
Introduction
Mammalian cell-based dual display and secretion (dualDS) technologies have emerged as powerful tools for therapeutic antibody development by integrating antibody display and secretion within the same cellular system. Traditional mammalian cell libraries designed for antibody display-only are typically paired with flow cytometry to screen for binding affinity and manufacturability, while secretion-only libraries are commonly used with microfluidics to evaluate diverse functionalities1,2,3,4,5. In contrast, dualDS technologies integrate both display and secretion within the same mammalian cells, enabling streamlined screening of antibody affinity, manufacturability, and functionality without re-cloning antibody genes6,7,8,9,10. Unlike microbial-based display systems—such as phage, bacterial, and yeast—which mainly screen scFv or Fab fragments. DualDS technologies support the expression of full-length antibodies with human-like post-translational modifications11,12. Although full-length IgGs have also been expressed in yeast, they typically carry non-mammalian glycosylation13. DualDS offers high-throughput screening capabilities while eliminating the need to re-clone genes, modify antibody formats, or switch host cells between discovery to early-stage production. This streamlining has the potential to shorten development timelines while reducing costs and risks compared with traditional antibody development.
Various dualDS technologies have been developed, which can be broadly categorized into two groups: (1) simultaneous display and secretion systems and (2) switchable display-to-secretion systems. Simultaneous display and secretion systems enable antibodies to be expressed both on the cell surface and in a secreted form concurrently. Methods achieving this include alternative splicing2,14,15,16,17, leaky stop codons18, 2A peptides19, and furin cleavage sequences20. Switchable display-to-secretion systems enable antibody expression to toggle between display-only and secretion-only modes. Examples include amber suppression7, nonsense codons21, chemically induced inhibition of furin cleavage22, inducible fusion of protein A dual Z-domain with the PDGFR transmembrane domain (ZZ-PDGFR)6, the enzyme-cleavable surface-tethered all-purpose screening system (ECSTASY)10, and “antibody-membrane switching” via recombinase-mediated DNA recombination8,9,23.
A variety of cell types, including COS24, BHK25, HEK293T2,22,26,27,28,29,30, lymphoma-derived B cells9,16,17,31,32, H129933, and Chinese Hamster Ovary (CHO)4,8,23,34,35,36, have been explored for developing mammalian cell-based library expression technologies. Among these, CHO cells have emerged as the most favorable choice due to their dominance as hosts for commercial therapeutic antibody production. CHO cells offer several advantages, such as the ability to produce antibodies at high titers and quality with human-compatible glycosylation patterns, low risk of human virus contamination, and robust growth conditions37,38,39. Additionally, CHO cells are easily cultured at high densities, highly transfectable, and amenable to genetic modifications, enabling the efficient expression of large antibody libraries.
To introduce antibody libraries into mammalian cells, common gene delivery strategies include transient expression14,35,40, virus- and transposon-mediated random genomic integration9,16,17,41, and recombinase- or nuclease-mediated targeted genomic integration23,27,30,31,32,34,36. Transient transfection is limited by its short duration, requiring repeated DNA rescue and multiple rounds of transfection, which reduces throughput and handling efficiency. Random genomic integration technologies can provide long-term expression; however, they lack control over integration sites and gene copy numbers, leading to variable expression levels across cells and the potential for incorrect heavy and light chain pairings. In contrast, targeted genomic integration technologies address these limitations by enabling precise integration at desired loci, ensuring uniform transcription, efficient single-gene insertion, and long-term stable expression levels.
A full-length IgG monoclonal antibody consists of two identical light chains (LC) and two identical heavy chains (HC). The relative expression levels of LC and HC genes affects both the expression level and quality of antibodies42. Reported mammalian cell library technologies have co-expressed LC and HC genes either in two separate vectors or a single vector, where the HC gene is linked to a membrane anchor25,26,36,43. However, the two-vector approach presents challenges of low and varied co-transfection efficiency across different cells. To overcome these limitations, single-vector designs for the construction of mammalian cell libraries have been explored, employing multiple promoters (MP)27,36, internal ribosomal entry sites (IRES)23,26, or 2A peptides21. The MP design uses two independent transcription units, each with its own promoter for LC and HC expression44. While this approach enables efficient co-expression, upstream promoter activity can suppress the transcription of downstream genes45. In the IRES strategy, the first gene undergoes cap-dependent translation, whereas the second gene is translated in a cap-independently manner, often leading to reduced expression of the latter gene46. The 2A peptide approach involves inserting a 2A sequence between the LC and HC genes, enabling co-expression from a single transcript. “Ribosomal skipping” occurs at the last two amino acids of the 2A peptide, glycine and proline, to separate the two polypeptides during translation and yield a similar amount of LC and HC polypeptides47,48. To prevent the attachment of residual 2A to the upstream gene, a minimal furin cleavage sequence (Fm), R-X-K/R-R, is often incorporated between the upstream gene and 2A49. Additionally, adding a GSG or SGSG linker between the furin cleavage site and 2A has been shown to enhance 2A cleavage efficiency49,50.
Previous studies comparing the MP, IRES, and 2A peptide approaches have demonstrated that MP and 2A produce higher expression levels than IRES in transient transfections, whereas IRES and 2A outperform MP in stable transfections51,52. Among the various 2A peptides tested, the combination of a minimal furin cleavage sequence (Fm), a GSG linker, and the 2A peptide from Thosea asigna virus (T2A) has been identified as the most effective for enhancing antibody expression levels49. However, beyond expression levels, dualDS applications require consistent expression of the same antibodies across different cells to avoid variability that could confound antibody screening results. Despite the application of various co-expression strategies for mammalian cell libraries, there has been no comprehensive comparison of these strategies for the construction of mammalian cell libraries.
In this study, we evaluated MP, IRES, and a combination of Fm, a SGSG linker, and T2A (Fm-T2A) for co-expressing LC and HC using vectors designed for the simultaneous display and secretion of monoclonal antibodies in CHO cells. The vectors were introduced into CHO cells via targeted integration, recombinase-mediated cassette exchange (RMCE). Our results showed that MP and IRES vectors produced low secretion levels and variable antibody display across cells, whereas the Fm-T2A vector achieved the highest secretion levels and uniform antibody display. However, the secreted antibodies contained residual 2A sequences, which were effectively minimized by engineering the furin cleavage sequence (FCS). These findings provide valuable insights into vector design, advancing mammalian cell-based antibody screening technologies and other applications in mammalian cell engineering that require the co-expression of multiple genes.
