
- Research
- Open access
- Published:
- Xiaotong Ji1,2,4 na1,
- Xixian Wang1,2 na1,
- Wenjun Zhou1,2,
- Lin Chen1,2,
- Tianzhong Liu1,2,
- Jian Xu1,3 &
- …
- Bo Ma1,3
Biotechnology for Biofuels and Bioproducts volume 18, Article number: 75 (2025) Cite this article
Abstract
Background
Palmitoleic acid, a valuable functional fatty acid, is notably scarce in traditional oil crops, with the exception of certain wild plants such as macadamia nuts and sea buckthorn. Recently, the lipid from Saccharomyces cerevisiae was found to contain approximately 50% palmitoleic acid. Consequently, S. cerevisiae has the potential to sustainably produce palmitoleic acid through fermentation, provided that the issue of promoting its lipid content is addressed.
Results
In this work, based on the previously isolated oleaginous wild strain of S. cerevisiae, the mutagenesis by zeocin combined with ARTP was carried out to generate S. cerevisiae mutants, and then the high lipid content mutants were isolated using the flow-mode Raman-activated cell sorting (FlowRACS) technique, which allowed for the high-throughput selection of these mutants in a label-free and non-invasive manner. The mutant MU2R48 was finally obtained and its lipid content was 40.26%, 30.85% higher than the original type. Transcriptome and targeted metabolome analysis revealed a coordinated interaction of fatty acid precursor biosynthesis, the pentose phosphate pathway, ethanol degradation, and amino acid metabolism, synergistically channeling carbon flux from acetyl-CoA and NADPH into lipid biosynthesis. Additionally, key transcriptional regulators within the lipid metabolism network were implicated in this enhanced lipid accumulation.
Conclusion
In this study, a mutant strain of Saccharomyces cerevisiae MU2R48 with 40.26% lipid content was successfully generated through zeocin-ARTP mutagenesis combined with Raman-activated cell sorting. Multi-omics analysis revealed that the enhanced lipid accumulation was driven by coordinated up-regulation of precursor biosynthesis, carbon flux redirection, and key transcriptional regulators, with increased acetyl-CoA and NADPH production fluxes likely serving as the pivotal determinants.
Background
Addressing chronic diseases through dietary interventions is an important research area, and the role of unsaturated fatty acids in maintaining human health is well recognized. It is widely known that unsaturated fatty acids like docosahexaenoic acid (DHA), eicosapentaenoic acid (EPA), arachidonic acid (ARA), and palmitoleic acid (POA) have been associated with various health benefits due to their potential effects on reducing inflammation, improving heart health, and supporting brain function [1, 2]. Consequently, there has been an escalating focus and research interest in natural functional dietary supplements that target unsaturated fatty acids in recent years [3,4,5,6,7].
At present, most unsaturated fatty acids can be produced stably on a commercial scale using microbial fermentation. POA, an important omega-7 monounsaturated fatty acid, has attracted widespread attention due to its biological activities and health benefits in conditions such as metabolic syndrome, diabetes, and inflammatory reactions [8,9,10,11]. However, its production is still largely dependent on a limited range of natural sources, such as wild plants or microorganisms. For example, sea buckthorn oil [12] contains about 25% of palmitoleic acid and macadamia seed oil [13] contains about 30%, in addition to which, the Ranunculus ternatus Thunb. contains about 64% [14]. Nevertheless, the accessibility of these wild resources is significantly constrained by climatic and geographical variables, posing a challenge to ensuring a sustainable supply. Conventional oil crops, such as peanuts, corn, and soybeans, exhibit minimal palmitoleic acid content, with oleic acid predominating as the primary unsaturated fatty acid in their oils [15]. Hence, there is an immediate need to identify a suitable microbial strain resource for palmitoleic acid production, especially by means of microbial fermentation which has been proven to be the most efficient way to produce functional fatty acids.
The majority of traditionally utilized oleaginous microbes rarely accumulate palmitoleic acid. Saccharomyces cerevisiae has been found to contain up to approximately 50% palmitoleic acid in its lipid composition, providing a potential novel microbial source of palmitoleic acid [16]. However, S. cerevisiae is not regarded as an industrial oleaginous microorganism due to its typically low lipid content (usually < 10%) [16, 17]. Therefore, increasing the lipid content is the prerequisite to facilitate the production of palmitoleic acid by S. cerevisiae. Fortunately, our laboratory had successfully isolated a strain of S. cerevisiae SC018 with the lipid content of about 30% through nitrogen-limited incubation assessment. Nevertheless, this lipid content remained insufficient to establish a cost-effective bioprocess for palmitoleic acid production.
