to untreated wells; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Dashed line indicates normalized values of no virus control for each dataset.
Next, we packaged each construct into AAV2/1 and then transduced these viruses onto Ube3am+/patYFP primary cortical neuron cultures at an MOI of 1.0 × 105 (Fig. 3H). We then collected genomic DNA (gDNA) and RNA for target site (Fig. 3I) and gene expression analyses. After performing qPCR on gDNA, we observed significant disruption of the 17/18 target site following treatment with active ZFN (+/+), but not with nickase or inactive ZFNs (Fig. 3I top; 3J). We and others previously observed AAV integration into DSBs produced by active Cas9, primarily through fusion of the AAV inverted terminal repeat (ITR) with the host genome [15, 32, 52,53,54]. While we observed AAV integration into DSBs produced by active ZFN, AAV did not integrate into nicked DNA, as others have corroborated [55], and we did not observe integration following treatment with inactive ZFN (Fig. 3I middle, bottom; 3K). Treatment with nickase ZFN marginally decreased Ube3a-ATS expression, though not statistically significant (Fig. 3L). Nickase ZFNs also significantly upregulated levels of patUbe3a, though not to levels seen when treating with active ZFN (Fig. 3M). FokI expression, indicative of AAV vector expression, was similar across conditions tested (Fig. 3N). Together, these results indicate that the active ZFN17/18 vector potently reduces Ube3a-ATS and unsilences patUbe3a when compared to inactive and nickase ZFNs targeted to the same genomic site.
Given that AAV integrates at on-target sites using active ZFN (Fig. 3K) and elsewhere in the genome where double-stranded breaks occur [56], we evaluated the extent to which the ZFN17/18 vector integrated at predicted off-target sites. We first examined the top six predicted off-target sites with the lowest number of mismatches total (determined by PROGNOS analysis [33]; see “Methods”; Supplementary Fig. 2A, Supplementary Table 2) and for which the ZF 17 and 18 arrays would heterodimerize. We observed no off-target AAV integration at measured predicted sites and only observed AAV integration at the on-target site (Supplementary Fig. 2B, C). While this zinc finger system contains mutations in the FokI domain to prevent homodimerization [50], we next performed qPCR to determine if AAV integrated at any of the sites where ZF17 or ZF18 homodimers were predicted to bind (Supplementary Fig. 2A, Supplementary Table 2). Again, we observed no off-target AAV integration (Supplementary Fig. 2B, C). These data further support the specificity of the ZFN system.
Multi-target ZFN17/18 unsilences paternal UBE3A in a mouse model of Angelman syndrome
We next sought to determine if the multi-target active AAV-ZFN17/18 vector could unsilence patUBE3A in the brain of a mouse model of AS. After packaging into AAV9, a commonly used serotype that transduces cells of the brain [57,58,59,60], we intracerebroventricularly (i.c.v; bilaterally) injected Ube3am+/p+ (Wt) and Ube3am−/p+ (AS) mice at P1 with 5.0 × 1010 viral particles (VP) per ventricle of ZFN17/18 AAV9 virus, or with an identical volume of vehicle (Veh; Fig. 4A; see “Methods”). AS-model mice (Ube3am−/p+) contain a maternal deletion of Ube3a, while the paternal Ube3a allele is intact [1]. We found that ZFN17/18 unsilenced patUBE3A in the cerebral cortex (Fig. 4B, left-most column), the hippocampus (Fig. 4C), and other brain regions (Supplementary Fig. 3A–C). The percentage of UBE3A-positive neurons in the cortex (Fig. 4D) and hippocampus (Fig. 4E) were 20.5% and 15.3%, respectively. UBE3A protein intensity levels in individual neurons from AS-ZFN-treated mice reached that of individual neurons in Wt-Veh-treated mice in both the cerebral cortex and hippocampus (Fig. 4F). Quantification of relative protein levels further demonstrates UBE3A unsilencing in the cortex of AS mice following ZFN treatment (Fig. 4G). ZFN17/18 treatment significantly downregulated Ube3a-ATS and Snord115 expression, but did not alter IPW, Snord116, or Snrpn, which are located upstream of the ZFN17/18 target sites (Fig. 4H). Treatment also did not affect NeuN expression, a marker for post-mitotic neurons not located within the Ube3a locus [61]. We also observed upregulated expression of Ube3a in the cortex, consistent with unsilenced patUbe3a (Supplementary Fig. 3D). FokI expression was statistically similar across all animal groups treated with the ZFN17/18 vector (Supplementary Fig. 3E). We also detected a positive correlation between FokI expression and patUbe3a unsilencing in AS-model mice, with variation possibly being from technical differences during injection (Supplementary Fig. 3F). These data highlight the potential of ZFN-based strategies for the treatment of AS in vivo.

A Schematic of Ube3am+/p+ (Wt) or Ube3am−/p+ (AS-model) mice injected i.c.v. at P1 with vehicle or AAV9:ZFN17/18 vector. Brain collected at P90. Representative images of B cortical column and C hippocampus immunostained with antibodies to UBE3A and B (bottom) NEUN. Magnification of boxed region. Scale bar, 200 μm. Quantification of the percentage of UBE3A+ neurons to total neurons in D cortical column and E hippocampus. Line indicates average of each dataset. n = 6–9 mice per condition. One-way ANOVA with Tukey’s multiple comparisons test; p-value reported if less than 0.05, ****p < 0.0001. F Mean UBE3A intensity per neuron in all mice. Line indicates average of each dataset. n = 6–9 mice per condition; n = 24,642–43,158 neurons per condition. One-way ANOVA, Tukey’s multiple comparisons test to Wt Veh condition, per brain region; ****p < 0.0001. G Western blot of (untagged) UEB3A protein levels in cortex of Wt and AS mice treated with Veh, or AS mice treated with AAV9:ZFN17/18. Loading control, GAPDH. The average (avg.) ratio of UBE3A to GAPDH protein per group is reported. H RT-qPCR to evaluate expression of Ube3a-ATS adjacent to Ube3a (1- ATS and 2 – ATS), Snord115, IPW, Snord116, Snrpn, and NeuN in AS mice following treatment with Veh or AAV9:ZFN17/18. Inset diagram illustrates primer binding sites along Ube3a-ATS transcript; numbering corresponds to numbering on graph x-axis. n = 10–12 mice per condition. Unpaired two-tailed Student’s t-test within each expression set; n.s. non-significant, **p < 0.01. For (D, H), whiskers, min and max; bounds of boxes, 25th and 75th percentiles; line within boxes, median.