Results
Design of RMCE and targeting vectors for evaluating different HC and LC co-expression strategies
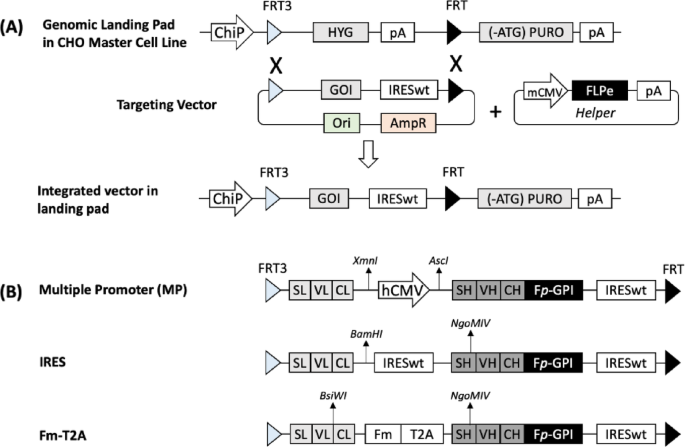
We evaluated different targeting vector designs for LC and HC co-expression in a CHO master cell line (MCL) using RMCE. The CHO MCL contains a single-copy genomic landing pad that expresses a hygromycin resistance gene (HYG) under a chimeric promoter (ChiP) and terminated by the SV40 polyadenylation signal (pA). This cassette is flanked by enhanced recombinase flippase (FLPe) target sites, FRT3 and FRT (Fig. 1A). Downstream of FRT is a start codon-deficient puromycin resistance gene ((ATG-)PURO) followed by pA, which remains inactive until RMCE occurs. The targeting vector incorporates the gene of interest (GOI) followed by a wild-type encephalomyocarditis virus (EMCV) internal ribosomal entry site (IRESwt), flanked by FRT3 and FRT sites identical to those in the landing pad. Upon co-transfection of a targeting vector and the helper vector expressing FLPe into the CHO MCL, FLPe mediates RMCE between the FRT3 and FRT sites of the landing pad and targeting vector. Successful RMCE replaces the HYG expression cassette with GOI and IRESwt, which activates PURO expression through the IRES. Cells that undergo successful RMCE survive puromycin selection and form stable transfected pools. As the targeting vector lacks a promoter and pA, only cells that undergo RMCE will survive selection, ensuring selective expression of the GOI.

An overview of recombinase-mediated-cassette-exchange (RMCE) and targeting vector designs for co-expression of antibody LC and HC genes. (A) Schematic representation of RMCE via FLPe/FRT recombination system. (B) Targeting vector designs for co-expression of antibody LC and HC genes in three configurations: multiple promoter (MP), internal ribosomal entry site (IRES), and combination of minimal furin recognition sequence and Thosea asigna virus 2A peptide (Fm-T2A). ChiP, chimeric promoter comprising murine CMV (cytomegalovirus) enhancer, human CMV core promoter and intron; FRT3 and FRT, mutant and wildtype recombinase flippase recognition sites; FLPe, enhanced recombinase flippase; HYG, hygromycin resistance gene; pA, simian virus 40 polyadenylation signal; (-ATG)PURO, puromycin resistance gene without start codon; GOI, gene-of-interest; IRESwt, wildtype encephalomyocarditis virus (EMCV) internal ribosome entry site; mCMV, murine CMV enhancer and promoter; LC, light chain cDNA; HC, heavy chain cDNA; Fp-GPI, furin cleavage sequence with partial cleavage (Fp) linked to human decay-accelerating factor glycosylphosphatidylinositol (GPI) membrane anchor; AscI, BamHI, BsiWI and NgoMIV restriction sites.
To co-express the LC and HC for simultaneous display and secretion of monoclonal antibodies, we designed three targeting vectors: MP, IRES, and Fm-T2A (Fig. 1B). In all three vectors, the HC gene is linked to a partial furin cleavage sequence (RRKRGTNFS) (Fp) and a membrane anchor (GPI), enabling simultaneous display and secretion53. This functionality leverages the native furin protease present in CHO cells to sporadically cleave the Fp region, generating both cell-surface-displayed and secreted antibodies. Each vector employs a distinct strategy for LC and HC co-expression. When the MP targeting vector integrates into the landing pad, LC expression is driven by the built-in ChiP promoter on the landing pad, while HC expression is controlled by the human cytomegalovirus (hCMV) promoter. Transcription of LC and HC occurs independently, producing separate mRNA transcripts that are translated into individual LC and HC polypeptides. When the IRES targeting vector integrates into the landing pad, both LC and HC genes are transcribed under the control of the same ChiP promoter into a single mRNA transcript. LC translation occurs via the conventional cap-dependent mechanism initiated by the promoter, while HC translation is driven by the downstream IRESwt sequence via a cap-independent mechanism. When Fm-T2A targeting vector integrates into landing pad, LC and HC genes are expressed in a single open reading frame, linked by R-R-K-R, a SGSG linker and the T2A peptide. Translation produces a single polypeptide that requires two mechanisms for co-expression: ribosomal skipping, triggered by the T2A peptide, separates HC from upstream sequences during translation, and furin cleavage, which removes residual 2A peptides attached to LC, ensuring proper functionality.
Comparison of different co-expression strategies for antibody display homogeneity, secretion levels and quality in stably transfected pools
We generated two stably transfected pools for each of the targeting vectors using RMCE. The dual display and secretion functionality in the vector designs allowed us to evaluate both antibody display homogeneity and secretion levels in CHO cell pools. To assess antibody display homogeneity, the surface-displayed antibodies was stained with FITC-labelled anti-HC IgG and analyzed via flow cytometry. Site-specific integration of targeting vectors into the landing pad was expected to result in uniform display across cells. However, MP-derived pools contained three heterogeneous cell populations (Fig. 2A), whereas the IRES and Fm-T2A vectors produced homogeneous cell populations. To compare secretion levels, 14-day fed-batch cultures were conducted for duplicate stable pools generated for each vector. The Fm-T2A vector achieved the highest specific productivity (qMab) of 2.95 pg/cell/day (pcd), whereas MP and IRES showed lower qMab values of 1.50 pcd and 1.39 pcd, respectively (Fig. 2B). By day 14, secreted antibody titers reached 169 mg/L for MP pools, 228 mg/L for IRES pools, and 356 mg/L for Fm-T2A pools.

Comparison of different targeting vectors for antibody display homogeneity, secretion levels and quality in stably transfected pools. Stably transfected pools were generated by co-transfecting CHO master cells with a specified targeting vector and a helper vector expressing FLPe recombinase, followed by selection in puromycin-containing medium. Each pool was characterized for antibody display levels (A) and productivity (B), and the secreted antibodies were purified and analyzed using SDS-PAGE (C) and intact mass spectrometry (MS) (D) under reducing conditions. For each targeting vector, duplicate pools were generated. As these duplicate pools exhibited identical characteristics, only one representative figure is shown for dot plot analysis, SDS-PAGE, and intact MS. To assess antibody levels displayed on the cell surface, flow cytometry analysis was performed by staining cells with FITC-conjugated IgG specific to the heavy chain, with non-transfected cells (blank) serving as controls. Specific antibody productivity (qMab) and titer of secreted antibodies were measured in fed-batch cultures, with each data point representing the average and standard deviation from two independent pools.
