Introduction
Type 2 Diabetes Mellitus (T2DM) is a chronic metabolic disorder characterized by insulin resistance and impaired insulin secretion, leading to elevated blood glucose levels1. This condition has reached epidemic proportions globally, significantly contributing to morbidity and mortality rates. In 2021, approximately 529 million people were living with diabetes globally, of which T2DM accounted for more than 80% of total diabetes cases2. Despite advancements in medical treatments, managing T2DM remains challenging due to its complex pathophysiology, and the limitations of current treatment options often rely on synthetic drugs and pharmacotherapies that may pose side effects or exhibit limited long-term efficacy over time and often have adverse effects, or fail to provide a precise therapeutic solution3,4,5. Although widely prescribed, standard antidiabetic drugs are associated with important side effects. Metformin often causes gastrointestinal intolerance and, in rare cases, lactic acidosis6,7. Sulfonylureas (e.g., gliclazide, tolbutamide, glimepiride, gliquidone, nateglinide) can induce hypoglycemia and weight gain8,9. Thiazolidinediones (rosiglitazone, pioglitazone, troglitazone) have been linked to cardiovascular risk, edema, and bone fractures10,11,12. Such limitations underscore the need to investigate safer, multi-targeted natural alternatives. The complexity of T2DM management and the need for novel therapeutic strategies are underscored by the global rise in incidence and the inadequacy of existing pharmacological modalities to halt disease progression10,11.
Natural compounds derived from botanical sources have garnered considerable attention for their potential therapeutic effects, attributed to their bioactive components and multi-targeted mechanisms of action13,14,15. Recent studies highlight the role of these compounds in modulating biochemical pathways involved in diabetes, offering a complementary approach to traditional pharmacotherapies. Studies have explored the impact of natural compounds such as resveratrol and curcumin on T2DM16,17,18. These compounds have demonstrated significant potential in modulating key biochemical pathways implicated in the disease; for example, resveratrol enhances the function of SIRT1 and AMPK pathways which is vital for cellular energy regulation and metabolic processes while curcumin targets the NF-κB pathway, known for its anti-inflammatory and antioxidative properties, which help in reducing oxidative stress and improving insulin sensitivity19,20,21,22. Such research findings support the therapeutic integration of these natural compounds into diabetes management, suggesting they can provide beneficial effects beyond conventional pharmacological treatments. Natural compounds offer a promising avenue for developing safer and more effective treatments for T2DM. However, identifying potent bioactive compounds that target specific T2DM-associated genes requires a systematic and comprehensive approach14,23.
Therefore, this study aimed to explore and identify natural compounds with high binding affinities to a wide range of key T2DM-related genes using advanced computational tools and extensive databases. Our research focuses on the relevance of these fourteen target genes to T2DM and availability in compound databases using molecular docking, pharmacokinetic, ADMET analyses, and molecular dynamics to identify lead molecule candidates with strong therapeutic potential. This methodology approach may help develop safer, more effective precision medicine strategies for chronic diseases like T2DM by integrating modern bioinformatics with traditional drug discovery. The results of this research contribute to valuable insights into the development of natural compound-based therapies for T2DM and pave the way for future experimental validations and clinical studies to advance personalized medicine strategies in diabetic care.
Materials and methods
Data collection
A comprehensive in-silico approach was adopted to identify potential bioactive natural compounds targeting genes involved in T2DM from botanical sources.
Gene targets
Identifying the genes associated with T2DM was done via the Online Mendelian Inheritance in Man (OMIM) database, STRING v.12.0 database (https://string-db.org), and research study claims. A search for “Type 2 Diabetes” and “Diabetes Mellitus” was conducted in OMIM in the Gene-Phenotype relationships section, specifically referencing the entry for Type 2 Diabetes Mellitus (#125853) to determine the associated genes. Same searching criteria was applied for the STRING database (122 items (human)). This provided an initial list of 30 gene targets in OMIM and 122 targets in STRING implicated in T2DM of which only nine of 30 from OMIM and three of 122 from STRING were selected for this study as they were the only ones available on compound databases, presenting active components for them. Apart from that, two targets AMPK (PRKAG3) and GAA claimed to be one of the integrated diabetic hormones in previous literature were also studied24,25, making the list consist of 14 total gene targets in this study (Table 1).
The proteins’ three-dimensional (3D) X-ray crystallographic structures were then retrieved from the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (http://www.pdb.org) and their structure identifiers can be found in Table 1.
Natural compounds sources and selection
Natural compounds targeting the fourteen T2DM-relevant genes were retrieved from the Natural Product Activity and Species Source Database (NPASS). Each gene was queried using standardized identifiers (common names, scientific names, other names, and abbreviations) to construct an initial compound library. NPASS entries include natural products from diverse biological origins (marine, microbial, animal, and plant); only compounds of plant origin were retained for this study. Source species were verified based on organism annotations and literature-linked evidence provided in the NPASS database, and compounds without clear explicit botanical attribution were excluded. The scientific names of all plant sources are listed in Supplementary File 1 (Table S1, “plant scientific name” column).
Compound selection followed a target-centric strategy, wherein each compound was retrieved in association with a specific protein target implicated in T2DM. This ensured initial functional relevance, which was further substantiated through molecular docking and molecular dynamics simulations. Compounds were pre-screened for drug-likeness using Lipinski’s Rule of Five alongside early-stage ADMET filters to refine the library toward pharmacologically viable candidates. Only compounds meeting these criteria advanced to downstream computational evaluation. These compounds were then subjected to docking analyses against the 14 T2DM-associated proteins. A final shortlist (consisting of 72 of the most promising ligands) was created based on their performance compared to reference antidiabetic drugs, as detailed in the Network Pharmacology Analysis section.
Library curation and compilation
Curating and Preparing 3D structures of genes Docking
The 3D structures of the fourteen selected identified target genes were located in the UniProt database, and the protein’s A chain PDB file for each gene was downloaded and saved with the corresponding entry name and ID (Table 1). The downloaded PDB file structures of the target genes were prepped for molecular docking using UCSF Chimera 1.17.3 software26,27. The pre-docking prep involved editing and optimizing the PDB files, ensuring their suitability and unity for docking procedures.
Curating natural product compound libraries
A curated library of natural product compounds was obtained from the ZINC12 database28,29. The database was searched for drug-like libraries using preset settings for each target gene, and the curated ZINC12 libraries were downloaded as SDF files for further analysis. The full list of natural compounds and their accession IDs from ZINC12 for each target protein is found in Supplementary File 2 (Table S2).
The 3D conformers of the selected NPASS compounds were obtained from PubChem. For the six compounds without the 3D conformers (C05, C06, C27, C87, C180, and C394), 3D structures were created using their Canonical Smiles via UCSF Chimera 1.17.3 software to ensure all compounds had uniformed format 3D representations.
Identification of marketed drugs for comparison purposes
Marketed drugs targeting the identified T2DM genes were screened, identified, and selected from the DRUGBANK database for future comparison with the characteristics and functionality of the curated botanical natural compounds. Each target gene was searched in the DRUGBANK database to identify existing approved and/or investigational drugs, and the most suitable ones, mainly hitting our reference target gene and relating to the condition T2DM, were selected as the comparison candidates. The 3D conformers of these handpicked marketed drugs were downloaded from PubChem to facilitate comparative analysis uniformity. For drugs without the 3D structures available on PubChem, the 3D conformer was simulated and built using their Canonical Smiles retrieved from PubChem by UCSF Chimera 1.17.3 software. Comparative analysis was conducted against selected drugs listed in Supplementary File 3 (Table S3).
Compilation of compounds and drugs libraries
A unified molecular library was created by combining NPASS, ZINC12, and DRUGBANK data (Supplementary File 4, Table S4). Molecular 3D structures from these sources were assembled using Open Babel GUI 2.3.2 to create unified molecular libraries for each target gene to build a comprehensive dataset for docking analyses30.
In-Silico analysis
Comparative analysis with approved drugs
A detailed comparative analysis was undertaken to evaluate the potential of selected natural compounds against approved drugs targeting T2DM-related genes. Molecular docking results, obtained using PyRx with Vina Wizard, provided binding affinity comparisons between natural compounds and corresponding approved drugs retrieved from the DrugBank database. This analysis assessed the efficacy, safety, and side effect profiles of approved drugs juxtaposed with the therapeutic potential of natural compounds. The aim was to identify natural compounds with comparable or superior binding affinities to highlight potential candidates for further investigation as potential alternatives or supplements to existing T2DM therapies.