Discussion
Genome editor-based therapeutics have the potential to enduringly treat life-long disorders such as AS [15, 27, 29]. CRISPR-Cas9 and ZF-ATFs are among several therapies to show promise in AS mouse models [15, 27], each presenting advantages and limitations. Our study used ZFNs, a previously unexplored gene editing tool for AS, as a paralleled approach that successfully unsilenced patUBE3A in neurons and in the brain.
Cas9 target recognition and induction of DSBs is distinct from induction by ZFNs [62,63,64,65]. Regardless, knockdown of Ube3a-ATS and patUbe3a unsilencing was comparable when using either Cas9 or ZFNs at multiple MOIs tested in culture (Fig. 2). This highlights the common principle that using an active nuclease editor could be a potent therapy for AS. Catalytically inactive Cas9 can impair transcriptional processes [46, 66, 67]. This roadblock potentially arises from stable hybridization of the guide RNA (gRNA) and the target DNA, forming an R-loop capable of interfering with the native transcribing RNA polymerase [67,68,69,70]. ZFNs would instead form a protein-DNA interaction with target DNA. While understudied, ZFN binding and searching kinetics may influence stability of a potential roadblock [71]. We found that a catalytically inactive ZFN, which still presumably binds to DNA and dimerizes the inactive FokI domains [49], was unable to knockdown Ube3a-ATS transcription (Fig. 3L). In addition to DSB repair leading to Ube3a-ATS transcription-blocking edits [15], transcriptional roadblocks specific to a Cas9-gRNA:DNA complex may contribute to knockdown with Cas9 therapeutics but not ZFN therapeutics.
We also tested whether nickase ZFNs were capable of disrupting Ube3a-ATS to unsilence patUbe3a. Researchers have explored repair mechanisms/outcomes following the use of dual-nickase Cas9 systems [55], use of nickase-based prime editing [72], or use of nickases in dividing cells [73]. However, it is unclear how the repair events of nicked DNA or persistent nicks in post-mitotic neurons may disrupt Ube3a-ATS transcription. We found that nickase ZFNs only modestly knocked down Ube3a-ATS (Fig. 3L) and unsilenced patUbe3a (Fig. 3M), regardless of the strand nicked. This suggests an inefficiency of nicks versus DSBs to introduce editing events capable of blocking transcription. Quantifying the editing outcomes at ZFN target sites following treatment of neurons with active, inactive, and nickase ZFNs, as well as exploring the binding and stability of ZFN protein on target DNA, could be useful in understanding the mechanism by which these various ZFN species are capable, or incapable, of knocking down Ube3a-ATS. It is important to note that we performed our studies by multi-targeting a highly repetitive region of within Ube3a-ATS, the Snord115 cluster, which we previously identify as potentially necessary to achieve maximal knockdown of Ube3a-ATS [15, 32]. Snord115 repeats are highly homologous, some indistinguishable or differing by only a few base-pairs [74]. This complicates editing analysis, making it difficult to isolate targetable versus non-targetable repeats and edited versus non-edited repeats. Future studies utilizing single-target ZFNs will be essential to characterize editing event frequencies.
Snord115 repeat-targeting strategies may be necessary to potently terminate Ube3a-ATS transcription to unsilence paternal UBE3A [15, 32]. Typical Snord115 function is elusive. In mice, Cas9 targeted to Snord115 repeats does not alter expression of Snord115 target genes [15], compromise putative function [75, 76], or lead to overt phenotypic changes [77]. In humans, SNORD115 deletion during differentiation may affect global transcription and translation [78]. Phenotypic consequences of disruption in post-mitotic neurons, as in this study, will be important to explore, particularly in a human-derived model system.
Potential treatments involving active Cas9, ZF-ATFs, and Cas13 achieved UBE3A protein unsilencing of 37%, 25%, and 35.9%, respectively, in the brains of AS-model mice (Supplementary Table 3). Treating AS-model mice with ZFNs led to 18.5% unsilencing in the cortex (Fig. 4G). While our treatment did not achieve unsilencing levels to those of previous treatments, differences may be due to protein quantification method used, timing of injection, length of treatment, total viral particles delivered, and delivery vector used (Supplementary Table 3). We observed a significant positive correlation between UBE3A unsilencing and vector expression in the cortex (Supplementary Fig. 3F), suggesting we may be able to develop a treatment to better unsilence UBE3A by increasing the number of viral particles delivered or by improving the efficiency of the delivery system used. Our treatment unsilences endogenous UBE3A and therefore has the potential to rescue AS-related phenotypes in vivo.
Active Cas9 delivered with AAV unsilenced UBE3A for at least 17 months [15], compared to the use of a non-nuclease ZF-ATF that only unsilenced up to 5 weeks [27]. AAV integration into DSBs in the host genome may contribute to long-term expression of a transgene [79], and this could enhance the durability of a nuclease-based therapy. We also observed AAV integration into ZFN target sites (Fig. 3K), as we did with an AAV-Cas9 vector [15, 32]. While such modifications to the genome are expected to be permanent, the durability of paternal UBE3A unsilencing in vivo could be evaluated in future studies. We did not observe signs of neurotoxicity when an AAV-active Cas9 vector was administered to mice[15]. Long-term complications of insertional mutagenesis are theoretically possible and will be important to evaluate when any AAV-genome editor vector is advanced clinically [80].