Mutagenesis, or adaptive laboratory evolution (ALE), has been widely employed to increase the production of target metabolites of microorganisms [18,19,20,21]. Employing an appropriate mutagenesis strategy for increasing the lipid content of S. cerevisiae is reasonable. Various mutagenesis techniques including physical mutagenesis using X-rays, heavy-ion beam irradiation [22] and Atmospheric and Room Temperature Plasma (ARTP) [18], chemical mutagenesis by ethyl methanesulfonate (EMS) [23], N-methyl-N-nitro-N-nitrosoguanidine (MNNG) [24], and nitrosoguanidine (NTG) [25], have been widely and effectively utilized. Zeocin is a water-soluble glycopeptide antibiotic capable of chelating copper and has the ability to bind and damage DNA. It can reduce intracellular Cu2+ to Cu+, which activates DNA double-stranded incisions, leading to amino acid shifts and genomic changes during repair [26]. Combining mutagenesis sequentially with different methods has been validated to be more effective in overcoming resistant mutagenesis compared to a single treatment, resulting in a superimposed effect [27, 28].
After mutagenesis, the identification of the target mutant poses a challenging task. High-throughput screening techniques have received considerable attention, with fluorescence-activated cell sorting (FACS) being a prevalent choice [29,30,31]. Nonetheless, FACS necessitates the use of fluorescent staining, and the availability of suitable fluorescent dyes is scarce, with some being potentially harmful to microorganisms. In addition, one disadvantage of Nile Red, a commonly used fluorescent dye, is that it requires a penetrant (e.g., DMSO), which will reduce cell viability. BODIPY505/515, as an emerging neutral lipid substitute dye, stains rapidly and without the need for a penetration agent, making it an ideal choice for FACS [32]. However, the BODIPY505/515 fluoresces strongly with non-polar materials such as plastics [33], which may lead to some measurement errors. A single-cell Raman spectrum (SCRS) provides the intrinsic biochemical profile of a cell in a given state and thus can be considered as a function-based instant portrait photo of the cell [34]. The majority of lipid molecules exhibit pronounced Raman characteristic peaks, such as at 1650 cm−1 (C=C stretching), 1440 cm−1 (–CH2 twisting), and 2800–3000 cm−1 (CH2 symmetric and asymmetric stretching). [35]. These peaks have facilitated Raman characterization of total lipids, triglycerides, and unsaturated fatty acids in model cells like yeasts and microalgae [36,37,38]. By sorting cells via SCRS, Raman-activated cell sorting (RACS) holds great promise as a powerful platform for screening target strains based on specific traits, as it is label free, culture free, non-invasive, and broadly applicable [34]. In our previous work, a high-throughput FlowRACS system was developed and the profiling and sorting of pigment-producing yeast cells and astaxanthin-producing microalgae cells were realized based on resonance Raman peaks [39,40,41]. Moreover, the feasibility of FlowRACS in sorting triacylglycerol-producing yeast cells based on non-resonance Raman peaks was also demonstrated [42]. Hence, FlowRACS is an ideal platform for sorting lipid-hyperproducing S. cerevisiae strains.
In this study, attempts were made to obtain a S. cerevisiae mutant with high lipid content by combining mutagenesis with high-throughput FlowRACS. Through zeocin compound ARTP mutagenesis and two consecutive rounds of FlowRACS enrichment sorting, the S. cerevisiae mutant MU2R48 with greatly increased lipid content was successfully isolated. Subsequently, genomic association analysis through whole-genome resequencing and transcriptome analysis provided valuable insights into the key elements contributing to lipid accumulation in mutant strains. This approach demonstrated the feasibility and effectiveness of combining zeocin-ARTP compound mutagenesis with FlowRACS sorting to develop lipid-hyperproducing S. cerevisiae strains. The findings will facilitate the enhancement of comprehension in the molecular genetic mechanisms underlying radically increased lipid accumulation in traditional non-oleaginous microorganisms, thereby laying the foundation for optimizing the lipid production platform utilizing S. cerevisiae.