Molecular Docking
Protein-ligand blind molecular docking was performed using PyRx Virtual Screening Tool 1.1 with AutoDock Vina Wizard31. Docking studies were conducted for each target gene and its associated ligands’ molecular library separately. Results were analyzed to identify potential bioactive compounds to identify promising natural compounds from botanical sources that target the genes involved in T2DM. For all docking runs, the “Maximize” option under the AutoGrid Dimensions tool in PyRx AutoDock was used to define the grid box, ensuring that the entire protein surface was encompassed for ligands to explore all potential binding sites and identify the most efficient binding poses. Additionally, the exhaustiveness parameter was set to 32 across all runs to enhance the reliability and reproducibility of the docking results. All docking simulations were performed in triplicate for each compound–protein pair to validate docking reproducibility, and the resulting binding affinities and poses were consistently reproduced across runs. Bond energies, including van der Waals interactions, hydrogen bonds, and electrostatic energy, were investigated, and the final comparison to pick the top-ranking promising compounds was decided and ranked based on their lower and upper RMSD, binding affinity values, and docked energy, with docking scores of the natural compounds compared to those of standard drugs. Only conformers that presented both upper and lower RMSD values of 0.0 were selected for the comparison; the binding affinity values were listed and ranked for all compounds and drugs. The top-ranked compounds were checked to detect whether they bind to the same binding pocket the drug fits into by assessing dock pose visual analysis using PyRx 1.1, PyMOL v3.0, and PockDrug (https://pockdrug.rpbs.univ-paris-diderot.fr) and the ones binding to different pockets were eliminated from the list. Following docking, the PockDrug server was used to confirm that each ligand bound within a predicted druggable pocket to enhance confidence in the biological relevance of the docking poses.
Protein-ligand interaction analysis
The interaction between the selected ligands and their responsive protein was evaluated at the binding site using the Protein-Ligand Interaction Profiler (PLIP)32,33 tool (https://plip-tool.biotec.tu-dresden.de/), and the involved amino acid residue, type of interaction bond, specific location of the atoms of both protein and ligand molecule, and the distance between the atoms forming the bond was recorded. The docking pose simulation illustration of each protein-ligand complex was visualized using PyMOL v.3.0.
Pharmacokinetic and ADMET analysis
Drug-likeness and pharmacokinetic (PK) properties of the candidate compounds were evaluated using a combination of established tools and predictive algorithms, with initial filtering based on Lipinski’s Rule of Five. Predictions of drug-likeness parameters—including molecular weight, hydrogen bond acceptors/donors, and LogP values—were conducted using MolSoft’s Mprop tool (https://molsoft.com/mprop/). While most compounds adhered to drug-likeness rules, seven substances (C04, C20, C80, C320, C353, C354, and C459) that failed Lipinski’s criteria but had prior experimental or clinical evidence for antidiabetic activity were retained to broaden the chemical diversity of the screening dataset (see Supplementary File 5, Table S5). Apart from these exceptions, all remaining compounds were subject to strict ADMET-based prioritization before inclusion in downstream docking and MD analysis.
Further ADMET and toxicity profiling was conducted using ProTox-3.0 (https://tox.charite.de/protox3/), ADMETlab (https://admet.scbdd.com/calcpre/index), admetSAR 2.0 (lmmd.ecust.edu.cn/admetsar2), and SwissADME (www.swissadme.ch). These tools were used to predict essential pharmacokinetic parameters, including human intestinal absorption (HIA), blood-brain barrier (BBB) permeability, caco-2 permeability (LogPapp, cm/s), half-life (T ½, hour), clearance (CL, mL/min/kg), LD50 (mg/kg), toxicity class (on the scale of 1 to 6 ̶ 1 equals highest toxicity and 6, the lowest), carcinogenicity toxicity endpoints (TEP), mutagenicity TEP, and nutritional toxicity TEP.
Compounds with HIA values < 0.8, low Caco-2 permeability, or classified as highly toxic (LD50 < 500 mg/kg or flagged as active carcinogens/mutagens) were deprioritized in the final compound selection to ensure pharmacological viability. The current multi-parameter screening strategy ensured only candidates with realistic pharmacokinetic and safety profiles were advanced to the docking and MD stages. Canonical SMILES representations were used as input for all prediction tools.
Network Pharmacology analysis
The network pharmacology analysis was conducted to explore the interactions between natural compounds, their target proteins, and the relevant signaling pathways implicated in T2DM. The analysis began with the construction of a compound-target-pathway network using Cytoscape 3.6.1. The network included 72 bioactive natural compounds identified through molecular docking studies, targeting 14 proteins associated with T2DM. These 72 compounds were systematically shortlisted from the initial large curated compound library based on their superior docking performance compared to reference antidiabetic drugs, and pre-screening filters (Lipinski’s Rule of Five and ADMET criteria). Only plant-derived compounds linked to the 14 selected T2DM genes were retained during the selection process to ensure that the network analysis proceeded with a pharmacologically viable and biologically relevant set of candidates, rather than an arbitrary collection of compounds.
The degree of connectivity for each node in the network was analyzed, with larger nodes indicating a higher level of interaction. The network visualized in Cytoscape allows identifying key interactions and multi-target effects that may contribute to the therapeutic potential of these natural compounds in T2DM management.
Gene set enrichment analysis
Pathway enrichment analysis was performed using the Kyoto encyclopedia of genes and genomes (KEGG) 2021 Human database via the Enrichr34,35,36,37,38,39 tool (https://maayanlab.cloud/Enrichr/) to identify the biological pathways modulated by the compounds. This analysis aimed to highlight critical pathways in T2DM. Gene ontology (GO)40,41 Biological Process (BP), GO Cellular Component (CC) functional, and GO Molecular Function37,38 enrichment analysis for the selected genes were performed via the above-mentioned tool.
Molecular dynamics simulation
Molecular dynamics (MD) simulations were employed to evaluate the structural stability, flexibility, and interaction behavior of 18 top-ranked protein-ligand complexes identified via molecular docking. One complex (Complex 5) was excluded due to structural instability and failure to reach equilibrium during initial tests. All simulations were executed on a Linux-based system running Ubuntu 22.04 using the Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) molecular dynamics package42.
The simulation process was conducted in five main stages:
- 1.
System Preparation.
Each protein-ligand complex was placed in a cubic simulation box solvated with TIP3P water molecules, extending 20 Å beyond the solute in all directions to ensure sufficient solvation. Na⁺ and Cl⁻ counterions were added to neutralize the system and replicate a physiological ionic strength of 0.15 M. Periodic boundary conditions and real units were applied for consistency with biomolecular simulations.
- 2.
Atom Definition and Structure Optimization.
Ligand and protein topologies were generated using the General AMBER Force Field (GAFF). Although a hybrid force field setup was originally proposed (GAFF for ligands and AMBER for proteins), it was not adopted due to stability issues. Hence, GAFF was applied uniformly to all atoms in the system. LAMMPS was selected for these simulations due to its high scalability on our Linux-based HPC system and its compatibility with the GAFF force field. While hybrid setups introduced instability during equilibration, applying GAFF uniformly maintained consistency across the system and minimized force field mismatches, ensuring stable and reproducible trajectories.
Initial energy minimization was conducted in two stages: (i) with positional restraints and (ii) without restraints, using the Conjugate Gradient method to ensure high accuracy. This was followed by equilibration using the NVT ensemble at 300 K for 50 ps, then NPT equilibration at 300 K and 1 atm for another 50 ps.
- 3.
Simulation Parameters.
A conservative time step of 0.1 fs was adopted to enhance accuracy and ensure stable trajectories of modeled structures. Non-bonded interactions were treated using a 10 Å cutoff, and the SHAKE algorithm was applied to constrain covalent bonds involving hydrogen atoms. Due to instability observed with PME, electrostatics were handled using conventional Coulombic methods.
- 4.
Production Run.
Production MD simulations were carried out for 500 ns under NPT conditions at 310 K and 1 atm pressure. Throughout the simulation, a list of key parameters—Potential Energy, Total Energy, Van der Waals Energy, Coulombic Energy, Enthalpy, Root-Mean-Square Deviation (RMSD), Root-Mean-Square Fluctuation (RMSF), Radius of Gyration (Rg), Solvent Accessible Surface Area (SASA), Buried SASA, Hydrogen Bond Analysis, and Free Energy—was monitored and recorded.
- 5.
Data Analysis.
Due to the limitations of LAMMPS in handling and performing the analyses of Principal Component Analysis (PCA) and Free Energy Landscape (FEL) calculations, thermodynamic properties such as van der Waals energy, enthalpy, and free energy were included to complement the simulation output. All numerical outputs were recorded, and graphical representations—including RMSD, Rg, and SASA plots—were included in the relative supplementary figures (Figures S28-S44). Each time step in the hydrogen-bond data output corresponds to 5 ns, with the final index (100) representing the 500 ns simulation endpoint.
