
Introduction
Prostate cancer (PCa), a malignancy originating from prostate tissue, remains a leading cause of cancer-related mortality among men worldwide. As an androgen-dependent disease, PCa is commonly treated with androgen deprivation therapy (ADT), which reduces serum androgen levels and suppresses androgen receptor (AR) signaling, typically with initial efficacy1. However, disease relapse frequently occurs, and patients often progress to metastatic castration-resistant prostate cancer (CRPC)2. A key mechanism underlying CRPC development is the persistent activation of AR in a ligand-independent manner3. Although significant advances have been made in understanding AR reactivation in CRPC, the specific molecular pathways, particularly those related to oxidative stress, warrant further investigation.
Initial research on oxidative stress and reactive oxygen species (ROS) focused on their harmful effects on cellular macromolecules, including DNA, proteins, and lipids. However, contemporary studies have shown that oxidative stress and ROS also function as signaling molecules that regulate diverse cellular behaviors4. Oxidative stress can aggravate tumor progression by inducing tumor cell proliferation, survival, and adaptation to hypoxia5. Notably, ADT has been shown to trigger oxidative stress in PCa6, and both ROS and oxidative stress are closely related to PCa progression and CRPC development, partly through the activation of AR signaling6. For instance, TXNDC9 has been identified as a key regulator of ROS levels, and the TXNDC9–PRDX1 pathway contributes significantly to ROS-mediated AR activation7. Additionally, Sharifi et al. demonstrated that reduced SOD2 expression leads to AR reactivation in CRPC by elevating ROS8. Despite these insights, the precise mechanisms by which oxidative stress drives AR reactivation are still not fully understood.
Previous reports indicate that treatment with 10 μM H2O2 elevates nuclear AR levels and promotes androgen resistance in PCa cells9. In the present study, we employed both the hormone-responsive LNCaP cell line and the CRPC-derived 22RV1 cell line to investigate the mechanisms through which oxidative stress, simulated by H2O2 exposure, influences AR–prostate-specific antigen (PSA) signaling and promotes androgen resistance. The use of these two PCa cell lines enabled us to assess the relevance of oxidative stress-induced AR activation in different phenotypes of PCa.
Materials and methods
Cell culture and drug treatment
LNCaP (Cat No. CL-0143) and 22RV1 (Cat No. CL-0004) cells (Procell, Wuhan, China) were maintained in low-glucose DMEM (Cat No. G4520–500ML; Servicebio, Wuhan, China) supplemented with 10% FBS (Cat No. 11011–8611; Solarbio, Beijing, China) in a humidified incubator (Thermo Fisher Scientific, Waltham, MA, USA). H2O2 was obtained from Shanghai Macklin Biochemical Technology (Macklin, Shanghai, China), and cycloheximide (CHX) was obtained from Selleck (Shanghai, China).
Cell counting kit 8 assay
Cell Counting Kit 8 (CCK8) assay (Cat No. C0041; Beyotime, Shanghai, China) was used to assess the cytotoxicity of H2O2 at different concentrations (0, 5, 10, 20, 40, 80, 160, and 320 μM). Briefly, PCa cells were seeded in a 96-well plate at a density of 5000 cells/well and treated with H2O2 for 24 h. Subsequently, CCK8 reagent was added to each well. After 4 h, the optical density (OD) was measured at the wavelength of 450 nm.
ROS activity analysis
ROS activity was assessed using a commercial ROS detection kit (Cat No. S0033S; Beyotime). Briefly, PCa cells were seeded in 6-well plates at a density of 100,000 cells/well and cultured overnight. The following day, H2O2 was added to each well and the cells were incubated for 24 h. DCFH-DA was diluted to 10 μmol/L at a ratio of 1:1000. The culture supernatant was discarded, and the cells were incubated with DCFH-DA for 20 min at 37 °C. The unbound probe was removed by washing the cells with a serum-free medium three times. Subsequently, the mean fluorescence intensity of ROS was evaluated to quantify intracellular ROS levels.
Western blotting
PCa cells were seeded in 6-well plates at a density of 500,000 cells/well. After 24 h of treatment with H2O2, the cells were washed with PBS, lysed with radioimmunoprecipitation assay (RIPA) buffer (Cat No. P0013D; Beyotime), and centrifuged at 12,000 × g for 15 min at 4 °C. The supernatants were collected as total protein extracts, and a Bradford protein assay kit (Cat No. G2001-250ML; Servicebio) was used to quantify the extracted proteins. A total of 30 μg of protein per lane was separated via sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) at 150 V for 1.5 h. The separated proteins were transferred to a PVDF membrane. After non-specific binding sites were blocked, the membrane was sequentially incubated with diluted primary and secondary antibodies. Subsequently, protein bands were visualized using an ECL kit, and the proteins were quantified using the ImageJ software (NIH, Bethesda, MD, USA). The following primary antibodies were used: anti-AR (Cat No. 66747-1-Ig; AB_2882094; Proteintech, Wuhan, China), anti-AR-V7 (Cat No. 19672, Cell Signaling Technology), anti-PSA (Cat No. 10501-1-AP; AB_2172597; Proteintech, Wuhan, China), anti-ubiquitin-specific peptidase 36 (anti-USP36) (Cat No. 14783-1-AP; AB_2213357; Proteintech, Wuhan, China), anti-Ub (Cat No. 10201-2-AP; AB_671515; Proteintech, Wuhan, China), and anti-β-actin (Cat No. GB15003; AB_3083699; Servicebio).