Results and discussion
Characterization of linear relationship and sorting accuracy
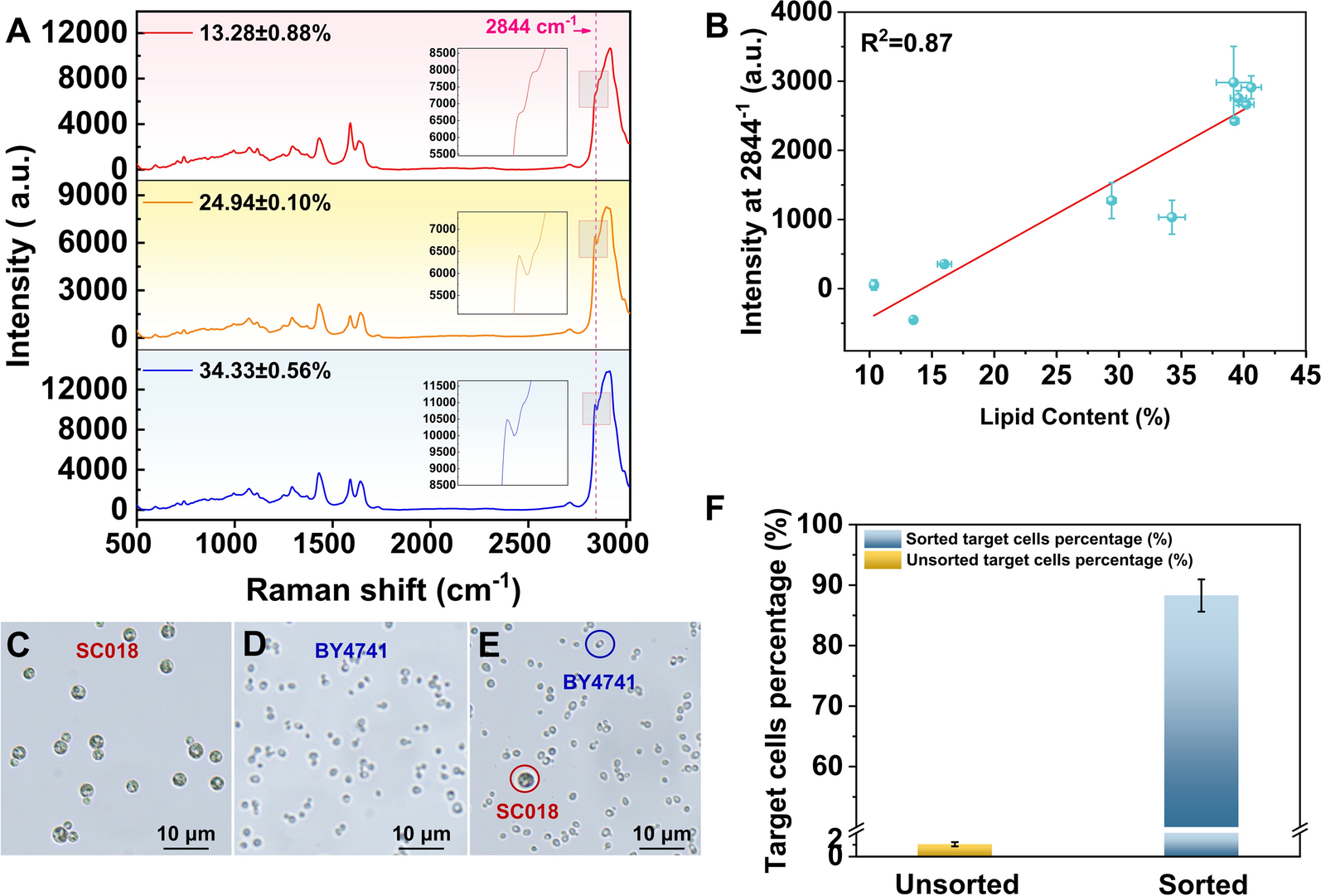
The intensity of the Raman spectrum at 2844 cm−1 (defined as I2844 – I2857) exhibited variation across S. cerevisiae samples with differing lipid contents (Fig. 1A). The correlation between the lipid content of the sample and its Raman intensity at 2844 cm−1 showed a positive linear relationship with R2 = 0.87 (Fig. 1B), which was better than the linear relationship between fluorescence intensity and lipid content (R2 = 0.65, Supporting information, Fig. S1), indicating a higher accuracy in profiling lipid content via FlowRACS than that of FACS. This superior correlation can be attributed to the specificity of Raman spectroscopy in detecting C-H stretching vibrations, which are directly associated with lipid molecules. The 2844 cm−1 peak is particularly indicative of symmetric CH2 stretching, a hallmark of lipid acyl chains, making it a robust marker for lipid quantification [43]. In contrast, fluorescence-based methods often suffer from non-specific binding and photobleaching, which can compromise accuracy. To further validate the sorting accuracy by FlowRACS, the cells with high lipid content (strain SC018, 3–5 μm in diameter; Fig. 1C) and cells with extremely low lipid content (strain BY4741, 0.5–1 μm in diameter; Fig. 1D) were mixed at a cell ratio of 1:99 (Fig. 1E) and underwent FlowRACS. Sorted cells were collected and cultured on YPD solid medium, and the purity of target cells (SC018) was measured via counting the colonies with larger cell size. It showed that the target cells percentage was greater than 85% (Fig. 1F). This high level of purity highlights the efficacy of FlowRACS in isolating specific cell populations based on lipid content, suggesting that it could potentially be applicable to other microorganisms where lipid content is a key parameter.