Results
Molecular Docking results
Molecular docking was conducted using the PyRx virtual screening tool integrated with AutoDock Vina, a highly efficient molecular docking and virtual screening software. The 3D structures of the target proteins were obtained from the RCSB Protein Data Bank, and the ligands (natural compounds and reference drugs) were sourced from the PubChem and ZINC databases. Before docking, the protein and ligand structures were prepared by adding polar hydrogens, assigning appropriate charges, and removing previously attached ligands, solvents, and water molecules. Grid boxes were defined around the active sites of the target proteins to ensure the correct positioning of the ligands during docking. The docking process involved simulating the binding of each ligand to the target protein’s active sites and calculating the binding affinities, expressed in kcal/mol. These binding affinities were used to predict the interaction strength between the ligands and the target proteins. In molecular docking, more negative binding affinity values (ΔG, in kcal/mol) indicate stronger predicted ligand-target interactions, with values below − 7.0 kcal/mol generally considered strong binders under in-silico conditions. The results were analyzed to identify natural compounds with higher or comparable binding affinities to the reference drugs, indicating their potential as bioactive drug candidates for treating T2DM. Supplementary File 6 (Table S6) encompasses the complete molecular docking record for all genes.
01. GPD2 Docking results
GPD2 was selected for due to its central role in hepatic gluconeogenesis, a process often dysregulated in T2DM. Moracin D and Moracin P bound to active site A with binding affinities of -9.8 and − 9.7 kcal/mol, respectively. These values are higher than the binding affinity of NADH (-9.0 kcal/mol), indicating that Moracin D and Moracin P may serve as better bioactive drug candidates than the drug, NADH. No compounds bound to the active site C that Metformin binds to; therefore, no comparisons could be made for this pocket.
02. IRS1 Docking results
IRS1 was chosen due to its critical involvement in insulin signal transduction. It is a known mediator of insulin action and resistance, making it a particularly relevant target in the context of T2DM. 5-[5-(4-Hydroxy-Benzyl)-4-(4-Methoxy-Benzyl)-1-Methyl-1 H-Imidazol-2-Ylamino]-3-Methyl-Imidazole-2,4-Dione showed a binding affinity of -7.1 kcal/mol. This is competitive with the drug [4-({5-(AMINOCARBONYL)-4-[(3-METHYLPHENYL)AMINO] PYRIMIDIN-2-YL}AMINO)PHENYL]ACETIC ACID (-7.2 kcal/mol), although they bind to different active sites, preventing a direct comparison. Knowing that the compound 5-[1-(4-Hydroxy-Benzyl)-4-(4-Methoxy-Benzyl)-1 H-Imidazol-2-Ylamino]-3-Methyl-Imidazole-2,4-Dione also binds to the other binding pocket than the reference drug, therefore, no comparison can also be made for this compound.
03. PPARG Docking results
Troglitazone, Icosapent, and AMG-131 bound to active site A with binding affinities of -7.9, -6.5, and − 9.1 kcal/mol, respectively. Pyrene, Guggulsterone, and Emodin exhibited stronger binding affinities of -8.6, -8.4, and − 8.1 kcal/mol, respectively, compared to Troglitazone. Ten compounds showed stronger binding affinities than Icosapent. Pyrene emerged as the most potential compound for pocket A; no compounds could present higher values than the reference drug AMG-131.
Pioglitazone and Rosiglitazone bound with affinities of -8.5 and − 6.9 kcal/mol, respectively.
Compounds such as Plantagineoside A (-9.3 kcal/mol), Isosilybin A (-9.1 kcal/mol), Amorphastilbol (-8.9 kcal/mol), Piperitol, Chrysin, and Plantagineoside B (-8.7 kcal/mol), (1R,2 S,5R,6R)-5’-O-Methylpluviatilol, Silibinin, and Bavachinin A (-8.6 kcal/mol) showed stronger binding affinities compared to Pioglitazone. In total, 35 compounds outperformed Rosiglitazone. Plantagineoside A was identified as the most potential compound for pocket B.
04. IAPP Docking results
Due to the 3D structure of Copper only containing an ion (Cu), it was unsuitable for docking. Thus, no comparisons could be made between the only compound found for IAPP (Curcumin (1E,4Z,6E)-5-hydroxy-1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-1,4,6-trien-3-one) with the reference drugs Copper. Despite being unable to compare the compound Curcumin (1E,4Z,6E)-5-hydroxy-1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-1,4,6-trien-3-one with Copper, this compound showed highly promising values of binding affinity (-35.6 kcal/mol) with the protein amylin, making it worth investigating further.
05. GCK Docking results
The drug 3-[(4-fluorophenyl)sulfanyl]-N-(4-methyl-1,3-thiazol-2-yl)-6-[(4-methyl-4 H-1,2,4-triazol-3-yl)sulfanyl]pyridine-2-carboxamide was found to be the most effective with a binding affinity of -8.9 kcal/mol. The compound, Rohitukine showed a higher binding affinity (-7.8 kcal/mol) compared to Beta-D-Glucose (-5.2 kcal/mol), indicating its potential as a more effective drug candidate.
06. ABCC8 Docking results
Glyburide (Glibenclamide) showed a binding affinity equal to Gliquidone (-10.7 kcal/mol), suggesting equal potential. Berberrubine (-8.5 kcal/mol) had a higher binding affinity than Nateglinide (-8.3 kcal/mol), making it a potential drug candidate. Also, Berberine Chloride (-8.2 kcal/mol) exhibited a higher binding affinity than Tolbutamide (-7.5 kcal/mol), indicating its potential efficacy.
07. MAPK8 Docking results
Both compounds Apigenin (-7.1 kcal/mol) and Emodin (-7.0 kcal/mol) displayed higher binding affinities than the reference drug Halicin (-5.8 kcal/mol), therefore, they could be more effective bioactive drug candidates.
08. MTNR1B Docking results
5-Meo-Dmt and Serotonin bound to active sites other than pocket A, thus, no direct comparisons could be made. The drug, Tasimelteon had the strongest binding affinity (-8.2 kcal/mol) among all studied ligands. Compounds N-[2-(5-methoxy-1 H-indol-3-yl)ethyl]cyclopropanecarboxamide (-7.7 kcal/mol), Propionamide (-7.6 kcal/mol), Z02 (-7.4 kcal/mol), and Melatonin (-7.3 kcal/mol) showed high binding affinities after Tasimelteon, respectively.
09. AKT2 Docking results
Chelerythrine (-10.4 kcal/mol) and Alvocidib (-10.2 kcal/mol) showed higher binding affinities than both drugs Capivasertib (-9.7 kcal/mol) and N-[(1 S)-2-amino-1-phenylethyl]-5-(1 H-pyrrolo[2,3-b]pyridin-4-yl)thiophene-2-carboxamide (-9.1 kcal/mol), indicating their strong potential as natural drug candidates. The compound, A-443,654 (-9.3 kcal/mol) exhibited a higher binding affinity than drug N-[(1 S)-2-amino-1-phenylethyl]-5-(1 H-pyrrolo[2,3-b]pyridin-4-yl)thiophene-2-carboxamide, suggesting its potential effectiveness.
10. PTPN1 Docking results
Compounds bound to 11 active sites (A-K). Both studied drugs Trodusquemine (-5.8 kcal/mol) and Ertiprotafib (-7.4 kcal/mol) bound to site A. Eighty-eight compounds exhibited binding affinities of -7.4 or lower, making them competitive with Ertiprotafib. Among these, nine compounds matched the − 7.4 affinity, while seventy-nine compounds had affinities ranging from − 7.5 to -9.0, indicating a stronger potential than Ertiprotafib. Only six compounds (Z02, 325, 263, 216, 394, and 232) showed binding affinities equal to or lower than Trodusquemine. Z02 – Caffeic acid matched the − 5.8 affinity, while the others exceeded this value, making them less comparable. The strongest binding affinities to pocket A were observed with Xambioona (-9.0 kcal/mol), 18Alpha-Glycyrrhetinic Acid (-8.8 kcal/mol), 18Beta-Glycyrrhetic Acid (-8.8 kcal/mol), Taiwaniaflavone (-8.8 kcal/mol), and Oleanderolide (-8.7 kcal/mol), highlighting these as the most potential drug candidates for PTPN1.