qRT-PCR
Total RNA was isolated from PCa cells using Trizol (Cat No. R0016; Beyotime). Reverse transcription was performed using the SweScript RT II First Strand cDNA Synthesis Kit (With gDNA Remover) (Cat No. G3333; Servicebio). Subsequently, qPCR was performed using the ArtiCanATM SYBR qPCR Mix (Cat No. TSE501; Tsingke, Beijing, China) on a fluorescent quantitative PCR instrument (ABI). The following primer sequences were used to amplify PSA: forward, 5’‐GCTGGGAGTGCGAGAAGCAT‐3’; reverse, 5’‐CGTAGAGCGGGTGTGGGAAG‐3’. β-actin was used as the internal reference gene, and the relative mRNA expression level of PSA was calculated using the 2−ΔΔCt method10.
Dual-luciferase reporter assay
The sequence of PSA promoter was amplified using qRT-PCR and inserted into the downstream region of the pGL3-basic vector (Miaoling Biotechnology). PCa cells (LNCaP and 22RV1) were transfected with the PSA-encoding vector using Lipofectamine 3000 reagent followed by indicated treatments. After 24 h of H2O2 exposure, luciferase activity was evaluated using a luciferase reporter assay kit (Cat No. RG029S; Beyotime) on a GloMax 20/20 Luminometer (Promega), with pRL-TK (Miaoling Biotechnology) serving as the reference. Firefly luciferase activity was normalized to Renilla luciferase activity for each sample.
TurboID-mediated proximity biotin labeling
LNCaP cells were seeded in 10-cm plates at a density of approximately 3 × 106 cells/plate. Upon reaching 70% confluence, the cells were transfected with the pLV3-CMV-AR(human)-EGFP-TurboID-NLS-3 × FLAG-Puro plasmid for 24 h, followed by an additional incubation of 24 h with or without 20-μM H2O2. Subsequently, the cells were incubated with 100-μM biotin for 1 h and collected. The cells were washed twice with ice-cold PBS to remove excess biotin, lysed with RIPA buffer for 30 min, and centrifuged. The supernatants were collected as total protein extracts. A total of 50 μL of streptavidin beads were washed thrice with 1 × TBS and separated on a magnetic stand. The beads were incubated with the extracted proteins for 12 at 4 °C with constant shaking. The beads were separated on the magnetic stand and washed thrice with a wash buffer. Subsequently, biotinylated proteins were eluted by incubating the beads with 5 × SDS loading buffer in a boiling water bath for 10 min.
Biotin labeling efficiency analysis: The eluted proteins were separated via SDS-PAGE and transferred to a PVDF membrane. After non-specific binding sites were blocked, the membrane was incubated with HRP-labeled streptavidin antibody for 1 h. Subsequently, the membrane was washed with 1 × TBST five times and protein bands were visualized using an ECL reagent.
Silver staining: The eluted proteins were separated via SDS-PAGE and subjected to silver staining using a standard protocol.
MS: MS was used to identify biotin-labeled proteins.
GO functional annotation: GO enrichment analysis was implemented using the DAVID database (https://david.ncifcrf.gov/).
Cell transfection
The pEnCMV-USP36(human)-3 × Myc plasmid (Miaolingbio, Wuhan, China) was used to overexpress USP36, whereas the pEnCMV empty vector (without target sequences) was used as the negative control. A FLAG tag was purchased from Solarbio (Cat No. P02839) to construct an AR-FLAG overexpression plasmid. An HA tag was purchased from Proteintech (Cat No. 51064-2-AP) to construct a Ub-HA overexpression plasmid. ShNC and shUSP36 were purchased from Tsingke (Beijing, China). Cell transfection was performed using lipo6000™ transfection reagent (Cat No. C0526; Beyotime).
Co-immunoprecipitation
For co-immunoprecipitation (Co-IP), PCa cells were seeded in 6-well plates at a density of 500,000 cells/well. To analyze the binding between exogenously expressed AR and USP36, the cells were co-transfected with the AR-Flag and pEnCMV-USP36(human)-3 × Myc plasmids. After 24 h of treatment with H2O2 (20 μM), the cells were lysed and incubated with an anti-FLAG antibody (Cat No. K200001m; Solarbio). Subsequently, the cells were incubated with BeyoMag™ Protein A + G beads (Cat No. P2108; Beyotime) for 1 h at 4 °C. The beads were washed with the lysis buffer 3 times, and the eluted proteins were analyzed using western blotting.