O’Geen et al. developed an epigenetic-based therapy for AS targeting the promoter of Snrpn/Snurf [27, 28]. However, this approach also leads to downregulation of Snrpn/Snurf and Snord116. Prenatal loss of Snord116 is associated with Prader-Willi syndrome [81]. O’Geen et al. treated adult AS mice, observed partial reduction in Snord116, and did not observe adverse phenotypes. However, reinstating or rescuing UBE3A early may be important, as UBE3A may play a critical role in neurodevelopment [16, 17, 82]. To this end, treating prenatally or embryonically with an epigenome editor targeted to the Snrpn/Snurf promoter may result in more dramatic phenotypic outcomes of Snord116 downregulation. Therefore, therapies delivered early in life (Fig. 4A), that are not promoter-targeting (Fig. 1A), and that avoid significant downregulation of upstream Snord116 expression, such as the ZFN vector we developed (Fig. 4H), may be ideal for development of an effective therapy for AS.
Methods
Expression plasmids
ZFN transfection experiments used pairs of ZFN plasmids generated by the CompoZr® Custom ZFN Service (MilliporeSigma, Burlington, MA, USA; pZFN-EF1α::3xFLAG-NLS-Binding domain-FokI domain-bGHpA); plasmids were custom-designed to exclude fluorescent tags and to drive expression via an EF1α promoter. We co-transfected pLenti-Camk2a-tdTomato (tdTom) to visualize positively transduced neurons, as previously described [15, 83]. Cas9 AAV experiments used pAAV-NmCas9-gRNA (pAAV-ITR-SapI-sgRNA::U6:hSyn::MVM-2xNLS-NmCas9-2xNLS-bGpA-ITR), as previously described [32]. We constructed ZFN AAV plasmids as follows: two monomers from a ZFN pair were cloned into a single bicistronic plasmid separated by a T2A peptide (pZFN-EF1α::3xFLAG-NLS-Binding domain1-FokI domain1-T2A-3xFLAG-NLS-Binding domain2-FokI domain2-bGHpA). We then used the backbone of pAAV-NmCas9-gRNA to create pAAV-ZFNs expressing from a single vector and driven by a neuron-specific human synapsin 1 (hSyn) promoter (pAAV-ITR-hSyn::MVM-3xFLAG-NLS-Binding domain1-FokI domain1-T2A-3xFLAG-NLS-Binding domain2-FokI domain2-bGpA-ITR). For nickase and inactive ZFN D450A mutants, we performed site-directed mutagenesis on pZFN17 and/or pZFN18 plasmids (F primer: cctgCcggcgccatctatacagtgg; R primer: ctttctgcttccgcccagg). We then performed cloning as above, to construct bicistronic plasmids of the following: inactive pAAV-ZFN17(D450A)-ZFN18(D450A), nickase pAAV-ZFN17(D450A)-ZFN18, and nickase pAAV-ZFN17-ZFN18(D450A). Plasmidsaurus performed whole plasmid sequencing using Oxford Nanopore Technology with custom analysis and annotation to confirm all plasmid sequences.
Mouse primary cortical neuron cultures
Ube3am+/pYFP E15.5 mouse primary cortical neuron cultures were prepared using previously described methods [36, 84, 85]. For immunocytochemistry experiments, we plated cortical neurons onto 384-well poly-D-lysine-coated plates at 3.5 × 104 cells per well. For all other neuron experiments, cells were plated onto 24-well plates at 3.5 × 105 cells per well. Neurons were maintained in neurobasal medium (12348017, Gibco, Boston, MA, USA) supplemented with B-27 Plus (A3582801, Gibco), GlutaMAX, and 5′-fluoro-2′-deoxyuridine (F0503, Sigma). For immunocytochemistry experiments, on days-in-vitro 3 (DIV3), neurons were transiently transfected using Lipofectamine 2000 transfection reagent (11668027, Invitrogen, Waltham, MA, USA) and 50 ng of each plasmid (two pZFNs, one of each monomer per pair, and tdTom plasmid). After 1 h, a complete media change was performed using conditioned neurobasal medium from adjacent, untreated wells. On DIV10, neurons were processed for immunocytochemistry experiments (see below). For AAV dosage experiments, neurons were transduced with either 0, 1.25 × 104, 2.5 × 104, or 5.0 × 104 virus particles/cell (VP/cell). For all other AAV experiments, neurons were transduced with 1.0 × 105 VP/cell. Cells were centrifuged at 4000 rpm for 5 min at 37 °C. On DIV10, we collected cells for downstream analyses.
Ethical approval
Use of all animals in these experiments was approved by the Institutional Animal Care and Use Committee and the University of North Carolina at Chapel Hill, and was used in accordance with the US NIH guidelines.
Animal use and brain injections
All culture experiments were performed using wild-type (Wt) and Ube3am+/pYFP mice. All in vivo experiments were performed using Wt and Ube3am−/p+ mice. All mice were on the C57BL/6J background. For brain injections, we diluted stock virus (BRAIN Initiative Viral Vector Core, Chapel Hill, NC, USA) in PBS with 5% D-Sorbitol and 0.5 mg/mL fast green dye. We placed P1 neonatal pups on ice to induce hypothermic anesthesia as previously described [86]. Each pup received bilateral i.c.v. injection of 1.0 × 1011 AAV9-ZFN17/18 VP total (1 μL per ventricle at 5.0 × 1010 VP/ μL) or 1 μL of vehicle (sorbitol, PBS, and fast green mixture identical to that of the ZFN mixture). Pups were placed on a heating pad for recovery and then returned to their dam. For each litter, half of the males and half of the females were randomly selected to receive ZFN17/18; the other half received vehicle. At weaning age, the mice were genotyped and separated by sex. Our study examined male and female mice, and similar findings are reported for both sexes. Collection of these mice was carried out blind to genotype and treatment. No animals were excluded from these studies. Sample size for animal use was based on previously published work.
Immunocytochemistry and analysis for UBE3A unsilencing screen in cultured neurons
To identify ZFN pairs capable of unsilencing patUBE3A-YFP, transiently transfected neurons were collected and fixed in 4% paraformaldehyde (PFA) in PBS, permeabilized/blocked in 10% normal donkey serum (NDS) in TBST, and immunostained with rabbit anti-GFP (NB600-308, Novus, Littleton, CO, USA; to identify patUBE3A-YFP signal) overnight at 4 °C with gentle shaking. The next day, cells were washed several times with PBS and incubated with secondary donkey anti-rabbit IgG AlexaFluor 647 (A31573, Thermo Fisher, USA) and DAPI (EN62248, Invitrogen). Images were acquired using a GE IN CELL Analyzer 2200 high-content imager, imaging at 10× magnification with 4 fields imaged per well. We quantified the percentage of YFP-positive tdTom-positive cells to total tdTom-positive cells, as well as the average mean YFP intensity of all tdTom-positive cells using a custom CellProfiler pipeline [87].