11. INSR Docking results
Linsitinib and the compound Altertoxin I exhibited the strongest binding affinity value of -9.3 kcal/mol, indicating that Altertoxin I has equal potential to Linsitinib. Furthermore, the compounds Altertoxin I, Alvocidib, A-443,654, Chelerythrine, Gefitinib, Ellagic Acid, Macrosporin, Alternariol monomethyl ether, Alternariol, Sb-202,190, 6-O-Methylalaternin, and Sp-600,125 demonstrated higher binding affinities than Fostamatinib, suggesting their superior potential compared to this drug.
12. AMPK Docking results
Compounds Chelerythrine, Sp-600,125, Rohitukine, and Sb-202,190 displayed binding affinity values of -10.2, -8.8, -8.4, and − 8.0 kcal/mol, respectively. These values are higher than those for drugs Fostamatinib (-7.8 kcal/mol) and Aspirin (-5.9 kcal/mol), indicating these compounds have higher binding potential.
13. GAA Docking results
The four ligands for the protein GAA bound to four different binding sites. Consequently, no direct comparison of their binding abilities can be made.
14. SLC2A4 Docking results
Nine compounds (C404, C168, C461, C462, C463, C296, C292, C338, and C193) exhibited better binding affinities than the drugs Ascorbic Acid and Glucosamine. Among these, Ursolic Acid and Oleanolic Acid showed the best binding affinities, highlighting their significant potential as alternatives to Ascorbic Acid and Glucosamine.
Table 2 summarizes the top compounds exhibiting potential antidiabetic properties, with superior docking values compared to existing reference drugs. These compounds show significant promise for further evaluation and potential use as alternative medications or natural therapeutics to currently approved or investigational drugs.
The list is later narrowed down from 72 potential ligands to 18 ligands (Table 3) that showed the most remarkable docking scores for each of the studied proteins. The selected 18 ligands were used as a smaller subset of the data for protein-ligand binding site interaction and molecular docking analyses.
Analysis of Docking Ligand-Protein interactions at the binding site
The molecular docking studies revealed specific interactions between the selected subset of 18 ligand molecules and targeted amino acid residues within the binding site of their responsive protein, listed in Table 3.
Supplementary File 7 (Figures S1-S24 and Tables S7-S58) illustrates the docking pose of each protein-ligand interaction complex number to highlight the key interactions with involved residues and show types of interactions and bonds at the binding site, which contribute to the stability and specificity of the ligand binding.
PK/PD and ADME/T analysis results
The PK/PD and ADME/T properties of the 72 selected natural compounds were evaluated and predicted and top ligands were selected for their superior ADME/T characteristics and potential to effectively traverse biological barriers which is essential for oral administration of antidiabetic drugs. The ADME/T results are provided in Supplementary File 8 (Tables S59-S62).
- 1.
Selection of Antidiabetic Compounds Based on ADME Profiles.
In the selection of potential antidiabetic compounds, a meticulous evaluation was conducted focusing on their Absorption, Distribution, Metabolism, and Excretion (ADME) profiles, with an emphasis on Human Intestinal Absorption (HIA), Caco-2 Permeability, and Blood-Brain Barrier (BBB) permeability. The rationale behind choosing each compound was grounded in their capability to ensure optimal therapeutic efficacy and patient compliance. Below is a detailed justification for the choice of each compound based on these ADME criteria:
Pyrene (C54) and Guggulsterone (C60) exhibit maximum HIA and significant Caco-2 permeability, suggesting excellent oral bioavailability. Their high BBB permeability also suggests potential utility in addressing diabetes-related neurological complications. Despite Pyrene showing favorable ADMET properties, it is flagged as both carcinogenic and mutagenic (Table S62) and was therefore excluded from the lead compound consideration. Melatonin (C103) and Gefitinib (C456) show perfect and near-perfect HIA scores respectively, with considerable BBB permeability, indicating potential benefits beyond glycemic control, particularly in neurological protection. Apigenin (C100), Rotenone (C73), and Curcumin (C96) are characterized by high HIA and Caco-2 scores, ensuring effective absorption and systemic availability, crucial for consistent therapeutic effects. Bavachinin A (C24), Bavachinin (C10), and Quinidine (C61), while displaying lower BBB permeability, maintain high HIA and Caco-2 permeability, focusing their action on peripheral organs involved in glucose metabolism. This selection presumes these compounds are safe, pending further pharmacological and toxicological evaluations to fully ascertain their therapeutic profiles.
- 2.
Evaluation of Toxicity Profiles for Antidiabetic Compounds.
In parallel with ADME profiling, an assessment of the toxicity profiles was crucial in selecting antidiabetic compounds to ensure patient safety, particularly relevant in chronic conditions like diabetes where long-term medication is common. Here’s the rationale for choosing compounds based on their toxicity characteristics:
2-Tert-Butyl-6-[(3-Tert-Butyl-2-Hydroxy-5-Methylphenyl)Methyl]-4-Methylphenol (C95) and 4-(2-Phenylpropan-2-Yl)Phenol (C45) both fall within Toxicity Class V, indicating they may be harmful if swallowed but possess a relatively low acute toxicity, which is an important consideration for medications intended for chronic use. Their inactive statuses across carcinogenicity, mutagenicity, and nutritional toxicity, coupled with high confidence values, suggest these compounds are associated with minimal long-term adverse effects.
Phenothiazine (C80), although minimally toxic (Class V), shows an excellent safety profile with inactive statuses in all three toxicity indicators. This compound’s high LD50 value and safety markers make it a promising candidate for diabetes treatment, especially considering the potential for long-term therapy. 2-Naphthalen-1-Ylacetic Acid (C26), categorized under Toxicity Class IV, offers a moderate safety margin. Despite its relatively higher acute toxicity, the inactive status in major toxicity categories underscores its potential as a safer alternative for diabetes management, provided that its efficacy and specific diabetic application are validated in clinical settings. The focus on compounds with high safety profiles is aimed at minimizing potential risks associated with long-term drug administration, thereby enhancing patient compliance and improving overall quality of life for diabetic patients. These compounds represent not only effective therapeutic options but also safer alternatives in the development of antidiabetic medications. Further experimental and clinical validation of these compounds could elucidate their efficacy and safety as potential treatments for diabetes.
Network Pharmacology analysis results
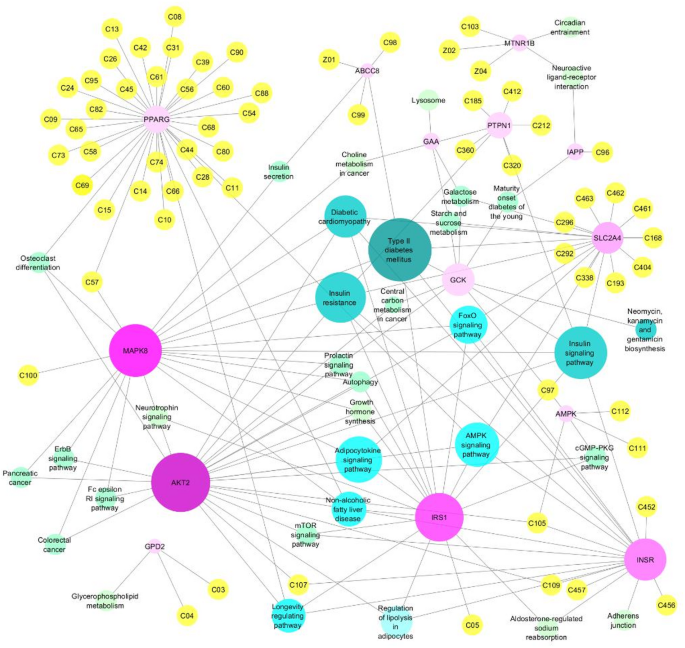
The compound-target-pathway network analysis revealed significant interactions between the selected 72 natural compounds and the 14 target proteins associated with T2DM (Fig. 1).

The systematic compound-target-pathway network in this study.
Key findings indicated that several compounds, such as Curcumin, Chelerythrine, and Ursolic Acid, demonstrated strong interactions with critical proteins involved in insulin signaling, such as PPARG, SLC2A4, and PTPN1. These proteins were identified as central hubs in the network due to their high degree of connectivity with multiple compounds.
The identified compounds indicated potential synergistic influences on effective T2DM management. The network visual representation underscored the importance of multi-target interactions of five compounds: Chelerythrine (C105) linked to three target genes AKT2, INSR, and AMPK, and compounds Emodin (C57), Rohitukine (C97), A-443,654 (C107), and Alvocidib (C109), each targeted two genes, simultaneously affecting multiple proteins within the same pathway.