Beyond secretion levels, antibody quality was also critical in determining the optimal design construct for co-expression of LC and HC. To assess antibody quality, supernatants from the fed-batch cultures of stable pools were purified using protein A and analyzed via SDS-PAGE under reducing conditions. The MP and IRES vectors produced accurate HC and LC bands at approximately 50 kDa and 25 kDa, respectively (Fig. 2C). In contrast, Fm-T2A produced antibodies exhibited a correctly sized HC polypeptide band but displayed two LC polypeptide bands with molecular weights exceeding 25 kDa. The uncropped SDS-PAGE image is shown in Supplementary Figure 1. Intact mass spectrometry (MS) analysis further confirmed that the LC and HC polypeptides expressed from MP and IRES targeting vectors had the correct molecular weight. In contrast, the HC polypeptides expressed from the Fm-T2A had correct molecular weight, while the LC polypeptides had higher molecular weights, likely due to the attachment of the RRKR-SGSG-T2A (EGRGSLLTCGDVEENPGP) and RRKR-SGSG-EG inferred from their molecular weights (Fig. 2D).
Comparison of different co-expression strategies for antibody expression levels and homogeneity in stably transfected clones
MP-generated stable pools exhibited three distinct cell populations with varying antibody expression levels: high-expressing, low-expressing, and non-expressing. To investigate the reasons behind this heterogeneity, fluorescence activate cell sorting (FACS) was performed on the two MP pools to isolate the three populations. Single-cell cloning was then conducted to obtain six clones from each sorted population. For simplicity, clones derived from the high-expressing populations of two pools were designated as AH and BH, from the low-expressing populations as AL and BL, and from the non-expressing populations as AN and BN. 14-day fed-batch cultures were performed to measure secreted antibody titers across the different clones. Supernatants were analyzed using a nephelometric method, which detects IgG via anti-human Fc region antibodies. Clones AH1-AH6 and BH1-BH6 exhibited significant variability in qMab, ranging from 0.78 to 2.9 pcd, with a mean value of 2.14 pcd across the 12 clones (Fig. 3A). In contrast, clones AL1-AL6 and BL1-BL6 showed similar but significantly lower titers, with a mean qMab of 0.38 pcd. All clones derived from the non-expressing population had undetectable levels of secreted antibodies. Since the nephelometer detection limit is 9.26 mg/L, an ELISA assay was performed to confirm the absence of secretion by detecting HC and LC in the supernatants of non-expressing clones. Except for AN3, which exhibited a negligible qMab of 0.01 in the HC-based ELISA, all other non-expressing clones showed no detectable antibody levels in either HC-based or LC-based ELISAs.

Specific productivity and junction PCR (jPCR) analysis of clones generated using different targeting vectors. (A) Multiple promoter (MP) vector, (B) IRES vector, and (C) Fm-T2A vector. Duplicate stably transfected pools were generated for each vector. The MP pools exhibited three distinct cell populations with high, low, and no display levels, which were sorted for single-cell cloning. From each of duplicate MP pools A and B, six clones were isolated from high (AH1-6 & BH1-6), low (AL1-6 & BL1-6), and non-displaying (AN1-6 & BN1-6) populations. The IRES and Fm-T2A pools exhibited homogeneous display levels, and six clones were randomly isolated from each pool. Specific productivity (qMab) of each pool and clone was determined in 14-day fed-batch cultures. To evaluate integration accuracy, genomic DNA (gDNA) was extracted from all isolated clones, and 5′ and 3′ jPCR was performed using one primer specific to the targeting vector and another binding to the genomic landing pad outside the flanking FRT sites. The untransfected CHO MCL was used as a negative control (-ve), while gDNA from MP-high sorted pools served as a positive control (+ ve). Correct integration at the 5′ site resulted in a 505 bp product, while accurate integration at the 3′ site produced a 959 bp product.
To investigate the heterogeneous expression levels observed in MP populations, 5′ and 3′ junction PCR (jPCR) were performed to assess integration accuracy in the clones. The primers for jPCR were designed with one binding site in the landing pad and the other in the targeting vector (Supplementary Figure 2). Results indicated that all MP high-expressing clones exhibited correct integration at both the 5′ and 3′ sites. Conversely, MP low-expressing clones showed correct 3′ integration but lacked 5′ integration. All MP non-expressing clones displayed no integration at the 5′ site, with some clones exhibiting integration at the 3′ site. The uncropped agarose gel images are shown in Supplementary Figure 3.
To further investigate the integration errors in MP clones, four MP low-expressing clones and four MP non-expressing clones were selected for sequencing analysis. Genomic DNA sequences spanning the FRT3 and FRT sites in each clone were amplified using a forward primer binding to the ChiP promoter and a reverse primer binding to the PURO gene within the landing pad. The PCR products were resolved on agarose gels (Supplementary Figure 4), excised, purified, and sequenced. Sequencing results revealed that in MP low-expressing clones (AL1, AL2, BL1, and BL2), the targeting vector had integrated via the FRT recognition site instead of through RMCE, resulting in the entire MP targeting vector (including the bacterial backbone) being inserted into the genomic landing pad (Fig. 4A). In contrast, MP non-expressing clones exhibited two types of integration errors. In clones AN1 and AN2, a substantial portion of the sequence between the 3′ end of the FRT3 site and the 5′ start of the HC gene was missing (Fig. 4B). For clones BN1 and BN2, a significant segment of the genomic landing pad region was lost between the 3′ end of the HYG site and the 5′ start of the defective PURO gene (Fig. 4C). PURO gene expression was activated in all these clones, either due to the integration of IRESwt upstream of FRT or gene deletion that shifted the PURO gene in-frame with the HYG start codon, explaining their survival in puromycin-containing medium. All the annotated sequences are presented in Supplementary Figure 5.