Characterization of linear relationship and sorting accuracy. A Averaged SCRS of S. cerevisiae cells with different lipid contents; B Linear relationship between the lipid content of S. cerevisiae cells and the Raman intensity at 2844 cm−1; Microscopy of morphology sizes of C strain SC018 cells, D strain BY4741 cells, and E 1:99 cells mixed sample of SC018 and BY4741 (Bar = 10 μm); F Sorting accuracy of SC018 cells
Selection of high lipid content mutant cells based on high-throughput FlowRACS
The direction and traits produced by mutagenesis are unknown, and the probability that a target strain may be identified from a single mutant library is low, so it is beneficial to construct multiple mutant pools and test the probability of the occurrence of the target strain in each mutant pool to improve the efficiency of screening. Among the fourteen mutant pools established through mutagenesis using various doses of zeocin, ARTP, and their combination, it was observed that the 9_ARTP 150 s-Zeocin 100 μg mL−1, 10_ARTP 30 s, and 14_Zeocin 150 μg mL−1-Zeocin 50 μg mL−1 exhibited a higher frequency of high Raman intensity at 2844 cm−1 (Supporting information, Fig. S2). These three mutant pools were mixed in equal proportions and subjected to FlowRACS sorting following lipid induction cultivation. Cells with high lipid content in the mutant pool were enriched by two consecutive rounds of FlowRACS to obtain Mut1R pool and Mut2R pool, respectively (Fig. 2A). As shown in Fig. 2B–D, after the 1st round of sorting enrichment, the frequency of cells with low TAG Raman intensity (0–3000 a.u.) decreased in Mut1R. In Mut2R (2nd-round sorting), 27.29% of the TAG Raman intensity ranging in 3000–4000, 2.92% in 4000–5000, and 0.39% in 5000–6000 were reached, which significantly increased the probability of screening cells with high lipid content. The iterative enrichment process is critical for isolating rare high-lipid variants from a heterogeneous population. The use of FlowRACS, which combines Raman spectroscopy with cell sorting, ensures that the selection is based on a direct and quantitative measure of lipid content, thereby improving the precision of the screening process. The shift in Raman intensity distribution after each round of sorting demonstrates the effectiveness of FlowRACS in progressively enriching the population for cells with high lipid content. The appearance of cells with Raman intensities above 5000 a.u. in the second round highlights the potential for identifying extreme lipid-accumulating phenotypes.

Enrichment process of targeted mutant pool and its Raman intensity frequency statistics at 2844cm−1. A Enrichment process of high lipid content mutant pool; Raman intensity frequency statistics at 2844cm−1 of the B initiating mutant pool, C Mut1R pool, and D Mut2R pool
Cells with high lipid content were rescreened by randomly selecting single clones from the Mut2R pool. The results of biomass and lipid accumulation evaluation are presented in Fig. 3A. 72% of the selected strains exhibited a higher lipid content than the original strain, among which MU2R48 exhibited the highest lipid content of 40.26%, with a relative increase by 30.85% compared to SC018, and a biomass of 11.94 g L−1, which was basically the same as the original strain (12.02 g L−1). The lipid content of MU2R48 was high and its biomass was comparable to that of the original strain, suggesting that the mutation leading to increased lipid accumulation did not affect cell growth. This is a crucial finding as it suggests that the strain can achieve higher lipid yields without sacrificing productivity, making it an ideal candidate for industrial applications. As depicted in Fig. 3B, the fatty acid composition of S. cerevisiae predominantly consisted of C16:0, C16:1, C18:0, and C18:1. Although the fatty acid composition of MU2R48 showed a slight decrease in the content of C16:1, the total fatty acid yield increased from 1.60 to 1.75 g L−1. The increase in total fatty acid production despite changes in fatty acid composition suggests that mutations in MU2R48 may enhance overall lipid biosynthesis rather than altering specific pathways for individual fatty acids. This may be due to changes in the expression levels of key enzymes in the lipid synthesis pathway or alterations in the regulatory mechanisms controlling lipid storage. Further multi-omics analysis of MU2R48 could provide insight into the specific genetic changes that lead to its enhanced lipid accumulation.