To validate the binding between endogenous USP36 and AR, PCa cells were exposed to H2O2, lysed, and incubated with an anti-USP36 polyclonal antibody (Cat No. 14783-1-AP; Proteintech) for IP. Subsequently, the cells were incubated with BeyoMag™ Protein A + G beads (Cat No. P2108; Beyotime) for 1 h at 4 °C. The beads were washed with the lysis buffer 3 times, and the eluted proteins were analyzed using western blotting.
Immunofluorescence assay
After indicated treatments, PCa cells were immobilized with 4% paraformaldehyde and permeabilized with 0.1% Triton. After non-specific binding sites were blocked with 1% BSA, the cells were incubated with an anti-AR antibody (Cat No. 66747-1-Ig; Proteintech, Wuhan, China) overnight at 4 °C. The following day, the cells were washed 3 times with PBS and incubated with a fluorescently labeled secondary antibody for 1 h. DAPI (Cat No. G1012; Servicebio) was used to stain nuclei, and immunofluorescence (IF) was observed using a fluorescence microscope.
Data analysis
GraphPad Prism 8.0 (GraphPad Prism, La Jolla, CA, USA) was used for statistical analysis, and all data were expressed as the mean ± standard deviation (SD). All assays were performed in triplicates unless otherwise mentioned. Differences between groups were analyzed using Student’s t‑test, whereas differences among three or more groups were analyzed using one-way analysis of variance (ANOVA). The association between USP36 expression and clinicopathological features (lymph node metastasis) or survival outcomes (Disease-Specific Survival and Progression-Free Interval) was analyzed using data from the TCGA prostate cancer (PRAD) cohort. A P value of < 0.05 indicated statistically significant differences.
Results
Low doses of H2O2 increased viability and induced oxidative stress in PCa cells
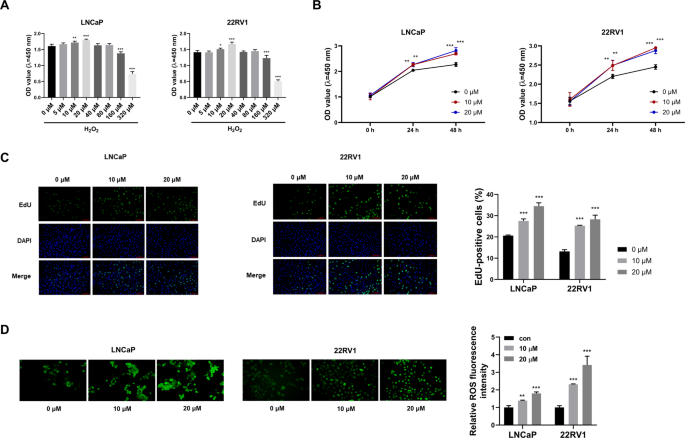
To determine the optimal concentration of H2O2, we first evaluated its cytotoxic effects on PCa cells using the CCK8 assay. Treatment with low concentrations of H2O2 (5, 10, and 20 μM) enhanced cell viability in a concentration-dependent manner (Fig. 1A). In contrast, high concentrations of H2O2 (160 and 320 μM) led to a marked decline in viability (Fig. 1A). Based on these results, 10 and 20 μM H2O2 were selected for subsequent experiments. To examine whether prolonged oxidative stress further enhances cell viability, we extended H2O2 treatment to 48 h with replenishment at 24 h to maintain consistent stress levels. The results demonstrated that cell viability increased only marginally after 48 h compared to 24 h in both LNCaP and 22Rv1 cells (Fig. 1B), indicating that the pro-survival effect of low-dose H2O2 had largely reached a plateau by 24 h. To further determine whether H2O2 directly stimulates cell proliferation, we performed EdU incorporation assays. After 24 h of treatment, both LNCaP and 22Rv1 cells exhibited a significant, dose-dependent increase in the proportion of EdU-positive cells following exposure to 10 μM or 20 μM H2O2 (Fig. 1C). These results confirm that low-dose H2O2 actively promotes DNA synthesis and cell proliferation within 24 h, providing functional evidence that supports the CCK-8 viability data. Androgen deprivation therapy has been shown to trigger oxidative stress in PCa. This oxidative stress induces reactivation of AR, leading to the development of CRPC6. A study reported that downregulation of SOD2 directly reactivated AR by increasing ROS levels in CRPC cells8. In this study, we used H2O2 as an exogenous oxidant to simulate oxidative stress in PCa cells. Treatment with low concentrations of H2O2 significantly increased ROS levels (Fig. 1D). Collectively, these findings indicate that low-dose H2O2 induces oxidative stress and promotes viability in PCa cells.

Low doses of H2O2 increases the viability and induces oxidative stress in PCa cells. (A) LNCaP and 22RV1 cells were administrated with gradient doses of H2O2 (0, 5, 10, 20, 40 ,80, 160, or 320 μM), and cell viability was determined via CCK8 assay. (B) Cell viability was assessed after 24 h or 48 h treatment with H2O2, with the medium and H2O2 being refreshed at the 24-h mark. (C) Proportion of EdU-positive LNCaP cells after 24-h treatment with H2O2 (0, 10, or 20 μM). (D) ROS abundance was assessed via a commercial ROS assay kit. ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001.