Gene set enrichment analysis
Pathway enrichment analysis further highlighted the involvement of key signaling pathways, including the type II diabetes mellitus pathway (MAPK8, IRS1, ABCC8, INSR, SLC2A4, and GCK), insulin signaling pathway (PTPN1, MAPK8, IRS1, INSR, AKT2, SLC2A4, and GCK), insulin resistance (PTPN1, MAPK8, IRS1, INSR, AKT2, and SLC2A4), adipocytokine signaling pathway (MAPK8, IRS1, AKT2, and SLC2A4), and AMPK signaling pathway (IRS1, INSR, AKT2, PPARG, and SLC2A4). While the pathways with highest combined scores were related to diabetes and obesity, some of the genes also incorporated within other pathways associated with other diseases (Fig. 2). For instance, based on overlap genes, Non-alcoholic fatty liver disease (MAPK8, IRS1, INSR, AKT2, and PPARG), pancreatic and colorectal cancer (MAPK8 and AKT2), Alzheimer disease (MAPK8, IRS1, INSR, and AKT2), and thyroid cancer (PPARG), endometrial and prostate cancer and melanoma (AKT2), tuberculosis and human immunodeficiency virus 1 (HIV) infection (MAPK8 and AKT2) were of other remarkable integrated pathways with these compounds and genes. Supplementary File 9 (Figures S25-S27) offers a detailed Gene Ontology (GO) functional pathway enrichment analysis for selected genes in 2023. In the GO Biological Process category, “Response to Insulin” is the most significantly enriched term, with a p-value of 5.25e-12. The top term for the GO Cellular Component is “Protein Kinase Complex,” having a p-value of 5.27e-05. Lastly, in the GO Molecular Function category, “Insulin-Like Growth Factor Receptor Binding” is highlighted as the most enriched function, with a p-value of 3.03e-05. GO results underscore critical biological functions, components, and interactions associated with the studied gene set to provide valuable insights into their roles in cellular processes in T2DM.

Molecular dynamics simulation of top-ranked protein-ligand complexes
To assess the dynamic behavior and structural stability of the 17 most promising protein-ligand complexes (Complex 05 excluded) identified via molecular docking (complexes are listed in Table 3), molecular dynamics (MD) simulations were performed for 500 ns under NPT ensemble conditions (310 K and 1 atm). Parameters including root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), solvent-accessible surface area (SASA), hydrogen bond formation, and thermodynamic energy profiles were evaluated (Table 4).
- 1.
Structural Stability and Flexibility (RMSD, Rg, RMSF).
RMSD values ranged from 4.39 Å (Complex 01: Moracin P–GPD2) to 5.33 Å (Complex 18: Oleanolic Acid–SLC2A4), suggesting that all complexes reached equilibrium and maintained overall structural integrity. The average radius of gyration (Rg) across all complexes remained between 30.41 Å and 31.01 Å, confirming that no major conformational collapse occurred. RMSF analysis revealed that most residues remained relatively rigid, with the lowest fluctuation in Complex 06, GCK–Rohitukine (0.67 Å) and highest in Complex 12, AKT2–Alvocidib (2.51 Å), the latter possibly indicating increased flexibility near loop or surface regions.
- 2.
Hydrogen Bond Dynamics.
Hydrogen bonding, critical for structural stability, varied across the studied complexes. The Complex 02 (Moracin D–GPD2) exhibited the highest average number of hydrogen bonds (439.9), while Complex 04 (Pyrene–PPARG) formed the fewest (249.98). Complexes involving Moracin compounds (Complexes 01 and 02), Berberrubine (Complex 08), and Ursolic Acid (Complex 17) consistently demonstrated high H-bond occupancy which reflects strong ligand-target interaction stability throughout the simulation.
- 3.
Solvent Accessibility and Buried Surface Area.
SASA analysis provided results on solvation effects and structural compactness. GPD2-bound complexes (Complexes 01 and 02) showed the largest solvent-accessible surface areas (17,273 Ų and 17,643 Ų), consistent with their extensive external contact regions. On the contrary, MAPK8 and PTPN1 complexes (Complexes 09 and 14) exhibited lower SASA values (10,781 Ų and 11,201 Ų), indicating a tighter solvation shell. Buried SASA, which reflects buried interface regions, was highest for Complex 02 (1,076 Ų) which explains its strong complexation potential. These observations were further supported by structural visualizations (Supplementary File 10, Figures S28-S44) illustrating noticeable conformational shifts and compact rearrangements across simulation endpoints.
- 4.
Energetic Profiles.
Potential energy values were most favorable for Moracin P–GPD2 (Complex 01: − 1.46 kcal/mol) and Moracin D–GPD2 (Complex 02: − 1.15 kcal/mol). Enthalpy and free energy values additionally confirmed these two complexes as energetically stable. Ursolic Acid–SLC2A4 (Complex 17) and Plantagineoside A–PPARG (Complex 03) also ranked among the top performers with free energy values of − 33.94 and − 36.89 kcal/mol, respectively.
- 5.
Structural and Surface Visualizations.
Visual inspection of initial and final simulation frames highlighted overall structural resilience across all 17 selected complexes, which showcases minimal unfolding and favorable compactness post-equilibration. The relevant visual data for all complexes (Supplementary File 11, Figures S45-S61) support quantitative findings on Rg, SASA, and RMSD stability, visually confirming the adaptive binding nature of the ligands without any major disruptions to the protein scaffold.
Discussion
T2DM is a complex metabolic disorder characterized by chronic hyperglycemia, insulin resistance, and impaired insulin secretion. In light of these complexities, discovering and developing novel antidiabetic agents require high-throughput screening and targeted mechanistic insights. This in-silico investigation systematically identified natural compounds from botanical sources that exhibit potential therapeutic efficacy against T2DM. This study provides a robust, systems-level understanding of how selected phytochemicals may interact with critical T2DM-associated targets by integrating molecular docking, MD simulations, PK and ADMET predictions, and network pharmacology analyses.
Our molecular docking analyses highlighted several potent natural compounds, notably the ligands Moracin D, Moracin P, Curcumin (1E,4Z,6E)-5-hydroxy-1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-1,4,6-trien-3-one, Chelerythrine, Alvocidib, Rohitukine, and Ursolic Acid which exhibited binding affinities superior to established antidiabetic drugs on the market; this suggests that these ligands can disrupt disease-related signaling pathways effectively. More specifically, Moracin D and Moracin P demonstrated higher binding affinities to glycerol-3-phosphate dehydrogenase 2 (GPD2; −9.8 and − 9.7 kcal/mol, respectively) compared to NADH (− 9.0 kcal/mol), proposing enhanced capacity to influence glycerol metabolism and gluconeogenesis. Under standard thermodynamic assumptions, a ΔG difference of ~ 0.8–0.9 kcal/mol corresponds to an estimated 5- to 8-fold increase in binding affinity, suggesting that these phytochemicals may engage GPD2 more effectively than its natural cofactor NADH and potentially inhibit its function with higher affinity. Since GPD2 is crucial in regulating the glycerol-phosphate shuttle and subsequently hepatic gluconeogenesis, inhibiting its dysregulated activity could help reduce hyperglycemia in T2DM. Curcumin emerged with an exceptionally strong affinity for islet amyloid polypeptide (IAPP; −35.6 kcal/mol) which considerably exceeded previously reported affinities for phytochemicals, such as Hydroxychavicol, Eugenol, and Chavibetol derived from Piper betle L. (Betle leaves), as well Genistein and Kaempferol from Annona muricata (Soursop plant)43,44. IAPP has been implicated in β-cell dysfunction via amyloid fibril formation, linked to T2DM progression. While Curcumin’s dramatically high docking score suggests that it may help modulate IAPP aggregation, our MD simulations revealed instability within the Curcumin–IAPP complex (Complex 5), meaning that additional factors beyond raw docking affinities, such as conformational flexibility, solvation effects, and protein-ligand adaptability, are therefore crucial to fully assess therapeutic viability. On the contrary, the Curcumin docking score (–35.6 kcal/mol) with the IAPP protein (Complex 05) is unusually strong, potentially supported by a dense network of hydrogen bonds and hydrophobic interactions, as illustrated in Supplementary Figure S7 and Tables S20–S21. However, this complex showed instability in molecular dynamics simulations, failing to reach equilibrium. This suggests the possibility of scoring artifacts resulting from excessive non-specific interactions or overpacking into flexible or non-physiological regions of the protein. Although Complex 05 was excluded from downstream MD-based evaluations for a realistic lead compound selection, experimental validation will be essential to confirm Curcumin’s binding behavior with IAPP and determine its true therapeutic potential.