Schematic representation of integration errors found in clones isolated from stably transfected pools generated using multiple promoter (MP) targeting vector design. To investigate the unexpected junction PCR (jPCR) results observed in MP low-expressing and non-expressing clones, representative clones were selected for molecular analysis. Two clones from each group—low-expressing (AL1, AL2, BL1, and BL2) and non-expressing (AN1, AN2, BN1, and BN2)—were analyzed. The entire integration region, flanked by FRT3 and FRT sites, was amplified using genomic DNA (gDNA) extracted from these clones as a template. PCR was performed using the ChiP-F primer, which binds upstream of the FRT3 region, and the PUR-R primer, which binds downstream of the FRT site. The amplified gene fragments were resolved on a 1% agarose gel (Supplementary Figure 2). The respective bands were excised, purified, and subjected to Sanger sequencing. (A) Low-expressing clones (AL1, AL2, BL1, and BL2) exhibited incorrect integration, where instead of undergoing recombinase-mediated cassette exchange (RMCE), the entire MP targeting vector was inserted into the landing pad region of the CHO master cell line (MCL). (B) Non-expressing clones from pool A (AN1 and AN2) showed gene truncation, leading to the loss of sequences between a portion of FRT3 and the heavy chain (HC) gene. (C) Non-expressing clones from pool B (BN1 and BN2) exhibited a deletion spanning the hygromycin resistance gene (HYG) and the puromycin resistance gene (PURO), including the PURO start codon (-ATG). This truncation, however, was maintained in-frame, resulting in the formation of a HYG-PURO fusion protein, which enabled cells to survive puromycin selection. ChiP, chimeric promoter comprising murine CMV (cytomegalovirus) enhancer, human CMV core promoter and intron; FRT3 and FRT, mutant and wild-type recognition sites for FLPe recombinase; pA, polyadenylation signal from simian virus), IRESwt, wildtype encephalomyocarditis virus (EMCV) internal ribosomal entry site; Fp-GPI, furin cleavage sequence with partial cleavage (Fp) linked to human decay-accelerating factor glycosylphosphatidylinositol (GPI) membrane anchor. Dotted lines indicate truncated gene regions.
For comparison, six clones were isolated from each of the two IRES and Fm-T2A stable pools to evaluate secretion levels and integration accuracy. The IRES clones exhibited moderate variation in qMab, ranging from 0.75 to 2.6 pcd, with a mean qMab of 1.38 pcd. jPCR analysis confirmed correct 5′ and 3′ integration for all IRES clones (Fig. 3B). In contrast, Fm-T2A clones demonstrated more consistent qMab, with a higher mean of 2.01 pcd. jPCR analysis verified correct 5′ and 3′ integration for all Fm-T2A clones (Fig. 3C). The uncropped agarose gel images are shown in Supplementary Figure 3.
Engineering FCS to enhance furin cleavage efficiency
The Fm-T2A targeting vector achieved the most homogeneous antibody display and the highest secretion levels among the three vector designs, both in stable pools and in single clones. However, the majority of the secreted antibodies from the Fm-T2A vector contained LC polypeptides with T2A residual still attached, suggesting inefficient furin cleavage at the Fm site. Previous studies have indicated that the amino acids flanking the Fm sequence significantly influence cleavage efficiency. In particular, the two amino acids upstream and five amino acids downstream of the furin cleavage site are highly conserved in sequences with high cleavage efficiency54,55.
To address this, six furin cleavage sequence variants (Fv) were designed, incorporating four highly conserved downstream amino acids (SVDT or SVDL) and one (R/K) or two (HR/GK) upstream amino acids of Fm (Fig. 5A). Stable pools generated using these newly designed Fv in combination with T2A were characterized for antibody display homogeneity, secretion levels, and product quality, then compared to the original Fm-T2A stable pools. All pools generated with the new Fv-T2A vectors exhibited homogeneous antibody display (Fig. 5B). Secretion levels varied slightly: F1-T2A and F5-T2A showed a small increase in antibody secretion, while all the other variants showed minor decreases compared to the Fm-T2A pool (Fig. 5C).


Evaluation of Fv-T2A variants for co-expression of antibody light chain and heavy chain genes in stably transfected pools. Stably transfected pools were generated by co-transfecting CHO master cells with a specified targeting vector containing a furin cleavage sequence variant (Fv)-T2A and a helper vector expressing FLPe recombinase, followed by selection in puromycin-containing medium. The amino acid sequences of each Fv-T2A variant are listed in the table (A). Each pool was characterized for antibody display levels (B) and productivity (C), and the secreted antibodies were purified using Protein A and analyzed by SDS-PAGE (D) and intact mass spectrometry (MS) (E) under reducing conditions. Antibody produced from the MP vector-generated pool was used as a standard (Std) in SDS-PAGE analysis. For each targeting vector, duplicate pools were generated. As these duplicate pools exhibited identical characteristics, only one representative figure is shown for dot plot analysis, SDS-PAGE, and intact MS. To assess antibody levels displayed on the cell surface, flow cytometry analysis was performed by staining cells with FITC-conjugated IgG specific to the heavy chain, with non-transfected cells (blank) serving as controls. Specific antibody productivities (qMab) of secreted antibodies in different pools were measured in fed-batch cultures, with each data point representing the average and standard deviation from two independent pools.
In contrast to the LC polypeptides from Fm-T2A, which exhibited two bands on SDS-PAGE under reducing conditions, all LC polypeptides from the newly designed Fv-T2A vectors displayed a single band. However, the band size was slightly larger than that of the LC polypeptide from the MP vector (Fig. 5D). The uncropped SDS-PAGE gel image is shown in Supplementary Figure 6. Intact MS analysis revealed the following insights (Fig. 5E). Consistent with the previously observed results (Fig. 1D), a small portion of the LC species expressed from the Fm-T2A vector had the T2A sequence removed, while the majority of LC species remained fused with T2A. For RRKR-SVDT and RRKR-SVDL, approximately 70% of the LC species had T2A successfully removed, while 30% retained T2A. The cleaved LC species had RRK attached as a residual tag. Adding R or K upstream of RRKR (e.g., R-RRKR-SVDT or K-RRKR-SVDT) resulted in complete T2A removal from all LC species. The predominant peak corresponded to LC with RRRK attached, with minor peaks showing LC attached to RRR, RR, or R. Adding two upstream amino acids (HR or GK) also produced LC species without T2A. However, the intact MS analysis revealed several peaks of similar intensity, corresponding to LC with residual sequences of two to four furin cleavage residues. All the uncropped intact mass spectrum results are shown in Supplementary Figure 7. These results suggest that modifying both upstream and downstream sequences of the Fm can improve cleavage efficiency and reduce the retention of T2A residues, offering a pathway for optimizing product quality in FCS-T2A-based co-expression systems.