H2O2 upregulates AR and activates PSA under both standard and castration-mimic conditions
AR is a member of the nuclear receptor superfamily and acts as a ligand-dependent nuclear transcription factor. PSA is one of the genes regulated by AR and is considered the most sensitive biomarker for prostate diseases, including PCa. Given the close relationship between ROS and reactivation of AR, we examined whether H2O2 exposure could upregulate the expression of AR and PSA. As shown in Fig. 2A, B, treatment with H2O2 increased the protein expression of AR and PSA in a dose-dependent manner. Furthermore, dual-luciferase reporter assay demonstrated that treatment with H2O2 enhanced the transcriptional activity of PSA promoter (Fig. 2C, D) possibly owing to the upregulation of AR. To further determine if oxidative stress can reactivate AR signaling under a clinically relevant, near-castration condition, we cultured cells in charcoal-stripped serum (CDT-FBS) supplemented with 0.5 nM DHT. Remarkably, H2O2 treatment still significantly increased the protein levels of both AR and PSA in this androgen-deprived setting (Fig. 2E–H), demonstrating that the reactivation of the AR-PSA axis by oxidative stress is effective even with minimal androgen support. To further validate the effect of H2O2 on AR signaling in a castration-resistant model endogenously expressing AR-V7, we performed Western blot analysis in 22RV1 cells. As shown in Fig. 2I, treatment with H2O2 markedly increased the protein levels of full-length AR (AR-FL) and its target PSA. Notably, the protein abundance of AR-V7 remained unchanged upon H2O2 exposure, suggesting that oxidative stress primarily activates canonical AR signaling through the full-length receptor rather than modulating the expression of this splice variant. Altogether, the results indicated that exposure to H2O2 activated the AR-PSA pathway in PCa cells.

H2O2 up-regulates AR and transcriptionally activates its target gene PSA. (A, B) LNCaP and 22RV1 cells were administrated with 10 or 20 μM H2O2, and Western blot assay was employed to measure AR and PSA protein abundance. (C, D) LNCaP and 22RV1 cells introduced with luciferase reporter plasmid containing PSA promoter were exposed to 20 μM H2O2 or not. Dual-luciferase reporter assay was carried out to determine the luciferase activities. (E–H) LNCaP and 22RV1 cells were cultured in medium supplemented with charcoal-stripped serum (CDT-FBS) and 0.5 nM DHT to mimic the castration-resistant physiological condition, followed by treatment with or without 20 μM H2O2. Western blot analysis was performed to assess the protein levels of AR and PSA. (I) Western blot analysis of full-length AR (AR-FL), PSA, and AR-V7 protein expression in 22RV1 cells treated with or without H2O2 (10 or 20 μM) for 24 h. **P < 0.01, ***P < 0.001.
To further investigate the interplay between oxidative stress and androgen signaling, we evaluated the effects of H2O2 alongside varying DHT concentrations. Low DHT (1–2 nM) significantly enhanced AR and PSA expression and cell viability (Fig. S1A–B, see supplementary material11). To confirm the central role of AR in mediating H2O2 effects, we performed AR knockdown and antagonism experiments. siRNA-mediated AR knockdown decreased PSA expression and reduced viability in H2O2−treated cells (Fig. S2A–D, see supplementary material11). Similarly, the AR antagonist enzalutamide suppressed PSA expression and viability without affecting AR protein levels (Fig. S3A–D, see supplementary material11). These results confirm that AR signaling is necessary for H₂O₂-induced growth promotion.
TurboID-mediated proximity biotin labeling revealed AR-interacting proteins upon H2O2 exposure
To investigate how H2O2 regulates AR expression, we employed TurboID-mediated proximity biotin labeling in LNCaP cells. Biotin labeling efficiency was high with or without H2O2 (Fig. 3A). Streptavidin pulldown and silver staining revealed distinct protein profiles between control and H2O2−treated groups (Fig. 3B). Mass spectrometry identified 546 H2O2−specific AR-interacting proteins (Fig. 3C, Table S1, see supplementary material12). GO analysis showed that these proteins were closely associated with “positive regulation of androgen receptor activity” and “proteasome-mediated ubiquitin-dependent protein catabolic process” (Fig. 3D, Table S2, see supplementary material12), suggesting H2O2 modulates AR via the ubiquitin–proteasome pathway. A total of 21 ubiquitination-associated proteins were found to interact with AR in H2O2−treated PCa cells (Table S3, see supplementary material12). These proteins included USP36, an important deubiquitinating enzyme of the ubiquitin-specific protease (USP) family.