Evaluation of lipid production capacity of mutant cells. A Evaluation of lipid content by randomly selecting 50 monoclones (numbered MU2R1-MU2R50, abbreviated as 1–50 in the figure) from Mut2R mutant pool. Data are presented as mean ± SD (n = 3). Statistical significance was determined by two-tailed unpaired Student’s t-test (p* ≤ 0.05; pns(n) > 0.05, not significant); B the fatty acid composition of MU2R48 mutant
Transcriptome and metabolome analysis of the mutant with increased lipid content
The transcriptome data revealed a total of 5302 genes expressed in both SC018 and MU2R48 under identical culture condition. In addition to these, 4 genes were uniquely expressed in SC018, while 8 genes were uniquely expressed in the mutant strain. Within the mutant strain, there were 231 differentially expressed genes, including 120 up-regulated genes and 111 down-regulated genes. These differentially expressed genes demonstrated significant enrichment in various pathways. To delve deeper into this, we selected the top 20 pathway exhibiting with the highest enrichment for further mapping (Supporting information, Fig. S3). The analysis revealed that the most significantly enriched pathways were secondary metabolites, valine, leucine, and isoleucine metabolism, glycolysis, and other metabolic pathways. Utilizing the sequencing results and referring to the lipid synthesis-related metabolic pathways available in the KEGG database, we successfully mapped the primary relevant metabolic pathways of the mutant strain. These pathways included glycolysis, the TCA cycle, fatty acid synthesis and degradation, the pentose phosphate pathway, and amino acid metabolic pathways. Combining transcriptomic data with metabolic pathway mapping will provide a comprehensive view of metabolic changes in MU2R48. A summary of these pathways for genes with significant expression levels is provided in Table 1.
The differentially expressed genes in the MU2R48 transcriptome were found to be enriched within several metabolic pathways. Specifically, the expression of certain genes within the glycolytic pathway, fatty acid synthesis pathway, methionine, and valine synthesis pathways was observed to be up-regulated. Conversely, the expression of certain genes in the TCA and fatty acid β-oxidation pathway was down-regulated. As shown in Fig. 4, the expression of genes such as TDH1 (coding glyceraldehyde-3-phosphate dehydrogenase), ENO2 (coding enolase), CDC19 (coding pyruvate kinase), PDC (coding pyruvate decarboxylase isozyme), ACS2 (coding acetyl-coenzyme A synthetase), ADH4 (coding alcohol dehydrogenase), and ALD5 (coding aldehyde dehydrogenase) was up-regulated in the glycolytic pathway. In particular, the expression of the genes of PDC1, PDC5, PDC6, ACS2, ADH4, and ALD5 was significantly up-regulated at the expression level. The up-regulation of genes within the glycolytic pathway would increase the flux of pyruvate. The up-regulation of these glycolytic genes indicates an enhanced glycolytic flux, which is consistent with the increased demand for pyruvate and acetyl-CoA in lipid biosynthesis. This metabolic rewiring is a common strategy in oleaginous microorganisms to support high lipid accumulation under conditions of nutrient imbalance or stress [44]. Subsequently, an increase in the expression of the pyruvate decarboxylase isoenzyme gene would also increase the flux of acetyl-CoA. In addition, the ethanol produced during fermentation would generate large amounts of acetic acid in response to the up-regulation of ADH4 and ALD5, which would be rapidly catalyzed by the intracellularly expressed ACS2 [45] to generate acetyl-CoA. Overexpression of genes encoding ethanol dehydrogenase, acetaldehyde dehydrogenase, and acetyl-CoA synthase had been reported to increase fatty acid content by more than three-fold [46]. This pathway not only provides an alternative route for acetyl-CoA generation but also recycles carbon from fermentation byproducts, enhancing overall carbon efficiency. The expression of the acetyl-CoA synthase gene ACS2 was up-regulated by 2.55-fold in the mutant, indicating an increased flux of acetyl-CoA synthesis, whereas the expression of the citrate synthase gene CIT1 was down-regulated by 2.18-fold, thereby decreasing the flux of acetyl-CoA into the TCA and subsequently to fatty acid synthesis. Based on the gene sequencing results, a mutation at base 825 in the exon region of the ACS2 gene and a mutation at base 1179 in the exon region of the CIT2 gene, which resulted in the substitution of the amino acid alanine by glycine, may be considered as primary factors for the significant changes in gene expression levels (Supporting information, Table S1), which need to be further verified.