Viral production for in vitro and in vivo experiments
AAV for culture experiments was produced using previously described methods [88, 89]. Briefly, HEK293-T cells (CRL-3216, ATCC; tested for mycoplasma and STR profiled by ATCC) were cultured in 12-well plates seeded at 3.5 × 105 cells per well and maintained in DMEM (11995065, Gibco) with 10% fetal bovine serum (FBS). After 24-h, when cells were about 50–80% confluent, cells were co-transfected with 400 ng of pAAV-Cas9-gRNA or pAAV-ZFN plasmid, 400 ng of AAV2/1 serotype plasmid (a gift from James M. Wilson; 112862, Addgene, Watertown, MA, USA), and 800 ng of pAdDELTAF6 helper plasmid (a gift from James M. Wilson; 112867, Addgene) using FuGENE 6 (E2691, Promega). 16–24 h post-transfection, media was replaced with DMEM (11960044, Gibco) with 10% FBS and 1% GlutaMAX (35050061, Gibco). 48 h after media change, media (containing AAV) was collected and centrifuged at 5000 rpm for 1 min through 0.45 μM filters (8162, Costar, USA). Virus was then stored at 4 °C and used within 2 weeks. For i.c.v. brain injections, the BRAIN Initiative Viral Vector Core at UNC Chapel Hill produced stock virus, as described [90, 91].
RNA extractions and RT-qPCR
For RNA extractions from cell culture experiments, cells were first washed with PBS and then collected in TRIzol (15596026, Invitrogen). For in vivo experiments, one hemisphere was isolated, microdissected for cortex and hippocampus, and flash-frozen on dry ice; the other hemisphere was processed for UBE3A staining analysis (see below). Upon extraction, frozen tissue was dounce homogenized with TRIzol. Total RNA was treated with ezDNase and reverse transcribed using SuperScript IV VILO (11766050, Invitrogen). All RT-qPCR experiments were performed using SsoAdvanced Universal SYBR Green Supermix (1725271, Bio-Rad, Hercules, CA, USA) on a QuantStudio5 (Applied Biosystems, CA, USA). Primers used are listed in Supplementary Table 4. We analyzed all data using the 2−ddCT method, normalized internally to Eif4a2, and normalized globally to untreated samples.
In vitro cleavage/nickase assay for ZFN mutation species
Crude protein production
Protein lysate was prepared, as described [92]. Briefly, HEK293-T cells were cultured in 12-well plates seeded at 3.0 × 105 cells per well and maintained in DMEM (11995065, Gibco) with 10% FBS. After 24-h, when cells were 50–80% confluent, cells were transfected with 2000 ng of active, inactive, or nickase ZFN plasmid using FuGENE 6 (E2691, Promega, Madison, WI, USA). After 24-h, 1 mL of DMEM was added, and allowed to incubate for 24 more hours. Cells were then washed with PBS and dissociated with TrypLE Express (12605-010, Gibco). After centrifugation (1000 × g for 1 min at room temperature), cell pellets were washed with PBS and then resuspended in 400 μL of PBS. Resuspensions were sonicated on ice for 8 s (30% amplitude), centrifuged for 20 min 16,000 × g at 4 °C, and reserved the supernatant as the lysate containing active, inactive, or nickase ZFN protein for downstream IVCA/IVNA.
Cleavage/nickase assay (IVCA/IVNA)
We prepared plasmid substrate, containing a target site for ZFN17/18, under 4 °C conditions to maximize production of supercoiled plasmid [93]. We then incubated protein lysate (produced above) with 200 ng of plasmid substrate in NEBuffer r3.1 (B6003S, New England Biolabs, Ipswich, MA, USA) for 18 h at 37 °C. Samples were then combined with 6x gel loading dye (B7024S, New England Biolabs) and run on a 0.7% agarose TAE gel with ethidium bromide for 1.5 h for visualization of supercoiled, nicked (relaxed), and linearized plasmid substrate.
DNA extractions and qPCR for detection of AAV integration and intact genome
For DNA extractions for on- and off-target analyses, DNA was collected using the DNeasy Blood & Tissue Kit (69504, Qiagen, Germantown, MD, USA). Primer sets spanning genome:genome target site, as well as different primer sets identifying genome:AAV fusion amplicons were designed to detect AAV integration. All primers are listed in Supplementary Table 4. qPCR and analyses were performed as described previously for RNA experiments.
Immunohistochemistry and analysis for UBE3A unsilencing in vivo
Following collection of mice as mentioned above, one cortical hemisphere was immersion fixed in 4% PFA and PBS for 72 h and then transferred into PBS. Samples were then sagittal sectioned to a thickness of 75 μm on a vibratome (Thermo Fisher Scientific), transferred to 24-well plates containing cryoprotectant solution (45% PBS, 30% Ethylene glycol, 25% Glycerol), and stored at −20 °C. Upon staining, sections were washed in PBS, washed in 0.1 M citrate buffer in distilled water (C9999, Sigma) for 30 min, permeabilized with 0.01 M PBS-T (PBS with 0.3% Triton X-100), and blocked with 10% NDS, 2% DMSO in PBS-T for 1 h at room temperature with gentle shaking. We then incubated blocked sections with mouse anti-UBE3A (1:600; SAB1404508, Sigma-Aldrich) and guinea pig anti-NEUN (1:600; ABN90P, Sigma-Aldrich) overnight at 4 °C with gentle shaking. The next day, we washed sections with PBS-T and incubated them with goat anti-mouse IgG2a AlexaFluor 488 (A21131, Invitrogen), donkey anti-guinea pig IgG AlexaFluor 647 (706-605-148, Jackson), and DAPI (EN62248, Invitrogen). Sagittal immunostained sections were then imaged via laser-scanning confocal microscopy (Zeiss LSM 710). Images were quantified (% UBE3A+ neurons and mean UBE3A intensity per neuron) with a custom CellProfiler pipeline [87].