TurboID-mediated proximity biotin labeling is employed to explore AR-interacted proteins upon H2O2 exposure. (A) Biotin labeling efficiency was analyzed in con and H2O2 groups. (B) Biotin-labeled proteins were enriched through streptavidin beads, and the pull-down samples were loaded onto SDS-PAGE followed by silver staining. (C) The enriched proteins in two groups were identified by mass spectrometry, and Venn diagram showed 546 proteins specific to H2O2 group. (D) GO annotation was utilized to functionally classify the 546 proteins based on their biological processes. (E) USP36 expression levels across different pathologic N stages in the TCGA-PRAD cohort, including Normal (n = 52), N0 (n = 351), N1 (n = 80). N1 (lymph node metastasis) tumors exhibit significantly higher USP36 expression than N0 (no metastasis) and normal tissues. (F) USP36 expression is significantly elevated in patients who experienced a progression-free interval (PFI) event (disease progression or recurrence, n = 94) compared to those who did not (n = 410). (G) Patients who died from prostate cancer (DSS event, n = 5) had significantly higher tumor USP36 expression than survivors (n = 497). Gene expression is presented as Log2(FPKM + 1). *P < 0.05, **P < 0.01.
To assess the clinical relevance of USP36, we analyzed TCGA-PRAD data. High USP36 expression was significantly correlated with aggressive clinicopathological features (Fig. 3E–G). High USP36 expression was significantly associated with aggressive clinicopathological features. USP36 was markedly elevated in tumors with lymph node metastasis (Pathologic N1 stage) compared to non-metastatic (N0) and normal tissues (Fig. 3E). Moreover, patients with higher USP36 expression exhibited significantly poorer progression-free interval (PFI, Fig. 3F) and worse disease-specific survival (DSS, Fig. 3G). These findings from a large clinical cohort strongly support our in vitro data, positioning USP36 as a key driver of aggressive disease and a promising prognostic biomarker. USP36 has been shown to play an oncogenic role in multiple malignancies, including glioblastoma and esophageal squamous carcinoma. For instance, Chang et al. reported that USP36 contributed to the progression of glioblastoma by deubiquitinating and stabilizing ALKBH513, whereas Zhang et al. demonstrated that USP36 aggravated esophageal squamous carcinoma by deubiquitinating and stabilizing YAP14. Therefore, we subsequently investigated whether H2O2 upregulated AR by promoting its interaction with USP36 in PCa cells.
USP36 overexpression enhanced AR stability
USP36 was overexpressed in PCa cells (Fig. 4A). While USP36 overexpression alone did not affect AR levels, it markedly enhanced AR protein expression in the presence of H2O2 (Fig. 4B, C), suggesting H2O2−dependent regulation. Subsequently, the stability of AR protein was evaluated using cycloheximide (CHX) chase assay, in which CHX prevents protein synthesis to assess the degradation rate of existing proteins. Overexpression of USP36 resulted in increased stability of AR protein as evidenced by the higher protein expression of AR in the USP36 group than in the Vec group at all time points (Fig. 4D).

USP36 overexpression enhances the protein stability of AR. (A) The overexpression efficiency of USP36 ectopic expression construct in LNCaP and 22RV1 cells was assessed by Western blot assay. (B, C) LNCaP and 22RV1 cells were introduced with Vec or USP36 ectopic expression construct followed by H2O2 exposure or not. The protein level of AR was determined by Western blot assay. (D) The effect of USP36 on AR protein stability was analyzed using protein synthesis inhibitor CHX. *P < 0.05, ***P < 0.001.
H2O2 inhibited AR ubiquitination via in a USP36-dependent manner
Given that USP36 is a deubiquitinating enzyme, we hypothesized that USP36 stabilized AR protein via deubiquitination in the presence of H2O2. The binding between USP36 and AR was examined using Co-IP. Co-IP confirmed that H₂O₂ promotes interaction between exogenous (Fig. 5A) and endogenous (Fig. 5B) AR and USP36. The knockdown efficiency of shUSP36 was validated using western blotting (Fig. 5C). As shown in Fig. 5D, exposure to H2O2 prominently suppressed the ubiquitination of AR, whereas silencing of USP36 reversed this phenomenon, indicating that H2O2 stabilized AR protein through USP36. These results collectively indicated that exposure to H2O2 enhanced and stabilized AR expression through the deubiquitinating effects of USP36.

H2O2 reduces AR ubiquitination via deubiquitinating enzyme USP36. (A) Co-IP assay was employed to verify the binding relation between exogenously expressed AR and exogenously expressed USP36 upon H2O2 exposure. (B) Co-IP was carried out to confirm the binding relation between endogenous USP36 and endogenous AR upon H2O2 exposure. (C) The knockdown efficiency of shUSP36 was verified by Western blot assay. (D) IP assay was employed to evaluate the impacts of H2O2 and USP36 on ubiquitination of AR.
USP36 mediates H2O2−induced activation of the AR-PSA pathway
We further investigated whether H2O2 could activate the AR–PSA pathway via USP36 in PCa. Western blotting showed that treatment with H2O2 increased the protein expression of AR and PSA, whereas silencing of USP36 counteracted this increase (Fig. 6A). Consistent with these results, IF assay showed that treatment with H2O2 increased the expression of AR, which was reversed by USP36 silencing (Fig. 6B, C). Consistent with protein-level changes, H2O2 upregulated PSA mRNA (KLK3) levels, whereas silencing of USP36 reversed this change (Fig. 6D). Treatment with H2O2 elevated the luciferase activity of PSA promoter, whereas silencing of USP36 counteracted this change (Fig. 6E), suggesting that H2O2 activated the transcription of PSA through USP36. Together, these results demonstrate that H2O2 stabilizes AR and activates PSA transcription through USP36.