Transcription levels of genes in central carbon metabolism, fatty acid synthesis, and degradation pathways in MU2R48 compared to SC018. The number next to the genes represents the log2 (transformed fold changes). Red genes indicate significant up-regulation, green genes indicate significant down-regulation, and blue genes indicate no significant difference in transcript levels (|log2 (FoldChange)|≥ 1 & padj ≤ 0.05). All transcript level differences are expressed as mean of log2 (fold change) values. All sample size n (biological replicates) is 3
The transcription levels of genes involved in the fatty acid synthesis pathway exhibited up-regulation, among which acetyl-coenzyme A carboxylase (ACC1) was up-regulated by 1.87-fold and fatty acid synthase gene 2 (FAS2) was up-regulated by 2.28-fold. The up-regulation of ACC1 and FAS2 directly correlates with the increased lipid content observed in MU2R48. ACC1 catalyzes the conversion of acetyl-CoA to malonyl-CoA, the first committed step in fatty acid synthesis, while FAS2 is responsible for the elongation of fatty acid chains [47]. Their increased expression suggests a heightened capacity for de novo lipid synthesis in the mutant strain. In addition, the delta-9 desaturase gene (OLE1), which desaturates C16:0 and C18:0 to form the monounsaturated fatty acids C16:1 and C18:1 [48], was up-regulated by 1.69-fold, suggesting that the mutant has an enhanced ability to produce unsaturated fatty acids that are required to maintain membrane necessary for membrane fluidity and functionality under various environmental conditions. These findings suggested that the increased lipid content of the mutant MU2R48 was associated with a possible link to the elevation of the precursor acetyl-CoA and up-regulation of the expression of genes in the fatty acid synthesis pathway (Fig. 4). Fatty acid elongation requires NADPH as reducing power supply, and in the PPP pathway, the generation of D-ribulose-5p from gluconate-6p via 6-phosphogluconate dehydrogenase gene (GND1) was accompanied by substantial synthesis of NADPH [44]. The expression level of GND1 was up-regulated by 1.89-fold in the mutant strain, thereby providing substantial reducing power for fatty acid synthesis (Fig. 5).

Transcription levels of genes in amino acid metabolism and PPP pathways in MU2R48 strain. The number next to the genes represents the log2 (transformed fold changes). Red genes indicate significant up-regulation, green genes indicate significant down-regulation, and blue genes indicate no significant difference in transcript levels (|log2 (FoldChange)|≥ 1 & padj ≤ 0.05). All transcript level differences were expressed as mean of log2 (fold change) values. All sample size n (biological replicates) is 3
In the amino acid metabolic pathway, there was a significant up-regulation in the expression of genes encoding 3-isopropylmalate dehydratase (LEU1, LEU2, and LEU9), dihydroxy-acid dehydratase (ILV3), and branched-chain amino acid aminotransferase (BAT1), which were associated with metabolism of leucine, isoleucine, and valine. The expression level of LEU2 was observed to be up-regulated by 3.01-fold, while BAT1 was up-regulated by 7.67-fold. They were associated with leucine metabolism and were considered to be involved in the regulation of lipid metabolism in oleaginous Yarrowia lipolytica [49]. Amino acids can indirectly support lipid accumulation by acting as precursors to acetyl-CoA or by becoming intermediates in the lipid synthesis pathway. Furthermore, fatty acid synthesis is an energy-consuming process, in which ATP serves as the primary source of energy for yeast cells. However, a decrease in the ATP/AMP ratio was detected in MU2R48 targeted metabolome, which would subsequently stimulate glycolysis to enhance energy provision (Supporting information, Table S2). In addition to this, transcriptional regulators were implicated in the lipid accumulation process in the mutant. POT1 (coding 3-ketoacyl-CoA thiolase) is a target gene of the ADR1 (coding regulatory protein) regulatory factor which can regulate fatty acid utilization [50]. In the MU2R48, there was a significant reduction in the expression of ADR1 to 0.7-fold, the expression of POT1 was correspondingly reduced by 0.3-fold, and the expression of POX1 was down-regulated by 0.6-fold compared with the control group, which would likely lead to an attenuation of the β-oxidation pathway. This metabolic rewiring, where lipid degradation is minimized while synthesis is maximized, is a key strategy for achieving high lipid yields in oleaginous microorganisms. In addition, the lipid droplet (LD) morphology and expression levels of LD assembly genes (CKB1 and SEI1, encoding a regulatory subunit of casein kinase and the seipin homologue, respectively) in the mutant strain remained similar to those in the parental strain, with negligible fold changes (CKB1: − 0.1; SEI1: + 0.24). This suggested that the observed lipid accumulation primarily resulted from metabolic flux redistribution rather than enhanced storage capacity. Future comparative studies with non-oleaginous yeast may reveal the impact of LD assembly on lipid accumulation in the mutant.