Discussion
In this study, we compared three co-expression strategies—MP, IRES, and Fm-T2A—evaluating their effects on antibody display homogeneity, secretion levels, and product quality. Both MP and IRES vectors exhibited limitations in secretion levels and display homogeneity. The MP vector led to low antibody secretion and the presence of non-expressing cells due to two types of integration errors: (1) partial exchange at a single FRT site instead of complete RMCE at both FRT3 and FRT sites and (2) gene deletions in either the integrated targeting vector or the original landing pad. Partial exchange at one FRT site may represent an intermediate form during RMCE56. Deletions in the targeting vector may result from fragmentation before integration, while landing pad deletions could arise from chromosomal instability triggered by transfection57,58,59,60. Notably, integration errors were absent in clones generated using IRES and Fm-T2A vectors. The high rate of incorrect integration in MP-derived clones is likely due to both vector design and size. In the MP vector, the hCMV promoter upstream of HC-Fp-GPI-IRESwt can activate PURO expression even when integration occurs at the FRT site, enabling clones with incorrect integration to survive selection (Fig. 4). In contrast, clones integrating IRES and Fm-T2A vectors via the same incorrect mechanism cannot activate PURO expression and thus do not survive selection. Vector size may also contribute to incorrect integration events. The cassette exchange region in the MP vector spans 4187 bp, compared to 3466 bp in the IRES vector and 2948 bp in the Fm-T2A vector. Larger vectors are more prone to fragmentation and exhibit reduced transfection efficiency61,62. These findings highlight the importance of optimizing vector size and design for dualDS applications.
Interestingly, even among clones with correct integration at the same genomic site, those generated using the MP vector still exhibited significant variation in secreted antibody levels. In contrast, IRES clones showed less variation, while Fm-T2A clones demonstrated the most uniform antibody secretion. Following transfection with each targeting vector, cells underwent puromycin selection to enrich for clones with correct integration. Antibiotic selection has been suggested to impose evolutionary pressure on cells, triggering genomic plasticity as they adapt to altered conditions56. The extent of genomic changes and their impact on gene expression likely vary across clones and depend on vector design, contributing to differences in secretion variability among clones generated using different vectors. The MP vector utilizes separate promoters for HC and LC, leading to independent transcriptional regulation. Although integration occurs at the same genomic site, chromatin accessibility and epigenetic modifications may differentially influence the two promoters, resulting in variations in HC and LC expression levels and ultimately affecting antibody secretion. The IRES vector reduces transcriptional variability by ensuring that HC and LC are transcribed as a single mRNA. However, translation is regulated independently—LC translation occurs via a cap-dependent mechanism, whereas HC translation relies on a cap-independent mechanism. Differences in translation efficiency between the two genes may still contribute to some variation in antibody secretion levels, though to a lesser extent than the MP vector. In contrast, the Fm-T2A vector links LC and HC within a single open reading frame, ensuring coordinated transcription and near-equimolar translation via ribosomal skipping and furin cleavage. This tightly regulated process may minimize variability in HC-to-LC ratios, leading to highly uniform antibody secretion across clones.
Although the Fm-T2A vector achieved the highest secretion levels and uniform antibody display, the presence of residual 2A sequences in the secreted antibodies indicated incomplete furin cleavage. It has been reported that sequences with high furin cleavage efficiency typically contain the motif R-X-R/K-R, with two upstream and five downstream highly conserved amino acids54,55. Our results demonstrated that including highly conserved amino acids flanking R-X-R/K-R significantly enhanced furin cleavage efficiency (Fig. 5). Interestingly, adding one or two conserved amino acids upstream of RRKR resulted in the secretion of heterogeneous LC species with different numbers of residual amino acids, and this heterogeneity increased with the number of amino acids added. For instance, constructs with HR-RRKR-SVDT and GK-RRKR-SVDL exhibited the highest level of LC heterogeneity, followed by R-RRKR-SVDT and K-RRKR-SVDL, while RRKR-SVDT and RRKR-SVDL had the lowest level of heterogeneity. Furin is a protease that recognizes and cleaves specific motifs, typically R-X-K/R-R. Adding one or two amino acids upstream of RRKR may alter the structure and accessibility of the cleavage site, potentially affecting cleavage efficiency and specificity. Adding one or two amino acids upstream of RRKR may alter the structure and accessibility of the cleavage site, potentially affecting cleavage efficiency and specificity.This could result in a mixture of LC species with different numbers of residual amino acids. Interestingly, adding highly conserved amino acids downstream of RRKR did not appear to impact LC heterogeneity. These findings highlight the importance of optimizing flanking sequences to achieve efficient furin cleavage and high-quality antibody production. By carefully engineering the amino acids surrounding the furin cleavage site, it is possible to enhance the uniformity and quality of secreted antibodies, thereby improving the overall efficiency of the dual display and secretion system.
We compared different LC and HC co-expression strategies to improve antibody display homogeneity, secretion levels, and product quality in the dualDS system for antibody library screening. The insights gained are also valuable for designing vectors used in generating stably transfected cell lines for large-scale antibody manufacturing. Currently, most industrial vector technologies rely on multiple promoters to co-express LC and HC genes. However, this approach often leads to a significant proportion of non-expressing clones within transfected pools due to vector fragmentation56. The Fv-2A design could mitigate this issue by enabling tightly controlled, balanced co-expression of LC and HC. In addition to monoclonal antibody library screening, the optimized dualDS platform can also be applied to bispecific antibody discovery. In our previous study, a three-chain Fab–scFv–Fc bispecific antibody was successfully expressed using the Fm-T2A peptide63. By contrast, expression of four-chain bispecific antibodies using the Fv-2A design may be challenging, as the long mRNA transcripts required could suffer from stability issues.
Materials and methods
Vector construction
The helper vector expressing FLPe and the IRES targeting vector were constructed in our previous studies52,64. The MP, Fm-T2A, and Fv-T2A targeting vectors were constructed by GenScript. The MP targeting vector was generated by inserting the synthesized hCMV promoter between BamHI and AscI in the IRES vector (Fig. 1). The Fm-T2A and Fv-T2A targeting vectors were constructed by inserting the synthesized CL-Fm (or Fv)-T2A-SH fragment between BsiWI and NgoMIV in the IRES vector. In all vectors, the antibody genes used were derived from trastuzumab, with LC and HC cDNAs designed based on the amino acid sequences available in the international ImMunoGeneTics information system (IMGT).
Generation and characterization of stable pools for productivity in fed-batch cultures
The CHO MCL was developed by integrating a landing pad vector into parental CHO K1 cells obtained from ATCC. The generation of the MCL, along with protocols for culturing the MCL and establishing stable cell pools via RMCE, has been described in a previous study64. The productivity of each stable pools was determined in 14-day fed-batch cultures, which were conducted by seeding 3 × 105 viable cells/mL into 30 mL of growth medium within a 50 mL TubeSpin® bioreactor (TPP). The growth medium consisted of an equal ratio of HyQ-PF (GE Healthcare Life Sciences) and CD-CHO (Thermo Fisher Scientific), supplemented with 0.05% Pluronic F-68 (Thermo Fisher Scientific), 1 g/L sodium bicarbonate (Sigma), and 6 mM L-glutamine (Sigma). Cultures were fed with Ex-Cell® Advanced CHO Feed 1 (with glucose) (SAFC, Sigma) at 10% culture volume on days 5, 7, 9, and 11. D-glucose (Sigma) was supplemented whenever the glucose concentration dropped below 2 g/L. Cell density, viability, and antibody titer were measured on days 3, 5, 7, 9, 11, and 14. Specifically, cell density and viability were monitored using a Vi-Cell XR viability analyzer (Beckman Coulter). Antibody titer was quantified using either IMMAGE 800 immunochemistry system (Beckman Coulter) or ELISA (for samples with antibody concentration < 9.26 mg/L). Detection antibodies against either HC or LC were used in ELISA, following protocols detailed in a previous study65. The specific productivity (qMab) was calculated by dividing the measured antibody concentration by the integrated viable cell density (IVCD), which was determined using the trapezoidal method.