H2O2 activates AR-PSA pathway through USP36. (A–E) LNCaP and 22RV1 cells were transfected with shNC or shUSP36 followed by H2O2 exposure. (A) The protein abundance of AR and PSA was examined via Western blot assay. (B, C) IF assay was carried out to examine AR abundance in LNCaP and 22RV1 cells upon the indicated treatment. (D) RT-qPCR was conducted to determine KLK3 abundance. (E) Dual-luciferase reporter assay was employed to validate the impacts of H2O2 and USP36 on the transcription of PSA. *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
Redox reactions function as a crucial second messengers in cancer cells and represent an emerging target for cancer therapy15. Oxidative stress promotes cancer cell survival, proliferation, and adaptation to hypoxic microenvironments5, and can activate multiple oncogenic signaling pathways, including MAPK, ERK1/2, and PI3K-AKT16. In PCa, oxidative stress plays an important role in both disease initiation and progression17. ADT has been shown to induce oxidative stress in PCa cells, leading to AR pathway activation through diverse mechanisms and ultimately contributing to the development of CRPC6,18. For instance, Shiota et al. demonstrated that H2O2 up-regulated Twist1 and AR expression, while Twist1 knockdown attenuates H2O2−induced AR elevation18. In addition, genetic polymorphisms in antioxidant enzymes have been linked to ADT response19, further supporting the role of oxidative stress in CRPC progression. Hormone deprivation disrupts cellular homeostasis, elevates ROS production, and increases oxidative stress, thereby driving CRPC development20. A detailed understanding of the mechanisms by which oxidative stress activates AR signaling and promotes tumor progression is essential for developing novel therapeutic strategies for CRPC.
In this study, we observed that low-dose H2O2 (10 or 20 μM) enhanced cell viability and increased ROS levels in PCa cells. These concentrations were therefore selected to simulate oxidative stress in subsequent experiments. AR is a ligand-dependent transcription factor that, upon androgen binding, translocates to the nucleus and activates target genes transcription. PSA, a key AR-regulated gene, serves as a sensitive biomarker for PCa21. We found that H2O2 exposure markedly enhanced PSA transcription and protein expression, likely through upregulation of AR. It is noteworthy that in our experiments, the PSA protein was readily detectable in 22RV1 cells under baseline conditions, which appears to contrast with the established knowledge that it is normally hard to detect under standard conditions. We attribute this discrepancy primarily to the distinct molecular characteristics of the 22RV1 cell line. 22RV1 cells are a well-established model of CRPC, particularly known for expressing truncated, constitutively active androgen receptor splice variants, most notably AR-V722,23. AR-V7 lacks the ligand-binding domain, leading to its constitutive activation and ligand-independent transcription of AR target genes, including KLK3/PSA23,24. The readily detectable basal PSA expression we observed is a hallmark of this AR-V7-driven, constitutively active signaling state inherent to the 22RV1 model. Our oxidative stress (H2O2) treatment served to further amplify this pre-existing signaling axis.
To investigate how H2O2 upregulates AR protein expression, we employed TurboID-mediated proximity biotin labeling and MS to identify AR-interacting proteins in H2O2−stimulated LNCaP cells. LNCaP cells were used to assess interactions between AR and other proteins under hormone-dependent conditions so as to obtain key insights into the AR signaling pathway. However, it is noteworthy that the findings obtained from LNCaP cells cannot be directly applied to CRPC cells, such as 22RV1 cells, without parallel assays. GO analysis showed that the AR-interacting proteins identified from H2O2−stimulated LNCaP cells, including USP36, were enriched in “proteasome-mediated ubiquitin-dependent protein catabolic process”. This finding suggested that H2O2 increased AR expression in PCa cells through the ubiquitin–proteasome pathway. Deubiquitinating enzymes stabilize proteins by removing ubiquitin from the proteins, thereby preventing the proteins from being degraded by proteasomes. The deubiquitinating enzyme USP36 promotes tumor progression by preventing the degradation of multiple proteins involved in tumorigenesis and inflammation25. USP36 is markedly upregulated and acts as an oncogene in various malignancies, such as esophageal carcinoma14, glioblastoma26, HCC27, colorectal cancer28, breast cancer29, and T cell lymphoma30. In this study, we found that endogenous USP36 could interact with endogenous AR in the presence of H2O2, leading to the deubiquitination and stabilization of AR. Furthermore, we found that H2O2 activated the AR–PSA pathway through USP36 in PCa cells. Both LNCaP and 22RV1 cells exhibited similar responses to oxidative stress, suggesting that AR–USP36 interaction induced by H2O2 was not limited to a specific PCa phenotype. These consistent findings highlight that oxidative stress plays an important role in AR signaling, potentially contributing to both hormone-responsive and castration-resistant prostate cancers.