Potential synergy with other standard T2DM therapies, such as metformin or SGLT2 inhibitors, remains another avenue for exploration since combining phytochemicals could yield more robust glycemic control or mitigate adverse effects compared to monotherapy.
Additionally, our findings align with and extend those of multiple recent studies that highlighted promising phytochemicals for T2DM therapy. Macalalad and Gonzales (2023) identified diverse plant-derived compounds targeting PTP1B (aka. PTPN1), Dipeptidyl Peptidase-4 (DPP-4), and Fructose 1,6-biphosphatase (FBPase) which paralleled our focus on PTPN1 with potential compounds Xambioona, 18Alpha-Glycyrrhetinic Acid, and Taiwaniaflavone45. Abdullah et al. (2023) similarly demonstrated the antidiabetic potential of Azadirachta indica (Neem plant) and emphasized stable molecular interactions similar to those observed in the current study for Chelerythrine–AKT2 and Ursolic Acid–SLC2A446. Balogun et al. (2023) strengthened this perspective by showcasing the potential of phytochemicals from Crescentia cujete (Calabash tree) in inhibiting carbohydrate-metabolizing enzymes associated with T2DM which supports the current study’s multi-target strategy47. Dar et al. (2024) investigated PPARG-targeting compounds from Gymnema sylvestre, including Quercetin and Rutin, which align with our identification of top PPARG-directed ligands, such as Plantagineoside A, Pyrene, and Emodin48. Also, Choudhury et al. (2024) explored Momordica charantia (Bitter melon) through precise protein-ligand interactions—reflected in the current study’s observations regarding compounds sourced from bitter melon, Moracin P and Moracin D’s strong affinity for GPD249. Finally, Halayal et al. (2024) and Singh et al. (2024) highlighted the efficacy of multi-target phytochemicals from Kalanchoe pinnata (Cathedral bells plant, stated to be effective in PI3-AKT, MAPK, and PPAR signaling pathways) and Cinnamomum tamala (Indian bay leaf, effective in PPAR and PI3K-AKT signaling pathways) respectively in modulating insulin signaling which is on par with our findings on AKT2, INSR, IRS, and PPARG50,51.
Molecular dynamics simulations served as a critical validation step to evaluate the stability, flexibility, and potential binding modes of each protein-ligand complex over time. The Moracin D–GPD2 and Moracin P–GPD2 complexes plateaued at an RMSD of 4.42 Å and 4.39 Å, respectively after ~ 10 ns of simulation which indicates that a stable binding conformation was rapidly achieved and maintained throughout the 100 ns run. The low RMSF values (1.52 Å and 1.62 Å) across GPD2’s active site residues showed minimal local fluctuations, and substantial hydrogen bond formations (averaging 439.9 and 429.11 H-bonds, respectively) suggested strong enthalpic contributions to the overall binding. These results align with Vijh and Gupta (2024) study’s reported stable RMSD (below 3 Å), an average Rg of 23.5 Å, and strong hydrogen bond formation (> 400 average occupancy) for natural compounds such as Soyasapogenol B and Corydine (from Dalbergia sissoo, aka. Indian rosewood) upon interaction with α-amylase (2QV4)52.
Our findings also provide a mechanistic basis for the potent docking affinities of Moracin D and Moracin P. The multi-target interactions in the current study observed for Chelerythrine, Alvocidib, and Rohitukine across proteins AKT2, INSR, and AMPK further support the therapeutic potential of polypharmacological strategies. Chelerythrine–AKT2 (RMSD: 4.68 Å, H-bonds: 469.01) and Alvocidib–AKT2 (RMSD: 4.71 Å, H-bonds: 464.36) displayed comparable stability to multi-target compounds studied by Saldívar-González et al. (2023) that identified chemical transformations of molecules to design multi-target compound libraries focused on targeting T2DM-related pathways particularly related to protein tyrosine phosphatase 1B (PTP1B) and aldose reductase (AR)53.
Apart from the top lead compounds based on MD results in the current study, Rohitukine–GCK (Complex 06) and Xambioona–PTPN1 (Complex 14) also showed reasonably stable final poses, though their free energies and RMSDs are slightly less favorable than top contenders; they still warrant interest but might rank slightly below the previously mentioned molecules. On the other hand, in the current study, with the Curcumin–IAPP complex (Complex 5)—initially hypothesized to be the most potential phytochemical in this study—exhibiting instability during MD simulations, it is confirmed how counting on high docking scores alone can be misleading in assessing ligand-protein compatibility. Comprehensive dynamic analyses are therefore essential to ensure that top-ranked poses translate into viable therapeutic candidates.
The potential clinical utility of these natural compounds is partly dependent on their ADMET characteristics. Our analyses identified Pyrene, Guggulsterone, and Curcumin as notable for favorable PK and ADMET profiles. Pyrene and Guggulsterone each displayed near-perfect intestinal absorption (~ 100%) and high BBB permeability, fulfilling clinical translation criteria outlined by Yanni (2015) while suggesting that many of the studied ligands might be orally bioavailable and potentially suitable for therapeutic development after in vivo confirmation54. Although Pyrene exhibited favorable pharmacokinetic properties in silico—including excellent intestinal absorption, strong BBB permeability (0.9728), and acceptable half-life—its predicted toxicity profile raises significant concerns. Specifically, Pyrene was flagged as both carcinogenic and mutagenic with high confidence scores (0.88 and 0.89, respectively; see Supplementary Table S62). Given this substantial toxicity risk, Pyrene was excluded from further consideration as a lead compound despite its computational performance and therefore, should not be considered a viable lead compound for further development. Its presence in the early-stage screening served only as a comparative data point and highlights the importance of integrating toxicity predictions into early drug discovery pipelines.
Network pharmacology analyses showed that T2DM is driven by interconnected pathways rather than isolated targets. Chelerythrine demonstrated multi-target capacity, interacting with AKT2, INSR, and AMPK—key mediators of insulin signaling and energy regulation. In parallel, pathway enrichment results identified insulin signaling, insulin resistance, and AMPK signaling as highly relevant T2DM pathways modulated by the compounds in this study. The broad distribution of gene targets hints that leveraging multiple plant-derived ligands could produce synergistic effects and yield more effective and comprehensive glycemic control. Additionally, these network interactions highlight the capacity for modulating inflammatory processes, oxidative stress pathways, and lipid metabolism linked with insulin action and T2DM progression. Several recent network pharmacology studies support these findings. Sahraeian et al. (2024) and Huang et al. (2024) emphasized that multi-target strategies can yield superior efficacy compared to single-target approaches in diabetic models, enhance metabolic regulation, and reduce side effects when employing conjugated phytochemical cocktails55,56. Similarly, Gu et al. (2024) reported that integrating network pharmacology with molecular docking and dynamics simulations provided deeper integrative pharmacology strategy into the cross-talk of pathways mediating insulin resistance and beta-cell function57. Patwardhan et al. (2010) supported this multi-target approach by theoretically advocating polypharmacology for achieving synergistic therapeutic outcomes in complex metabolic disorders58. Huang et al. (2024), Ahmed et al. (2022), and Kausar et al. (2022) demonstrated stable MD profiles for Apigenin derivatives interacting with metabolic regulators such as PPARγ43,56,59. Similarly, Yuan et al. (2024) reported stable values for Tirzepatide interacting with glucagon-like peptide-1 receptor (GLP-1R) and glucose-dependent insulinotropic polypeptide receptor (GIPR), analogous to our Altertoxin I–INSR complex60. Comparatively, Ahmed et al. (2022) identified apigenin-7-O-glucoside as a potent inhibitor with binding energies notably higher than standard inhibitors, highlighting the broad spectrum of natural compounds efficacy in managing T2DM43. Omiyale et al. (2024) also reported significant interactions between compounds from Annona muricata and carbohydrate-digesting enzymes, which complements our broader multi-target approach involving GPD2, AKT2, INSR, and AMPK44. Future studies may explore combinatorial approaches or co-administration of these phytochemicals, aiming to maximize network-based synergy while minimizing adverse effects and offering a more integrative therapeutic strategy for enhanced glycemic control and overall metabolic health.
Limitations of the study
Despite robust computational methodologies, inherent limitations of in-silico analyses persist. Molecular docking and MD simulations, although informative, cannot entirely replicate the complexity of in vivo biological interactions. PK and toxicity predictions require empirical validation. Additionally, Principal Component Analysis (PCA) and Free Energy Landscape (FEL) calculations, initially planned, could not be performed due to the limitations of LAMMPS in handling these specific analyses. The absence of PCA and FEL analyses restricts a comprehensive interpretation of the dynamic behavior and conformational landscapes of the studied complexes. Future work may consider using GROMACS, AMBER, or other compatible tools to address this limitation. Nonetheless, thermodynamic properties such as van der Waals energy, enthalpy, and free energy were included to complement the simulation output. Moreover, the potential synergistic or antagonistic interactions among selected natural compounds were not assessed and warrant further exploration.