Off-target analysis for ZFN17/18
We first identified in silico predicted off-target sites for ZFN17/18 using PROGNOS [33]. We defined our search to output all possible predicted off-target sites, regardless of number of mismatches, that would allow for an optimal 5–7 bp spacer length [94, 95]. A full list of off-target sites can be found in Supplementary Table 5. 86 sites were identified as exact targets (the expected 86 Snord115 repeat targets). 50 off-target sites were identified within the Snord115 cluster region, up to but not including Ipw, containing 1–7 mismatches total per heterodimer. These sites were not interrogated due to the repetitive, highly homologous sequence of the Snord115 repeats, and would add to the multi-target nature of this approach should ZFN17/18 target them. For off-target AAV integration analysis, off-target sites were selected based on the lowest number of mismatches total and based on the output “ZFN score” [33]. Three sites were selected where ZF monomers would homodimerize (17/17 or 18/18), and six sites were selected where ZF monomers would heterodimerize (17/18).
Western blot for detection of UBE3A in cortex
Frozen cortices from one hemisphere were homogenized in radioimmunoprecipitation assay (RIPA) buffer (25 mM Tris pH: 8.0, 150 mM NaCl, 2% sodium dodecyl sulfate (SDS), 0.5% sodium deoxycholate, 1% Triton X-100) with protease (78429, Halt, Thermo Fisher Scientific) and phosphatase (78426, Halt, Thermo Fisher Scientific) inhibitors. Equal amounts of protein, determined by BCA protein assay (23225, Pierce, Thermo Fisher Scientific), were separated on an SDS gel (4568084, Bio-Rad) and transferred onto a nitrocellulose membrane (1620115, Bio-Rad). Membranes were blocked in 5% milk (sc-2324, Chem Cruz, Dallas, TX, USA) in TBS-T and incubated in rabbit anti-UBE3A (1:500; 10344-1-AP, Proteintech, Rosemont, IL, USA) and rabbit anti-GAPDH (1:2000; 2118, Cell Signaling, MA, USA) overnight at 4 °C with gentle shaking. Proteins were detected by incubation with HRP-conjugated secondary antibody (1:1000; 18-8816-31, TrueBlot, Rockland, Victoria, BC, Canada) and SuperSignal ECL Substrate (34577, Thermo Fisher Scientific). Membranes were imaged using an Amersham Imager 680.
Statistical analyses
All data are presented as mean ± S.E.M. For data comparing two groups with one varying factor, two-tailed unpaired Student’s t-test was used. For data comparing more than two groups with one varying factor, one-way analysis of variance (ANOVA) with Dunnett’s multiple comparisons post-hoc test was used. For data comparing groups with two varying factors, two-way ANOVA with Šídák’s multiple comparison post-hoc test was used. To compare the main effect of treatment across dosage, two-way ANOVA with Tukey’s or Dunnett’s multiple comparisons test was used (depending on the number of groups compared). To compare the effect of dosage within treatment groups, two-way ANOVA with Dunnett’s multiple comparisons post-hoc test was used. Data was considered significant if P < 0.05.
Data availability
All values for all data points in graphs are reported in the Supporting Raw Data file.
References
-
Jiang YH, Armstrong D, Albrecht U, Atkins CM, Noebels JL, Eichele G, et al. Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron. 1998;21:799–811.
-
Kishino T, Lalande M, Wagstaff J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet. 1997;15:70–3.
-
Matsuura T, Sutcliffe JS, Fang P, Galjaard RJ, Jiang YH, Benton CS, et al. De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat Genet. 1997;15:74–7.
-
Sutcliffe JS, Jiang YH, Galijaard RJ, Matsuura T, Fang P, Kubota T, et al. The E6-Ap ubiquitin-protein ligase (UBE3A) gene is localized within a narrowed Angelman syndrome critical region. Genome Res. 1997;7:368–77.
-
Williams CA, Driscoll DJ, Dagli AI. Clinical and genetic aspects of Angelman syndrome. Genet Med. 2010;12:385–95.
-
Williams CA, Beaudet AL, Clayton-Smith J, Knoll JH, Kyllerman M, Laan LA, et al. Angelman syndrome 2005: updated consensus for diagnostic criteria. Am J Med Genet A. 2006;140:413–8.
-
Clayton-Smith J, Pembrey ME. Angelman syndrome. J Med Genet. 1992;29:412–5.
-
Nicholls RD. Genomic imprinting and candidate genes in the Prader-Willi and Angelman syndromes. Curr Opin Genet Dev. 1993;3:445–56.
-
Ledbetter DH, Ballabio A. Molecular cytogenetics of contiguous gene syndromes: Mechanisms and consequences of gene dosage imbalance. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. New York, NY: McGraw-Hill; 1995. pp. 811–39.
-
Clinical Trials. NCT04428281: https://clinicaltrials.gov/study/NCT04428281; Hoffmann-La Roche.
-
Clinical Trials. NCT05127226: https://clinicaltrials.gov/study/NCT05127226; Ionis Pharmaceuticals, Inc.
-
Clinical Trials. NCT06415344: https://clinicaltrials.gov/study/NCT06415344; Ultragenyx Pharmaceutical Inc.
-
Clinical Trials. NCT04259281: https://clinicaltrials.gov/study/NCT04259281; Ultragenyx Pharmaceutical Inc.
-
Dindot SV, Christian S, Murphy WJ, Berent A, Panagoulias J, Schlafer A, et al. An ASO therapy for Angelman syndrome that targets an evolutionarily conserved region at the start of the UBE3A-AS transcript. Sci Transl Med. 2023;15:eabf4077.
-
Wolter JM, Mao H, Fragola G, Simon JM, Krantz JL, Bazick HO, et al. Cas9 gene therapy for Angelman syndrome traps Ube3a-ATS long non-coding RNA. Nature. 2020;587:281–4.