Flow cytometry analysis
Once the stable cell pool viability recovered to above 95%, surface-displayed antibody analysis was performed using flow cytometry. For each cell pool, 1 × 107 cells were collected, washed with 1X PBS, and stained with Anti-Human IgG (γ-chain specific) − FITC (Sigma) diluted 1:100. The stained cells were incubated on ice in the dark for 30 min. After staining, the cell pellets were washed twice with 1X PBS and resuspended to a final concentration of 5 × 10⁶ cells/mL for flow cytometry analysis. Flow cytometry was conducted using a MACSQuant® Analyzer (Miltenyi Biotec), and data analysis was performed using FlowJo™ 10.7.2 (Tree Star Inc.).
Pro A purification and SDS-PAGE analysis
The harvested culture supernatants were purified using a Tricon 10/50 chromatography column (Cytiva) packed with TOYOPEARL® AF-rProtein A-650F resin (Tosoh Bioscience) on a GE ÄKTA Purifier 100 FPLC System (GE Healthcare). The purified samples were analyzed by SDS-PAGE under reducing conditions. Protocols for Protein A purification and SDS-PAGE have been described in a previous study49.
Intact MS analysis
Intact mass analysis was performed using LC–MS as previously described66. In brief, purified antibody variants were reduced with 50 mM DTT in 6 M GnHCl at 50 °C for 30 min. The samples were then diluted in 0.3% formic acid before MS analysis. A total of 200 ng of protein was injected into a BioResolve RP mAb Polyphenyl column (1 × 150 mm) on a nanoACQUITY UPLC (Waters) system, coupled to a TripleTOF 6600 MS (SCIEX). Mass spectra were deconvoluted and annotated using the PMI-Byos v5.2.31 Intact Mass™ workflow (Protein Metrics Inc.).
Single cell cloning
Approximately 3–4 days before limiting dilution, cell pools were seeded in 125 mL shake flasks (Corning) at 3 × 105 cells/mL. Conditioned medium was prepared by collecting spent medium and filtering it through a 0.22 µm filter. The single-cell cloning medium consisted of 20% conditioned medium, 80% EX-CELL® CHO cloning medium (Sigma), and 4 mM L-glutamine (Gibco). After quantifying the cell density, serial dilution was performed using the prepared single-cell cloning medium to obtain a final concentration of 5 cells/mL. A 200 µL aliquot of the diluted cell suspension was pipetted into individual wells of a non-treated 96-well plate (Nunc) and incubated at 37 °C in a humidified Kuhner shaker (Adolf Kühner AG) with 8% CO2 for 14 days. Wells containing more than one colony were identified and eliminated by microscopic examination. Confluent wells were expanded into non-treated 24-well plates (Nunc) and subsequently transferred into 10 mL of growth media within a 50 mL TubeSpin® bioreactor (TPP) for each clone. These clones were then maintained under standard cell passaging procedures.
Junction PCR (jPCR) and sequencing analysis
Genomic DNA (gDNA) was extracted from 5 × 10⁶ cells using the PureLink® Genomic DNA Kit (Thermo Fisher Scientific) following the manufacturer’s protocol. jPCR was performed using 2X Platinum™ SuperFi™ II Green PCR Master Mix (Thermo Fisher Scientific) with 2 µL of gDNA as the template. The primer designs and sequences used to verify 5′ and 3′ integration are listed in Supplementary Figure 2. PCR cycling conditions were as follows: initial denaturation at 98 °C for 30 s, followed by 30 cycles of 98 °C for 10 s, 60 °C for 10 s, and 72 °C for 30 s, with a final extension at 72 °C for 5 min. PCR products were resolved by gel electrophoresis on a 1% agarose gel at 100 V for 45 min, and images were captured using the ChemiDoc™ Imaging System (Bio-Rad Laboratories). After resolving the PCR product on a 1% agarose gel containing ethidium bromide, the desired band was excised and purified using the QIAquick Gel Extraction Kit (Qiagen). Sanger sequencing was performed by an external vendor (1st BASE, Singapore), and sequencing results were analyzed using BioEdit Sequence Alignment Editor67. All the annotated sequences are presented in Supplementary Figure 5.
Data availability
The datasets generated and/or analyzed during the current study are available in the supplementary file and upon request from the corresponding author, Yuansheng Yang.
Abbreviations
- CHO:
-
Chinese Hamster Ovary
- DualDS:
-
Dual display and secretion
- ECSTASY:
-
Enzyme-cleavable surface-tethered all-purpose screening system
- EMCV:
-
Encephalomyocarditis virus
- FCS:
-
Furin cleavage sequence
- Fm:
-
Minimal furin cleavage sequence
- Fm-T2A:
-
Minimal furin cleavage sequence and 2A peptide from thosea asigna virus
- FRT:
-
Flippase recognition target
- GPI:
-
Glycosylphosphatidylinositol
- HC:
-
Heavy chain
- HYG:
-
Hygromycin resistance gene
- IRES:
-
Internal ribosomal entry site
- IVCD:
-
Integrated viable cell density
- jPCR:
-
Junction PCR
- LC:
-
Light chain
- MCL:
-
Master cell line
- MP:
-
Multiple promoters
- pA:
-
Simian virus 40 polyadenylation signal
- PURO:
-
Puromycin resistance gene
- qMab:
-
Specific productivity
- RMCE:
-
Recombinase-mediated cassette exchange
- SGSG:
-
Serine-glycine-serine-glycine linker
- SV40:
-
Simian virus 40 polyadenylation signal
- ZZ-PDGFR:
-
Protein A dual Z-domain with the PDGFR transmembrane domain
References
-
King, D. J., Bowers, P. M., Kehry, M. R. & Horlick, R. A. Mammalian cell display and somatic hypermutation in vitro for human antibody discovery. Curr Drug Discov. Technol. 11, 56–64 (2014).
-
Huhtinen, O., Salbo, R., Lamminmäki, U. & Prince, S. Selection of biophysically favorable antibody variants using a modified Flp-In CHO mammalian display platform. Front. Bioeng. Biotechnol. 11, 1170081 (2023).
-
Luo, R. et al. Simultaneous maturation of single chain antibody stability and affinity by CHO cell display. Bioengineering 9, 360 (2022).