Future prospects
Future research should prioritize experimental and empirical validation of computational findings through rigorous in vitro and in vivo experiments, beginning with in vitro assays to confirm predicted binding affinities and functional impacts on glucose metabolism and insulin signaling. Subsequent in vivo evaluations using diabetic animal models will be crucial to determine therapeutic efficacy and safety profiles. Investigating synergistic compound combinations might enhance therapeutic outcomes. Additionally, advancements in precision medicine may enable personalized therapeutic regimens tailored to individual genetic and metabolic profiles. Employing alternative simulation software, such as GROMACS or AMBER, to perform PCA and FEL analyses will provide deeper insights into the conformational dynamics and energetic landscapes of the protein-ligand interactions identified. Additionally, studies should explore potential synergistic effects among the identified natural compounds to develop multi-target therapeutic strategies for enhanced efficacy in T2DM management.
Conclusion
This computational study employed an in-silico approach to systematically identify novel antidiabetic agents and promising bioactive natural compounds from diverse botanical sources that target 14 key genes implicated in T2DM. By integrating molecular docking, pharmacokinetic and pharmacodynamic evaluations, and molecular dynamics simulations, we demonstrated that Moracin D, Moracin P, Plantagineoside A, Chelerythrine, Alvocidib, and Ursolic Acid possess robust binding affinities, exhibit stable protein-ligand interactions under simulated physiological conditions, and display favorable PK/ADMET characteristics which makes them promising therapeutic candidates against T2DM. Despite the encouraging results, the translational success of these compounds ultimately requires extensive experimental validation to clarify their exact biochemical mechanisms and effects on insulin signaling and glucose metabolism and verify their therapeutic efficacy, safety margins, and long-term tolerability in diabetic animal models before advancing to clinical trials.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
-
DeFronzo, R. A. et al. Type 2 diabetes mellitus. Nat. Rev. Dis. Primers. 1, 15019. https://doi.org/10.1038/nrdp.2015.19 (2015).
-
Ong, K. L. et al. Global, regional, and National burden of diabetes from 1990 to 2021, with projections of prevalence to 2050: a systematic analysis for the global burden of disease study 2021. Lancet 402, 203–234. https://doi.org/10.1016/S0140-6736(23)01301-6 (2023).
-
Owens, D. R., Monnier, L. & Barnett, A. H. Future challenges and therapeutic opportunities in type 2 diabetes: changing the paradigm of current therapy. Diabetes Obes. Metab. 19, 1339–1352. https://doi.org/10.1111/dom.12977 (2017).
-
DeMarsilis, A. et al. Pharmacotherapy of type 2 diabetes: an update and future directions. Metabolism 137, 155332. https://doi.org/10.1016/j.metabol.2022.155332 (2022).
-
Ansari, M. A. et al. Emerging therapeutic options in the management of diabetes: recent trends, challenges and future directions. Int. J. Obes. 47, 1179–1199. https://doi.org/10.1038/s41366-023-01369-3 (2023).
-
Bonnet, F. & Scheen, A. Understanding and overcoming Metformin Gastrointestinal intolerance. Diabetes Obes. Metab. 19, 473–481. https://doi.org/10.1111/dom.12854 (2017).
-
DeFronzo, R., Fleming, G. A., Chen, K. & Bicsak, T. A. Metformin-associated lactic acidosis: current perspectives on causes and risk. Metabolism 65, 20–29. https://doi.org/10.1016/j.metabol.2015.10.014 (2016). https://doi.org/https://doi.org/
-
Douros, A. et al. Pharmacologic differences of sulfonylureas and the risk of adverse cardiovascular and hypoglycemic events. Diabetes Care. 40, 1506–1513. https://doi.org/10.2337/dc17-0595 (2017).
-
Sahin, I., Bakiner, O., Demir, T., Sari, R. & Atmaca, A. Current position of Gliclazide and sulfonylureas in the contemporary treatment paradigm for type 2 diabetes: A scoping review. Diabetes Therapy. 15, 1687–1716. https://doi.org/10.1007/s13300-024-01612-8 (2024).
-
Della-Morte, D. et al. Pharmacogenomics and pharmacogenetics of thiazolidinediones: role in diabetes and cardiovascular risk factors. Pharmacogenomics 15, 2063–2082. https://doi.org/10.2217/pgs.14.162 (2014).
-
Sunder, M., Chang, A. R., Henry, R. R. & Thiazolidinediones Peripheral Edema, and type 2 diabetes: Incidence, Pathophysiology, and clinical implications. Endocr. Pract. 9, 406–416. https://doi.org/10.4158/EP.9.5.406 (2003).
-
Taylor, C. & Hobbs, F. D. R. Type 2 diabetes, thiazolidinediones, and cardiovascular risk. Br. J. Gen. Pract. 59, 520. https://doi.org/10.3399/bjgp09X453440 (2009).
-
Dzobo, K. The role of natural products as sources of therapeutic agents for innovative drug discovery. Compr. Pharmacol. 408–422. https://doi.org/10.1016/B978-0-12-820472-6.00041-4 (2022).
-
Mosaddeghi, P. et al. A systems Pharmacology approach to identify the autophagy-inducing effects of traditional Persian medicinal plants. Sci. Rep. 11, 336. https://doi.org/10.1038/s41598-020-79472-y (2021).
-
Chaachouay, N. & Zidane, L. Plant-Derived natural products: A source for drug discovery and development. Drugs Drug Candidates. 3, 184–207. https://doi.org/10.3390/ddc3010011 (2024).
-
Li, C. et al. The effect of resveratrol, Curcumin and Quercetin combination on immuno-suppression of tumor microenvironment for breast tumor-bearing mice. Sci. Rep. 13, 13278. https://doi.org/10.1038/s41598-023-39279-z (2023).
-
McCubrey, J. A. et al. Effects of berberine, curcumin, Resveratrol alone and in combination with chemotherapeutic drugs and signal transduction inhibitors on cancer cells—Power of nutraceuticals. Adv. Biol. Regul. 67, 190–211. https://doi.org/10.1016/j.jbior.2017.09.012 (2018).
-
Banaszak, M., Górna, I., Woźniak, D., Przysławski, J. & Drzymała-Czyż, S. The impact of Curcumin, Resveratrol, and cinnamon on modulating oxidative stress and antioxidant activity in type 2 diabetes: moving beyond an Anti-Hyperglycaemic evaluation. Antioxid. (Basel). 13. https://doi.org/10.3390/antiox13050510 (2024).
-
Um, J-H. et al. AMP-activated protein kinase-deficient mice are resistant to the metabolic effects of Resveratrol. Diabetes 59, 554–563. https://doi.org/10.2337/db09-0482 (2010).
-
Fullerton, M. D. & Steinberg, G. R. SIRT1 takes a backseat to AMPK in the regulation of insulin sensitivity by Resveratrol. Diabetes 59, 551–553. https://doi.org/10.2337/db09-1732 (2010).
-
Hussain, Y. et al. How Curcumin targets inflammatory mediators in diabetes: therapeutic insights and possible solutions. Molecules 27 https://doi.org/10.3390/molecules27134058 (2022).
-
Zamanian, M. Y. et al. NF-κB pathway as a molecular target for Curcumin in diabetes mellitus treatment: focusing on oxidative stress and inflammation. Cell. Biochem. Funct. 42, e4030. https://doi.org/10.1002/cbf.4030 (2024).
-
Rao, M. M. V. & Hariprasad, T. P. N. In Silico analysis of a potential antidiabetic phytochemical erythrin against therapeutic targets of diabetes. Silico Pharmacol. 9, 5. https://doi.org/10.1007/s40203-020-00065-8 (2021).
-
Lee, S-H. et al. Octaphlorethol A: a potent α-glucosidase inhibitor isolated from ishige foliacea shows an anti-hyperglycemic effect in mice with streptozotocin-induced diabetes. Food Funct. 5, 2602–2608. https://doi.org/10.1039/C4FO00420E (2014).
-
Sivajothi, V. & Dakappa, S. S. In vitro and in Silico antidiabetic activity of Pyran ester derivative isolated from tragia Cannabina. Asian Pac. J. Trop. Biomed. 4, S455–S459. https://doi.org/10.12980/APJTB.4.2014C1049 (2014).