-
Silva-Santos S, van Woerden GM, Bruinsma CF, Mientjes E, Jolfaei MA, Distel B, et al. Ube3a reinstatement identifies distinct developmental windows in a murine Angelman syndrome model. J Clin Investig. 2015;125:2069–76.
-
Sonzogni M, Hakonen J, Bernabe Kleijn M, Silva-Santos S, Judson MC, Philpot BD, et al. Delayed loss of UBE3A reduces the expression of Angelman syndrome-associated phenotypes. Mol Autism. 2019;10:23.
-
Kimura S, Harashima H. Current status and challenges associated with CNS-targeted gene delivery across the BBB. Pharmaceutics. 2020;12:1216.
-
Rincon MY, de Vin F, Duque SI, Fripont S, Castaldo SA, Bouhuijzen-Wenger J, et al. Widespread transduction of astrocytes and neurons in the mouse central nervous system after systemic delivery of a self-complementary AAV-PHP.B vector. Gene Ther. 2018;25:83–92.
-
Burger C, Gorbatyuk OS, Velardo MJ, Peden CS, Williams P, Zolotukhin S, et al. Recombinant AAV viral vectors pseudotyped with viral capsids from serotypes 1, 2, and 5 display differential efficiency and cell tropism after delivery to different regions of the central nervous system. Mol Ther. 2004;10:302–17.
-
Castle MJ, Turunen HT, Vandenberghe LH, Wolfe JH. Controlling AAV tropism in the nervous system with natural and engineered capsids. Methods Mol Biol. 2016;1382:133–49.
-
Hocquemiller M, Giersch L, Audrain M, Parker S, Cartier N. Adeno-associated virus-based gene therapy for CNS diseases. Hum Gene Ther. 2016;27:478–96.
-
Liguore WA, Domire JS, Button D, Wang Y, Dufour BD, Srinivasan S, et al. AAV-PHP.B Administration Results in a Differential Pattern of CNS Biodistribution in Non-human Primates Compared with Mice. Mol Ther. 2019;27:2018–37.
-
Tal J. Adeno-associated virus-based vectors in gene therapy. J Biomed Sci. 2000;7:279–91.
-
Charlesworth CT, Deshpande PS, Dever DP, Camarena J, Lemgart VT, Cromer MK, et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat Med. 2019;25:249–54.
-
Gough V, Gersbach CA. Immunity to Cas9 as an obstacle to persistent genome editing. Mol Ther. 2020;28:1389–91.
-
O’Geen H, Beitnere U, Garcia MS, Adhikari A, Cameron DL, Fenton TA, et al. Transcriptional reprogramming restores UBE3A brain-wide and rescues behavioral phenotypes in an Angelman syndrome mouse model. Mol Ther. 2023;31:1088–105.
-
Bailus BJ, Pyles B, McAlister MM, O’Geen H, Lockwood SH, Adams AN, et al. Protein delivery of an artificial transcription factor restores widespread Ube3a expression in an Angelman syndrome mouse brain. Mol Ther. 2016;24:548–55.
-
Li J, Shen Z, Liu Y, Yan Z, Liu Y, Lin X, et al. A high-fidelity RNA-targeting Cas13 restores paternal Ube3a expression and improves motor functions in Angelman syndrome mice. Mol Ther. 2023;31:2286–95.
-
Sasaki-Honda M, Akatsuka K, Sawai T. Is epigenome editing non-inheritable? Implications for ethics and the regulation of human applications. Stem Cell Rep. 2023;18:2005–9.
-
Roth GV, Gengaro IR, Qi LS. Precision epigenetic editing: technological advances, enduring challenges, and therapeutic applications. Cell Chem Biol. 2024;31:1422–46.
-
Bazick HO, Mao H, Niehaus JK, Wolter JM, Zylka MJ. AAV vector-derived elements integrate into Cas9-generated double strand breaks and disrupt gene transcription. Mol Ther. 2024;32:4122–37.
-
Fine EJ, Cradick TJ, Zhao CL, Lin Y, Bao G. An online bioinformatics tool predicts zinc finger and TALE nuclease off-target cleavage. Nucleic Acids Res. 2014;42:e42.
-
Dindot SV, Antalffy BA, Bhattacharjee MB, Beaudet AL. The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum Mol Genet. 2008;17:111–8.
-
Judson MC, Sosa-Pagan JO, Del Cid WA, Han JE, Philpot BD. Allelic specificity of Ube3a expression in the mouse brain during postnatal development. J Comp Neurol. 2014;522:1874–96.
-
Huang HS, Allen JA, Mabb AM, King IF, Miriyala J, Taylor-Blake B, et al. Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature. 2011;481:185–9.
-
Vihma H, Li K, Welton-Arndt A, Smith AL, Bettadapur KR, Gilmore RB, et al. Ube3a unsilencer for the potential treatment of Angelman syndrome. Nat Commun. 2024;15:5558.
-
Lee HM, Clark EP, Kuijer MB, Cushman M, Pommier Y, Philpot BD. Characterization and structure-activity relationships of indenoisoquinoline-derived topoisomerase I inhibitors in unsilencing the dormant Ube3a gene associated with Angelman syndrome. Mol Autism. 2018;9:45.
-
Hurh S, Cho B, You DJ, Kim H, Lee EM, Lee SH, et al. Expression analysis of combinatorial genes using a bi-cistronic T2A expression system in porcine fibroblasts. PLoS ONE. 2013;8:e70486.
-
Kugler S, Kilic E, Bahr M. Human synapsin 1 gene promoter confers highly neuron-specific long-term transgene expression from an adenoviral vector in the adult rat brain depending on the transduced area. Gene Ther. 2003;10:337–47.
-
Kugler S, Meyn L, Holzmuller H, Gerhardt E, Isenmann S, Schulz JB, et al. Neuron-specific expression of therapeutic proteins: evaluation of different cellular promoters in recombinant adenoviral vectors. Mol Cell Neurosci. 2001;17:78–96.