Building upon the findings reported by de las Perez et al.31, who examined the impact of multi-DUB inhibition on AR, we investigated the specific mechanism of USP36 under oxidative stress. The results enhanced the understanding of AR regulation by highlighting the role of USP36 under oxidative stress. Unlike previous studies that broadly addressed the effects of DUB inhibition on AR, this study specifically showed that oxidative stress induces AR expression via USP36 in both hormone-responsive and castration-resistant PCa cells. Although we validated the interaction between USP36 and AR under oxidative stress, the precise mechanisms facilitating this interaction remain unclear. Future studies should focus on investigating the potential post-translational modifications of AR or USP36 and the resulting alterations in protein interactions and localization in response to oxidative stress. In addition, while our bioinformatic analysis of the TCGA cohort revealed a significant association between high USP36 expression and poor clinical outcomes, the precise biological mechanisms underlying this association remain to be fully elucidated beyond the AR stabilization pathway we identified in vitro.
In conclusion, our findings indicate that castration-induced oxidative stress activates the AR–PSA pathway via USP36-mediated deubiquitination and stabilization of AR, ultimately driving CRPC progression. Targeting castration-induced oxidative stress and the USP36-AR-PSA axis may therefore represent a promising therapeutic strategy for both hormone-sensitive and castration-resistant prostate cancer.
Data availability
Some or all datasets generated during and/or analyzed during the current study are not publicly available but are available from the corresponding author on reasonable request.
References
-
Liu, J. M., Chen, Y. T., Wu, C. T., Hsu, W. L. & Hsu, R. J. Androgen deprivation therapy for prostate cancer and the risk of thyroid diseases. Prostate 82(7), 809–815. https://doi.org/10.1002/pros.24323 (2022).
-
Morote, J., Aguilar, A., Planas, J. & Trilla, E. Definition of castrate resistant prostate cancer: New insights. Biomedicines https://doi.org/10.3390/biomedicines10030689 (2022).
-
Scher, H. I. & Sawyers, C. L. Biology of progressive, castration-resistant prostate cancer: Directed therapies targeting the androgen-receptor signaling axis. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 23(32), 8253–8261. https://doi.org/10.1200/jco.2005.03.4777 (2005).
-
Forman, H. J. & Zhang, H. Targeting oxidative stress in disease: promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 20(9), 689–709. https://doi.org/10.1038/s41573-021-00233-1 (2021).
-
Jelic, M. D., Mandic, A. D., Maricic, S. M. & Srdjenovic, B. U. Oxidative stress and its role in cancer. J. Cancer Res.Ther. 17(1), 22–28. https://doi.org/10.4103/jcrt.JCRT_862_16 (2021).
-
Shiota, M., Yokomizo, A. & Naito, S. Oxidative stress and androgen receptor signaling in the development and progression of castration-resistant prostate cancer. Free Radic. Biol. Med. 51(7), 1320–1328. https://doi.org/10.1016/j.freeradbiomed.2011.07.011 (2011).
-
Feng, T. et al. TXNDC9 regulates oxidative stress-induced androgen receptor signaling to promote prostate cancer progression. Oncogene 39(2), 356–367. https://doi.org/10.1038/s41388-019-0991-3 (2020).
-
Sharifi, N., Hurt, E. M., Thomas, S. B. & Farrar, W. L. Effects of manganese superoxide dismutase silencing on androgen receptor function and gene regulation: Implications for castration-resistant prostate cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 14(19), 6073–6080. https://doi.org/10.1158/1078-0432.Ccr-08-0591 (2008).
-
Gonzalez-Menendez, P. et al. GLUT1 protects prostate cancer cells from glucose deprivation-induced oxidative stress. Redox Biol. 17, 112–127. https://doi.org/10.1016/j.redox.2018.03.017 (2018).
-
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25(4), 402–408. https://doi.org/10.1006/meth.2001.1262 (2001).
-
Zhou, Bo. Supplementary figures. Figshare. Fig. https://doi.org/10.6084/m9.figshare.28407956.v1 (2025).
-
Zhou, Bo. Supplementary tables. Figshare. Dataset. https://doi.org/10.6084/m9.figshare.28407959.v1 (2025).
-
Chang, G. et al. USP36 promotes tumorigenesis and drug sensitivity of glioblastoma by deubiquitinating and stabilizing ALKBH5. Neuro. Oncol. 25(5), 841–853. https://doi.org/10.1093/neuonc/noac238 (2023).
-
Zhang, W. et al. USP36 facilitates esophageal squamous carcinoma progression via stabilizing YAP. Cell Death Dis. 13(12), 1021. https://doi.org/10.1038/s41419-022-05474-5 (2022).
-
Perillo, B. et al. ROS in cancer therapy: the bright side of the moon. Exp. Mol. Med. 52(2), 192–203. https://doi.org/10.1038/s12276-020-0384-2 (2020).