-
Fischer, K. et al. Rapid discovery of monoclonal antibodies by microfluidics-enabled FACS of single pathogen-specific antibody-secreting cells. Nat. Biotechnol. 43, 1–11 (2024).
-
Gaa, R. et al. Mammalian display to secretion switchable libraries for antibody preselection and high throughput functional screening. In Mabs vol. 15 2251190 (Taylor & Francis, 2023).
-
Chakrabarti, L. et al. Amber suppression coupled with inducible surface display identifies cells with high recombinant protein productivity. Biotechnol. Bioeng. 116, 793–804 (2019).
-
Yu, B., Wages, J. M. & Larrick, J. W. Antibody-membrane switch (AMS) technology for facile cell line development. Protein Eng. Des. Sel. 27, 309–315 (2014).
-
Breous-Nystrom, E. et al. Retrocyte Display® technology: generation and screening of a high diversity cellular antibody library. Methods 65, 57–67 (2014).
-
Chen, C.-P. et al. ECSTASY, an adjustable membrane-tethered/soluble protein expression system for the directed evolution of mammalian proteins. Protein Eng. Des. Sel. 25, 367–375 (2012).
-
Wang, S. et al. Precision engineering of antibodies: A review of modification and design in the Fab region. Int. J. Biol. Macromol. 275, 133730 (2024).
-
Simons, J. F. et al. Affinity maturation of antibodies by combinatorial codon mutagenesis versus error-prone PCR. MAbs 12, 1803646 (2020).
-
Jiang, Y. Yeast surface display of full-length igg for engineering monoclonal and bispecific antibodies. Antib Ther. 6, tbad014.022 (2023).
-
Horlick, R. A. et al. Simultaneous surface display and secretion of proteins from mammalian cells facilitate efficient in vitro selection and maturation of antibodies. J. Biol. Chem. 288, 19861–19869 (2013).
-
Aebischer-Gumy, C. et al. SPLICELECT™: An adaptable cell surface display technology based on alternative splicing allowing the qualitative and quantitative prediction of secreted product at a single-cell level. MAbs 12, 1709333 (2020).
-
Waldmeier, L. et al. Transpo-mAb display: Transposition-mediated B cell display and functional screening of full-length IgG antibody libraries. In MAbs vol. 8 726–740 (Taylor & Francis, 2016).
-
Hellmann, I. et al. Novel antibody drug conjugates targeting tumor-associated receptor tyrosine kinase ROR2 by functional screening of fully human antibody libraries using Transpo-mAb display on progenitor B cells. Front Immunol. 9, 2490 (2018).
-
Lang, S. et al. Surface display vectors for selective detection and isolation of high level antibody producing cells. Biotechnol. Bioeng. 113, 2386–2393 (2016).
-
Yu, K. K. et al. Use of mutated self-cleaving 2A peptides as a molecular rheostat to direct simultaneous formation of membrane and secreted anti-HIV immunoglobulins. PLoS ONE 7, e50438 (2012).
-
Zhou, Y. et al. Simultaneous expression of displayed and secreted antibodies for antibody screen. PLoS ONE 8, e80005 (2013).
-
Bouquin, T., Rasmussen, P. B., Bertilsson, P.-A. & Okkels, J. S. Regulated readthrough: A new method for the alternative tagging and targeting of recombinant proteins. J. Biotechnol. 125, 516–528 (2006).
-
Chuang, K. H. et al. High-throughput sorting of the highest producing cell via a transiently protein-anchored system. PLoS ONE 9, e102569 (2014).
-
Tomimatsu, K. et al. A rapid screening and production method using a novel mammalian cell display to isolate human monoclonal antibodies. Biochem. Biophys. Res. Commun. 441, 59–64 (2013).
-
Higuchi, K. et al. Cell display library for gene cloning of variable regions of human antibodies to hepatitis B surface antigen. J. Immunol. Methods 202, 193–204 (1997).
-
Beerli, R. R. et al. Isolation of human monoclonal antibodies by mammalian cell display. Proc. Natl. Acad. Sci. USA 105, 14336–14341 (2008).
-
Akamatsu, Y., Pakabunto, K., Xu, Z., Zhang, Y. & Tsurushita, N. Whole IgG surface display on mammalian cells: Application to isolation of neutralizing chicken monoclonal anti-IL-12 antibodies. J. Immunol. Methods 327, 40–52 (2007).
-
Parthiban, K. et al. A comprehensive search of functional sequence space using large mammalian display libraries created by gene editing. MAbs 11, 884–898 (2019).
-
Taube, R. et al. Lentivirus display: stable expression of human antibodies on the surface of human cells and virus particles. PLoS ONE 3, e3181 (2008).
-
Zhang, J. et al. Mammalian cell display for rapid screening scFv antibody therapy. Acta Biochim. Biophys. Sin. (Shanghai) 46, 859–866 (2014).
-
Dyson, M. R. et al. Beyond affinity: selection of antibody variants with optimal biophysical properties and reduced immunogenicity from mammalian display libraries. MAbs 12, 1829335 (2020).
-
Mason, D. M. et al. High-throughput antibody engineering in mammalian cells by CRISPR/Cas9-mediated homology-directed mutagenesis. Nucleic Acids Res. 46, 7436–7449 (2018).
-
Parola, C. et al. Antibody discovery and engineering by enhanced CRISPR-Cas9 integration of variable gene cassette libraries in mammalian cells. In Mabs vol. 11 1367–1380 (Taylor & Francis, 2019).
-
Chen, S. et al. Affinity maturation of anti-TNF-alpha scFv with somatic hypermutation in non-B cells. Protein Cell 3, 460–469 (2012).
-
Chen, C., Li, N., Zhao, Y. & Hang, H. Y. Coupling recombinase-mediated cassette exchange with somatic hypermutation for antibody affinity maturation in CHO cells. Biotechnol. Bioeng. 113, 39–51 (2016).
-
Nguyen, A. W., Le, K. C. & Maynard, J. A. Identification of high affinity HER2 binding antibodies using CHO Fab surface display. Protein Eng. Des. Sel. 31, 91–101 (2018).
-
Zhou, C., Jacobsen, F. W., Cai, L., Chen, Q. & Shen, D. Development of a novel mammalian cell surface antibody display platform. In Mabs vol. 2 508–518 (Taylor & Francis, 2010).
-
Dhara, V. G., Naik, H. M., Majewska, N. I. & Betenbaugh, M. J. Recombinant antibody production in CHO and NS0 cells: Differences and similarities. BioDrugs 32, 571–584 (2018).
-
Andersen, D. C. & Reilly, D. E. Production technologies for monoclonal antibodies and their fragments. Curr. Opin. Biotechnol. 15, 456–462 (2004).