-
Shapovalov, M. V. & Dunbrack, R. L. J. A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure 19, 844–858. https://doi.org/10.1016/j.str.2011.03.019 (2011).
-
Wang, J., Wang, W., Kollman, P. A. & Case, D. A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph Model. 25, 247–260. https://doi.org/10.1016/j.jmgm.2005.12.005 (2006).
-
Irwin, J. J., Sterling, T., Mysinger, M. M., Bolstad, E. S. & Coleman, R. G. ZINC: A free tool to discover chemistry for biology. J. Chem. Inf. Model. 52, 1757–1768. https://doi.org/10.1021/ci3001277 (2012).
-
Irwin, J. J. & Shoichet, B. K. ZINC–a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 45, 177–182. https://doi.org/10.1021/ci049714+ (2005).
-
O’Boyle, N. M. et al. Open babel: an open chemical toolbox. J. Cheminform. 3, 33. https://doi.org/10.1186/1758-2946-3-33 (2011).
-
Trott, O. & Olson, A. J. AutoDock vina: improving the speed and accuracy of Docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461. https://doi.org/10.1002/jcc.21334 (2010).
-
Salentin, S., Schreiber, S., Haupt, V. J., Adasme, M. F. & Schroeder, M. PLIP: fully automated protein–ligand interaction profiler. Nucleic Acids Res. 43, W443–W447. https://doi.org/10.1093/nar/gkv315 (2015).
-
Adasme, M. F. et al. PLIP 2021: expanding the scope of the protein–ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 49, W530–W534. https://doi.org/10.1093/nar/gkab294 (2021).
-
Chen, E. Y. et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 14, 128. https://doi.org/10.1186/1471-2105-14-128 (2013).
-
Xie, Z. et al. Gene set knowledge discovery with enrichr. Curr. Protoc. 1, e90. https://doi.org/10.1002/cpz1.90 (2021).
-
Kuleshov, M. V. et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97. https://doi.org/10.1093/nar/gkw377 (2016).
-
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462. https://doi.org/10.1093/nar/gkv1070 (2016).
-
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
-
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53, D672–D677. https://doi.org/10.1093/nar/gkae909 (2025).
-
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat. Genet. 25, 25–29. https://doi.org/10.1038/75556 (2000).
-
The Gene Ontology Consortium. Expansion of the gene ontology knowledgebase and resources. Nucleic Acids Res. 45, D331–D338. https://doi.org/10.1093/nar/gkw1108 (2017).
-
Thompson, A. P. et al. LAMMPS – a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comput. Phys. Commun. 271, 108171. https://doi.org/10.1016/j.cpc.2021.108171 (2022).
-
Ahmed, S. et al. Molecular Docking and dynamics simulation of natural compounds from betel leaves (Piper Betle L.) for investigating the potential Inhibition of Alpha-Amylase and Alpha-Glucosidase of type 2 diabetes. Molecules 2022;27. https://doi.org/10.3390/molecules27144526
-
Omiyale, B. O., Oyinloye, B. E., Ajiboye, B. O. & Ubah, C. S. Curated phytochemicals of Annona muricata modulate proteins linked to type II diabetes mellitus: molecular Docking studies, ADMET and DFT calculation. Inf. Med. Unlocked. 47, 101511. https://doi.org/10.1016/j.imu.2024.101511 (2024).
-
Macalalad, M. A. B. & Gonzales, A. A. In Silico screening and identification of antidiabetic inhibitors sourced from phytochemicals of Philippine plants against four protein targets of diabetes (PTP1B, DPP-4, SGLT-2, and FBPase). Molecules 28 https://doi.org/10.3390/molecules28145301 (2023).
-
Abdullah, A. et al. Molecular dynamics simulation and pharmacoinformatic integrated analysis of bioactive phytochemicals from Azadirachta indica (Neem) to treat diabetes mellitus. J. Chem. 2023, 4170703. https://doi.org/10.1155/2023/4170703 (2023).
-
Balogun, F. O. et al. Cheminformatics identification of modulators of key carbohydrate-metabolizing enzymes from C. cujete for type-2 diabetes mellitus intervention. J. Diabetes Metab. Disord. 22, 1299–1317. https://doi.org/10.1007/s40200-023-01249-7 (2023).
-
Dar, M. I. et al. Exploring the anti-diabetic mechanism of selective phytochemicals identified from Gymnema sylvestre using TLC-UPLC-MS, complemented by in Silico studies. Phytomedicine Plus. 4, 100606. https://doi.org/10.1016/j.phyplu.2024.100606 (2024).
-
Choudhury, A. AA et al. Anti-diabetic drug discovery using the bioactive compounds of momordica Charantia by molecular Docking and molecular dynamics analysis. J. Biomol. Struct. Dyn. 1–15. https://doi.org/10.1080/07391102.2024.2313156 (n.d.).
-
Singh, R. et al. Integrative network Pharmacology, molecular Docking, and dynamics simulations reveal the mechanisms of cinnamomum Tamala in diabetic nephropathy treatment: an in Silico study. Curr. Issues Mol. Biol. 46, 11868–11889. https://doi.org/10.3390/cimb46110705 (2024).
-
Halayal, R. Y., Bagewadi, Z. K., Aldabaan, N. A., Shaikh, I. A. & Khan, A. A. Exploring the therapeutic mechanism of potential phytocompounds from Kalanchoe pinnata in the treatment of diabetes mellitus by integrating network pharmacology, molecular Docking and simulation approach. Saudi Pharm. J. 32, 102026. https://doi.org/10.1016/j.jsps.2024.102026 (2024).
-
Vijh, D. & Gupta, P. GC–MS analysis, molecular docking, and Pharmacokinetic studies on dalbergia Sissoo barks extracts for compounds with anti-diabetic potential. Sci. Rep. 14, 24936. https://doi.org/10.1038/s41598-024-75570-3 (2024).
-
Saldívar-González, F. I., Navarrete-Vázquez, G. & Medina-Franco, J. L. Design of a multi-target focused library for antidiabetic targets using a comprehensive set of chemical transformation rules. Front. Pharmacol. 14, 1276444. https://doi.org/10.3389/fphar.2023.1276444 (2023).
-
Yanni, S. & Translational ADMET for Drug Therapy: Principles, Methods and Pharmaceutical Applications (Wiley-Interscience, 2015). https://doi.org/10.1002/9781118838440
-
Sahraeian, S., Rashidinejad, A. & Golmakani, M-T. Recent advances in the conjugation approaches for enhancing the bioavailability of polyphenols. Food Hydrocoll. 146, 109221. https://doi.org/10.1016/j.foodhyd.2023.109221 (2024).
-
Huang, X. et al. Combination of plant metabolites hinders starch digestion and glucose absorption while facilitating insulin sensitivity to diabetes. Front. Pharmacol. 15 https://doi.org/10.3389/fphar.2024.1362150 (2024).
-
Gu, H. et al. Exploring the mechanism of Jinlida granules against type 2 diabetes mellitus by an integrative Pharmacology strategy. Sci. Rep. 14, 10286. https://doi.org/10.1038/s41598-024-61011-8 (2024).
-
Patwardhan, B. & Vaidya, A. D. B. Natural products drug discovery: accelerating the clinical candidate development using reverse Pharmacology approaches. Indian J. Exp. Biol. 48, 220–227 (2010).
-
Kausar, A. et al. Identifying natural therapeutics against diabetes via Inhibition of dipeptidyl peptidase 4: molecular Docking and MD simulation study. Indian J. Pharm. Educ. Res. 56, s21–31. https://doi.org/10.5530/ijper.56.1s.39 (2022).
-
Yuan, J. et al. Molecular dynamics-guided optimization of BGM0504 enhances dual-target agonism for combating diabetes and obesity. Sci. Rep. 14, 16680. https://doi.org/10.1038/s41598-024-66998-8 (2024).
Acknowledgements
The authors would like to thank Dr. Maryam Farajpour Mojdehi and the Iranian Institute of New Sciences (IINS) for sharing their valuable knowledge and support throughout this study.
Funding
This study received no financial funding.
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This study did not involve human participants or animals and solely utilized publicly available data; therefore, ethical approval and consent to participate are not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Rahmani, B., Akbari, H., Esmaeili, H. et al. Discovering bioactive pharmaceuticals from natural products for type 2 diabetes mellitus using network pharmacology, molecular docking, and molecular dynamics. Sci Rep 15, 43205 (2025). https://doi.org/10.1038/s41598-025-27173-9
-
Received:
-
Accepted:
-
Published:
-
Version of record:
-
DOI: https://doi.org/10.1038/s41598-025-27173-9