-
Zhang, J. et al. ROS and ROS-Mediated cellular signaling. Oxid. Med. Cell. Longev. 2016, 4350965. https://doi.org/10.1155/2016/4350965 (2016).
-
Mondal, D. et al. Oxidative stress and redox signaling in CRPC progression: therapeutic potential of clinically-tested Nrf2-activators. Cancer Drug Resist. 4(1), 96–124. https://doi.org/10.20517/cdr.2020.71 (2021).
-
Shiota, M. et al. Castration resistance of prostate cancer cells caused by castration-induced oxidative stress through Twist1 and androgen receptor overexpression. Oncogene 29(2), 237–250. https://doi.org/10.1038/onc.2009.322 (2010).
-
Shiota, M. et al. Gene polymorphisms in antioxidant enzymes correlate with the efficacy of androgen-deprivation therapy for prostate cancer with implications of oxidative stress. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 28(3), 569–575. https://doi.org/10.1093/annonc/mdw646 (2017).
-
Bassey, I. E. et al. Impact of androgen deprivation on oxidative stress and antioxidant status in nigerian patients with prostate cancer undergoing androgen deprivation therapy. JCO. Glob. Oncol. 6, 1481–1489. https://doi.org/10.1200/go.20.00290 (2020).
-
Attard, G., Cooper, C. S. & de Bono, J. S. Steroid hormone receptors in prostate cancer: A hard habit to break?. Cancer Cell 16(6), 458–462. https://doi.org/10.1016/j.ccr.2009.11.006 (2009).
-
Khurana, N. et al. Multimodal actions of the phytochemical sulforaphane suppress both AR and AR-V7 in 22Rv1 cells: Advocating a potent pharmaceutical combination against castration-resistant prostate cancer. Oncol. Rep. 38(5), 2774–2786. https://doi.org/10.3892/or.2017.5932 (2017).
-
Szafran, A. T. et al. High-content screening identifies Src family kinases as potential regulators of ar-v7 expression and androgen-independent cell growth. Prostate 77(1), 82–93. https://doi.org/10.1002/pros.23251 (2017).
-
Djusberg, E. et al. High levels of the AR-V7 splice variant and co-amplification of the golgi protein coding YIPF6 in AR amplified prostate cancer bone metastases. Prostate 77(6), 625–638. https://doi.org/10.1002/pros.23307 (2017).
-
Niu, M. Y. et al. The emerging role of ubiquitin-specific protease 36 (USP36) in cancer and beyond. Biomolecules https://doi.org/10.3390/biom14050572 (2024).
-
Qiu, W. et al. PRL1 promotes glioblastoma invasion and tumorigenesis via activating USP36-mediated snail2 deubiquitination. Front. Oncol. 11, 795633. https://doi.org/10.3389/fonc.2021.795633 (2021).
-
Sun, W. et al. Gene signature and prognostic value of ubiquitin-specific proteases members in hepatocellular carcinoma and explored the immunological role of USP36. Front. Biosci. 27(6), 190. https://doi.org/10.31083/j.fbl2706190 (2022).
-
Ling, H. et al. CEP63 upregulates YAP1 to promote colorectal cancer progression through stabilizing RNA binding protein FXR1. Oncogene 41(39), 4433–4445. https://doi.org/10.1038/s41388-022-02439-y (2022).
-
Wu, H. et al. miR-140–3p/usp36 axis mediates ubiquitination to regulate PKM2 and suppressed the malignant biological behavior of breast cancer through Warburg effect. Cell Cycle 22(6), 680–692. https://doi.org/10.1080/15384101.2022.2139554 (2023).
-
Li, B. et al. MELK mediates the stability of EZH2 through site-specific phosphorylation in extranodal natural killer/T-cell lymphoma. Blood 134(23), 2046–2058. https://doi.org/10.1182/blood.2019000381 (2019).
-
de Las, P. A., Reiner, T., De Cesare, V., Trost, M. & Perez-Stable, C. Inhibiting multiple deubiquitinases to reduce androgen receptor expression in prostate cancer cells. Sci. Rep. 8(1), 13146. https://doi.org/10.1038/s41598-018-31567-3 (2018).
Acknowledgements
I would like to express my gratitude to the funding bodies that supported this research, including the Henan Province Medical Science and Technology Research Program, the Henan Medical Science and Technology Research and Development Program, and the Young and Middle-aged Innovative Talents in Health Science and Technology in Henan Province.
Funding
This work was supported by grants from Henan Province Medical Science and Technology Research Program (Joint Co-construction) (No. LHGJ20230352, LHGJ20240314), Henan Medical Science and Technology Research and Development Program (No. 201702078) and Young and middle-aged innovative talents in health science and technology in Henan Province (No. 201004118).
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fan, C., Huang, Z., Gao, J. et al. Oxidative stress reactivates androgen receptor signaling via USP36 to drive castration resistance in prostate cancer. Sci Rep 15, 42185 (2025). https://doi.org/10.1038/s41598-025-25964-8
-
Received:
-
Accepted:
-
Published:
-
Version of record:
-
DOI: https://doi.org/10.1038/s41598-025-25964-8