-
Valldorf, B. et al. Antibody display technologies: Selecting the cream of the crop. Biol. Chem. 403, 455–477 (2022).
-
Bowers, P. M. et al. Mammalian cell display for the discovery and optimization of antibody therapeutics. Methods 65, 44–56 (2014).
-
Robertson, N. et al. Development of a novel mammalian display system for selection of antibodies against membrane proteins. J. Biol. Chem. 295, 18436–18448 (2020).
-
Ho, S. C. L. et al. Comparison of internal ribosome entry site (IRES) and furin-2A (F2A) for monoclonal antibody expression level and quality in CHO cells. PLoS ONE 8, e63247 (2013).
-
Ho, M., Nagata, S. & Pastan, I. Isolation of anti-CD22 Fv with high affinity by Fv display on human cells. Proc. Natl. Acad. Sci. 103, 9637–9642 (2006).
-
Kim, K. et al. Two-promoter vector is highly efficient for overproduction of protein complexes. Protein Sci. 13, 1698–1703 (2004).
-
Szymczak, A. L. & Vignali, D. A. A. Development of 2A peptide-based strategies in the design of multicistronic vectors. Expert Opin. Biol. Ther. 5, 627–638 (2005).
-
Mizuguchi, H., Xu, Z., Ishii-Watabe, A., Uchida, E. & Hayakawa, T. IRES-dependent second gene expression is significantly lower than cap-dependent first gene expression in a bicistronic vector. Mol. Ther. 1, 376–382 (2000).
-
Ahier, A. & Jarriault, S. Simultaneous expression of multiple proteins under a single promoter in Caenorhabditis elegans via a versatile 2A-based toolkit. Genetics 196, 605–613 (2014).
-
Daniels, R. W., Rossano, A. J., Macleod, G. T. & Ganetzky, B. Expression of multiple transgenes from a single construct using viral 2A peptides in Drosophila. PLoS ONE 9, e100637 (2014).
-
Chng, J. et al. Cleavage efficient 2A peptides for high level monoclonal antibody expression in CHO cells. In MAbs vol. 7 403–412 (Taylor & Francis, 2015).
-
Yang, S. et al. Development of optimal bicistronic lentiviral vectors facilitates high-level TCR gene expression and robust tumor cell recognition. Gene Ther. 15, 1411–1423 (2008).
-
Ho, S. C. L. et al. Comparison of internal ribosome entry site (IRES) and Furin-2A (F2A) for monoclonal antibody expression level and quality in CHO cells. PLoS ONE 8, e63247 (2013).
-
Ho, S. C. L. et al. IRES-mediated Tricistronic vectors for enhancing generation of high monoclonal antibody expressing CHO cell lines. J. Biotechnol. 157, 130–139 (2012).
-
WO2024186270A1—Engineered furin cleavage sequences for co-expression of multiple genes in mammalian cells—Google Patents. https://patents.google.com/patent/WO2024186270A1/en?oq=WO2024186270A1.
-
Izidoro, M. A. et al. A study of human furin specificity using synthetic peptides derived from natural substrates, and effects of potassium ions. Arch. Biochem. Biophys. 487, 105–114 (2009).
-
Weldon, J. E. et al. Designing the furin-cleavable linker in recombinant immunotoxins based on Pseudomonas exotoxin A. Bioconjug. Chem. 26, 1120–1128 (2015).
-
Kim, M.-S. & Lee, G.-M. Use of Flp-mediated cassette exchange in the development of a CHO cell line stably producing erythropoietin. J. Microbiol. Biotechnol. 18, 1342–1351 (2008).
-
Barnes, L. M., Bentley, C. M., Moy, N. & Dickson, A. J. Molecular analysis of successful cell line selection in transfected GS-NS0 myeloma cells. Biotechnol. Bioeng. 96, 337–348 (2007).
-
Chusainow, J. et al. A study of monoclonal antibody-producing CHO cell lines: What makes a stable high producer?. Biotechnol. Bioeng. 102, 1182–1196 (2009).
-
Ng, S. K., Lin, W., Sachdeva, R., Wang, D. I. C. & Yap, M. G. S. Vector fragmentation: Characterizing vector integrity in transfected clones by Southern blotting. Biotechnol. Prog. 26, 11–20 (2010).
-
Stepanenko, A. A. & Heng, H. H. Transient and stable vector transfection: Pitfalls, off-target effects, artifacts. Mutat. Res./Rev. Mutat. Res. 773, 91–103 (2017).
-
Molnar, M. J. et al. Factors influencing the efficacy, longevity, and safety of electroporation-assisted plasmid-based gene transfer into mouse muscles. Mol. Ther. 10, 447–455 (2004).
-
Hornstein, B. D., Roman, D., Arévalo-Soliz, L. M., Engevik, M. A. & Zechiedrich, L. Effects of circular DNA length on transfection efficiency by electroporation into HeLa cells. PLoS ONE 11, e0167537 (2016).
-
Grav, L. M. et al. Minimizing clonal variation during mammalian cell line engineering for improved systems biology data generation. ACS Synth. Biol. 7, 2148–2159 (2018).
-
Ong, H. K., Nguyen, N. T. B., Bi, J. & Yang, Y. Vector design for enhancing expression level and assembly of knob-into-hole based FabscFv-Fc bispecific antibodies in CHO cells. Antib. Ther. 5, 288 (2022).
-
Nguyen, N. T. B. et al. Multiplexed engineering glycosyltransferase genes in CHO cells via targeted integration for producing antibodies with diverse complex-type N-glycans. Sci. Rep. 11, 12969 (2021).
-
Lin, J. et al. Impact of signal peptides on Furin-2A mediated monoclonal antibody secretion in CHO cells. Biotechnol. J. 12, 1700268 (2017).
-
Ingavat, N. et al. Harnessing ceramic hydroxyapatite as an effective polishing strategy to remove product-and process-related impurities in bispecific antibody purification. Bioresour. Bioprocess 10, 93 (2023).
-
Hall, T. A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. In Nucleic Acids Symposium Series vol. 41 95–98 (Oxford, 1999).
Acknowledgements
This work was supported by the Biomedical Research Council (BMRC) of the Agency for Science, Technology and Research (A*STAR), Singapore and Bioprocessing Technology Institute, A*STAR, Singapore. J.P.Z.N was supported by A*STAR Graduate Scholarship (AGS) from A*STAR Graduate Academy (AGA).
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ng, J.P.Z., Mariati, M., Ong, H.K. et al. Optimizing co-expression strategies for the simultaneous display and secretion of monoclonal antibodies in CHO cells. Sci Rep 15, 42508 (2025). https://doi.org/10.1038/s41598-025-26682-x
-
Received:
-
Accepted:
-
Published:
-
Version of record:
-
DOI: https://doi.org/10.1038/s41598-025-26682-x