-
Anderlid BM, Lundin J, Malmgren H, Lehtihet M, Nordgren A. Small mosaic deletion encompassing the snoRNAs and SNURF-SNRPN results in an atypical Prader-Willi syndrome phenotype. Am J Med Genet A. 2014;164A:425–31.
-
Bieth E, Eddiry S, Gaston V, Lorenzini F, Buffet A, Conte Auriol F, et al. Highly restricted deletion of the SNORD116 region is implicated in Prader-Willi Syndrome. Eur J Hum Genet. 2015;23:252–5.
-
de Smith AJ, Purmann C, Walters RG, Ellis RJ, Holder SE, Van Haelst MM, et al. A deletion of the HBII-85 class of small nucleolar RNAs (snoRNAs) is associated with hyperphagia, obesity and hypogonadism. Hum Mol Genet. 2009;18:3257–65.
-
Kim E, Kim S, Kim DH, Choi BS, Choi IY, Kim JS. Precision genome engineering with programmable DNA-nicking enzymes. Genome Res. 2012;22:1327–33.
-
Zukher I, Dujardin G, Sousa-Luis R, Proudfoot NJ. Elongation roadblocks mediated by dCas9 across human genes modulate transcription and nascent RNA processing. Nat Struct Mol Biol. 2023;30:1536–48.
-
Ramirez CL, Certo MT, Mussolino C, Goodwin MJ, Cradick TJ, McCaffrey AP, et al. Engineered zinc finger nickases induce homology-directed repair with reduced mutagenic effects. Nucleic Acids Res. 2012;40:5560–8.
-
Waugh DS, Sauer RT. Single amino acid substitutions uncouple the DNA binding and strand scission activities of Fok I endonuclease. Proc Natl Acad Sci USA. 1993;90:9596–600.
-
Bitinaite J, Wah DA, Aggarwal AK, Schildkraut I. FokI dimerization is required for DNA cleavage. Proc Natl Acad Sci USA. 1998;95:10570–5.
-
Doyon Y, Vo TD, Mendel MC, Greenberg SG, Wang J, Xia DF, et al. Enhancing zinc-finger-nuclease activity with improved obligate heterodimeric architectures. Nat Methods. 2011;8:74–9.
-
Garner MM, Chrambach A. Resolution of circular, nicked circular and linear DNA, 4.4 kb in length, by electrophoresis in polyacrylamide solutions. Electrophoresis. 1992;13:176–8.
-
Simpson BP, Yrigollen CM, Izda A, Davidson BL. Targeted long-read sequencing captures CRISPR editing and AAV integration outcomes in brain. Mol Ther. 2023;31:760–73.
-
Dalwadi DA, Calabria A, Tiyaboonchai A, Posey J, Naugler WE, Montini E, et al. AAV integration in human hepatocytes. Mol Ther. 2021;29:2898–909.
-
Hanlon KS, Kleinstiver BP, Garcia SP, Zaborowski MP, Volak A, Spirig SE, et al. High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat Commun. 2019;10:4439.
-
Torella L, Klermund J, Bilbao-Arribas M, Tamayo I, Andrieux G, Chmielewski KO, et al. Efficient and safe therapeutic use of paired Cas9-nickases for primary hyperoxaluria type 1. EMBO Mol Med. 2024;16:112–31.
-
Breton C, Clark PM, Wang L, Greig JA, Wilson JM. ITR-Seq, a next-generation sequencing assay, identifies genome-wide DNA editing sites in vivo following adeno-associated viral vector-mediated genome editing. BMC Genom. 2020;21:239.
-
McLean JR, Smith GA, Rocha EM, Hayes MA, Beagan JA, Hallett PJ, et al. Widespread neuron-specific transgene expression in brain and spinal cord following synapsin promoter-driven AAV9 neonatal intracerebroventricular injection. Neurosci Lett. 2014;576:73–8.
-
Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27:59–65.
-
Haddad MR, Donsante A, Zerfas P, Kaler SG. Fetal brain-directed AAV gene therapy results in rapid, robust, and persistent transduction of mouse choroid plexus epithelia. Mol Ther Nucleic Acids. 2013;2:e101.
-
Chakrabarty P, Rosario A, Cruz P, Siemienski Z, Ceballos-Diaz C, Crosby K, et al. Capsid serotype and timing of injection determines AAV transduction in the neonatal mice brain. PLoS ONE. 2013;8:e67680.
-
Mullen RJ, Buck CR, Smith AM. NeuN, a neuronal specific nuclear protein in vertebrates. Development. 1992;116:201–11.
-
Feng Y, Liu S, Chen R, Xie A. Target binding and residence: a new determinant of DNA double-strand break repair pathway choice in CRISPR/Cas9 genome editing. J Zhejiang Univ Sci B. 2021;22:73–86.
-
Chandrasegaran S, Carroll D. Origins of programmable nucleases for genome engineering. J Mol Biol. 2016;428:963–89.
-
Hu JH, Davis KM, Liu DR. Chemical biology approaches to genome editing: understanding, controlling, and delivering programmable nucleases. Cell Chem Biol. 2016;23:57–73.
-
Richardson CD, Ray GJ, DeWitt MA, Curie GL, Corn JE. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat Biotechnol. 2016;34:339–44.
-
Whinn KS, Kaur G, Lewis JS, Schauer GD, Mueller SH, Jergic S, et al. Nuclease dead Cas9 is a programmable roadblock for DNA replication. Sci Rep. 2019;9:13292.
-
Doi G, Okada S, Yasukawa T, Sugiyama Y, Bala S, Miyazaki S, et al. Catalytically inactive Cas9 impairs DNA replication fork progression to induce focal genomic instability. Nucleic Acids Res. 2021;49:954–68.
-
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–21.
-
Jiang Y, Chen B, Duan C, Sun B, Yang J, Yang S. Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system. Appl Environ Microbiol. 2015;81:2506–14.
-
Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014;507:62–7.
-
Iwahara J, Levy Y. Speed-stability paradox in DNA-scanning by zinc-finger proteins. Transcription. 2013;4:58–61.
-
Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–57.
-
Davis L, Maizels N. Homology-directed repair of DNA nicks via pathways distinct from canonical double-strand break repair. Proc Natl Acad Sci USA. 2014;111:E924–32.