As a transcriptional regulator, CAT8 played a role in the regulation of gluconeogenesis and the glyoxylate cycle [50, 51]. The observed 1.9-fold down-regulation of CAT8 in the mutant would lead to a decrease in gluconeogenesis and acetate assimilation, shifting carbon flow toward fatty acid biosynthesis. This shift is consistent with the overall metabolic strategy of the strain, which prioritizes lipid accumulation over energy storage or alternative metabolic pathways. By limiting competing pathways, MU2R48 ensures that a larger proportion of available carbon is channeled into lipid production, further enhancing its lipid-accumulating phenotype. Almost all of these genes with significant expression levels were found to have meaningful SNP mutations in exonic regions in the gene resequencing results (Supporting information, Table S1). The mutagenesis process may have induced specific genetic changes that directly affect lipid metabolism, and these mutations may affect enzyme activity, protein stability, or regulatory interactions, leading to the observed changes in gene expression and metabolic fluxes. Consequently, further investigation of these mutations is crucial to verify whether they are associated with the increased accumulation of lipid in the mutant strains. Functional validation of these mutations, through techniques such as site-directed mutagenesis or CRISPR/Cas9 editing, could provide definitive evidence of their role in enhancing lipid accumulation. Additionally, these mutations could serve as valuable targets for metabolic engineering in other strains, offering a pathway to further optimize lipid production in industrial applications.
Conclusion
This study successfully isolated a mutant strain of S. cerevisiae, MU2R48, utilizing high-throughput FlowRACS. The lipid content of MU2R48 was determined to be 40.26%, marking a relative increase of 30.85% in comparison to the wild-type strain. Multi-omics analysis revealed transcriptional reprogramming in the expression of numerous genes implicated in this increased lipid content in the mutant strain. Notably, the up-regulation of acetyl-CoA supply plays a pivotal role in these changes. This work demonstrated the effectiveness of obtaining high lipid-producing yeast mutants by combining zeocin-ARTP mutagenesis with high-throughput FlowRACS technique, providing a valuable microbial strain resource for the production of microbial lipids and palmitoleic acid, and laid a theoretical foundation for the molecular genetic modification of lipid production in S. cerevisiae.
Materials and methods
The strain and cultivation conditions
The original strain of S. cerevisiae SC018, which was selected based on species characteristics and proven to exhibit a high lipid content [52], and type strain BY4741 was preserved in Microbial Manufacturing Engineering Center, Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences. The S. cerevisiae cells were cultured in YPD medium containing 20 g L−1 glucose, 20 g L−1 peptone, and 10 g L−1 yeast extract and incubated at 28℃ with shaking at 180 rpm for 24–48 h. Yeast cells were cultured to produce large amounts of lipid by induction medium (LIM) containing 4 g L−1 yeast extract, 0.4 g L−1 (NH4)2SO4, 1 g L−1 KH2PO4, 0.25 g L−1 Na2HPO4, 1.5 g L−1 MgSO4·7H2O, 0.15 g L−1 FeCl3·6H2O, 0.15 g L−1 CaCl2·2H2O, 0.02 g L−1 ZnSO4·7H2O, 0.06 g L−1 MnSO4·H2O, and 20 g L−1 glucose. They were incubated at 28℃ with shaking at 180 rpm for 3–4 days. Additionally, 30 g L−1 glucose was added at the time point of 24 h, 48 h, and 72 h during the incubation period.