-
Cavaille J, Buiting K, Kiefmann M, Lalande M, Brannan CI, Horsthemke B, et al. Identification of brain-specific and imprinted small nucleolar RNA genes exhibiting an unusual genomic organization. Proc Natl Acad Sci USA. 2000;97:14311–6.
-
Doe CM, Relkovic D, Garfield AS, Dalley JW, Theobald DE, Humby T, et al. Loss of the imprinted snoRNA mbii-52 leads to increased 5htr2c pre-RNA editing and altered 5HT2CR-mediated behaviour. Hum Mol Genet. 2009;18:2140–8.
-
Kishore S, Stamm S. The snoRNA HBII-52 regulates alternative splicing of the serotonin receptor 2C. Science. 2006;311:230–2.
-
Hebras J, Marty V, Personnaz J, Mercier P, Krogh N, Nielsen H, et al. Reassessment of the involvement of Snord115 in the serotonin 2c receptor pathway in a genetically relevant mouse model. eLife. 2020;9:e60862.
-
Helwak A, Turowski T, Spanos C, Tollervey D. Roles of SNORD115 and SNORD116 ncRNA clusters during neuronal differentiation. Nat Commun. 2024;15:10427.
-
Greig JA, Martins KM, Breton C, Lamontagne RJ, Zhu Y, He Z, et al. Integrated vector genomes may contribute to long-term expression in primate liver after AAV administration. Nat Biotechnol. 2024;42:1232–42.
-
Sabatino DE, Bushman FD, Chandler RJ, Crystal RG, Davidson BL, Dolmetsch R, et al. Evaluating the state of the science for adeno-associated virus integration: an integrated perspective. Mol Ther. 2022;30:2646–63.
-
Mendiola AJP, LaSalle JM. Epigenetics in Prader-Willi Syndrome. Front Genet. 2021;12:624581.
-
Rotaru DC, Wallaard I, de Vries M, van der Bie J, Elgersma Y. UBE3A expression during early postnatal brain development is required for proper dorsomedial striatal maturation. JCI Insight. 2023;8:e166073.
-
Mabb AM, Simon JM, King IF, Lee HM, An LK, Philpot BD, et al. Topoisomerase 1 regulates gene expression in neurons through cleavage complex-dependent and -independent mechanisms. PLoS ONE. 2016;11:e0156439.
-
King IF, Yandava CN, Mabb AM, Hsiao JS, Huang HS, Pearson BL, et al. Topoisomerases facilitate transcription of long genes linked to autism. Nature. 2013;501:58–62.
-
Dickerson AS, Rahbar MH, Bakian AV, Bilder DA, Harrington RA, Pettygrove S, et al. Autism spectrum disorder prevalence and associations with air concentrations of lead, mercury, and arsenic. Environ Monit Assess. 2016;188:407.
-
Kim JY, Grunke SD, Levites Y, Golde TE, Jankowsky JL. Intracerebroventricular viral injection of the neonatal mouse brain for persistent and widespread neuronal transduction. J Vis Exp. 2014;51863.
-
Stirling DR, Swain-Bowden MJ, Lucas AM, Carpenter AE, Cimini BA, Goodman A. CellProfiler 4: improvements in speed, utility and usability. BMC Bioinform. 2021;22:433.
-
Uezu A, Kanak DJ, Bradshaw TW, Soderblom EJ, Catavero CM, Burette AC, et al. Identification of an elaborate complex mediating postsynaptic inhibition. Science. 2016;353:1123–9.
-
Gao Y, Hisey E, Bradshaw TWA, Erata E, Brown WE, Courtland JL, et al. Plug-and-play protein modification using homology-independent universal genome engineering. Neuron. 2019;103:583–97.e8.
-
Judson MC, Shyng C, Simon JM, Davis CR, Punt AM, Salmon MT, et al. Dual-isoform hUBE3A gene transfer improves behavioral and seizure outcomes in Angelman syndrome model mice. JCI Insight. 2021;6:e144712.
-
Kim H, Gao EB, Draper A, Berens NC, Vihma H, Zhang X, et al. Rescue of behavioral and electrophysiological phenotypes in a Pitt-Hopkins syndrome mouse model by genetic restoration of Tcf4 expression. eLife. 2022;11:e72290.
-
Hou Z, Zhang Y, Propson NE, Howden SE, Chu LF, Sontheimer EJ, et al. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc Natl Acad Sci USA. 2013;110:15644–9.
-
Carbone A, Fioretti FM, Fucci L, Ausio J, Piscopo M. High efficiency method to obtain supercoiled DNA with a commercial plasmid purification kit. Acta Biochim Pol. 2012;59:275–8.
-
Maeder ML, Thibodeau-Beganny S, Osiak A, Wright DA, Anthony RM, Eichtinger M, et al. Rapid “open-source” engineering of customized zinc-finger nucleases for highly efficient gene modification. Mol Cell. 2008;31:294–301.
-
Bibikova M, Carroll D, Segal DJ, Trautman JK, Smith J, Kim YG, et al. Stimulation of homologous recombination through targeted cleavage by chimeric nucleases. Mol Cell Biol. 2001;21:289–97.
Acknowledgements
The authors thank Eric S. McCoy for preparing primary neuron cultures for these studies. The authors also thank James L. Krantz and Trinity E. Rust for assistance in cloning and mutating the ZFN plasmids, respectively.
Funding
This work was supported by the Angelman Syndrome Foundation, the Simons Foundation (SFARI, award ID 631904 to MJZ), the National Institute of Neurological Disorders and Stroke (NINDS; 1R01NS109304 to MJZ and T32NS007431 to HOB), and the UNC Royster Society of Fellows (to LMJ).
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bazick, H.O., James, L.M., Taylor-Blake, B. et al. Multi-targeting zinc finger nuclease vector unsilences paternal UBE3A in a mouse model of Angelman syndrome. Gene Ther (2025). https://doi.org/10.1038/s41434-025-00582-1
-
Received:
-
Revised:
-
Accepted:
-
Published:
-
Version of record:
-
DOI: https://doi.org/10.1038/s41434-025-00582-1