Establishment of the mutant pools
Mutant pools were established through mutagenesis using varying doses of zeocin, Atmospheric and Room Temperature Plasma (ARTP) with different bombardment times, and a combination of both methods, as described below: 1_Zeocin 100 μg mL−1, 2_Zeocin 150 μg mL−1, 3_ARTP 60 s, 4_ARTP 90 s, 5_ARTP 120 s, 6_ARTP 150 s, 7_ARTP 90 s-Zeocin 100 μg mL−1, 8_ARTP 120 s-Zeocin 100 μg mL−1, 9_ARTP 150 s-Zeocin 100 μg mL−1, 10_ARTP 30 s, 11_ARTP 90 s-Zeocin 100 μg mL−1-Zeocin 50 μg mL−1, 12_ARTP 150 s-Zeocin 100 μg mL−1-Zeocin 50 μg mL−1, 13_Zeocin 100 μg mL−1-Zeocin 50 μg mL−1, and 14_ Zeocin 150 μg mL−1-Zeocin 50 μg mL−1. The mutagenized cells were cultured in the LIM and incubated at 28 ℃ with shaking at 180 rpm for 3 days. After incubation, the cells were washed twice with ultrapure water, and the cell concentration was diluted to an OD600 of 2 for further analysis. The Raman intensity of TAG peak (2844 cm−1) in the mutant pools was measured by the FlowRACS system. The mutant pools with higher Raman intensity at 2844 cm−1 were selected for mixing and sorting.
Characterization of the correlation between lipid content and Raman or fluorescence intensity
S. cerevisiae SC018 was cultured in either LIM or YPD medium, with cell samples of varying lipid content procured through different incubation durations. The cells were resuspended twice with ultrapure water and the OD600 was diluted to 2. The SCRS were acquired under 200 mW 532 nm laser power and 0.2 s acquisition time. A linear correlation was established between the lipid content of each sample and its Raman intensity at 2844 cm−1. Determination of total lipid content was done by the following gravimetric method and fluorescence intensity of samples was measured by Enzyme-labeled instrument [53].
Sorting accuracy validation
The sorting process was conducted using a FlowRACS instrument (Qingdao Single-cell Biotech, CN). The FlowRACS instrument was utilized Nd:YAG 532 nm laser emitter as the excitation light source (200 mW was used in all the experiments), with a 60 × water objective (NA = 1.0, Olympus, JP) to focus the laser beam on the sample. An electron-multiplying charge-coupled device (EMCCD; Newton DU970N-BV, Andor, UK) was used for collecting SCRS. The cells were centrifuged at 2000 g and washed three times with sterile deionized water, collected, and resuspended in 1% Pluronic F-127 (Sigma Aldrich, US) for FlowRACS. An alternating current (AC) of 18 V peak to peak (Vp-p) at 1 MHz frequencies was applied to generate positive dielectrophoresis (pDEP) force to trap the cells and an acquisition time of 200 ms was used for obtaining SCRS. According to our previous work [41], the velocity of 40 μL min−1 and the cell concentration of ~ 6.75 × 103 cells mL−1 were used for cell loading. Sorted cells were collected and cultured on YPD solid medium, and the purity of target cells (SC018) was measured via counting the colonies with different cell sizes in diameter (3–5 μm for target cells and 0.5–1.0 μm for non-target cells, discriminated under microscope). The sorting accuracy is calculated by (number of SC018 colonies) / (number of total colonies) × 100%.
Enrichment of high-lipid content cells by FlowRACS
The mutant pool cells were induced to produce lipid by LIM for 3 days, and then were subjected to FlowRACS within a predetermined threshold to isolate the Mut1R mutant pool. Subsequently, the Mut1R was induced to synthesize lipid for 3 days and then re-sorted to obtain the Mut2R mutant pool. Finally, the sorted cells were plate-cultured and 50 colonies were selected randomly for further liquid culture, lipid induction and extraction, and GC-based analysis.
Evaluation of lipid production capacity of S. cerevisiae strains
The dry cell weight (DCW) was determined by filtering 5 mL of fermented sample through 0.45 μm cellulose nitrate filters and drying to constant weight.
To analyze the fatty acid composition, 100 mg of freeze-dried biomass was dissolved in 3 mL of 4 mol L−1 HCl solution and boiled for 30 min. Subsequently, a 6 mL chloroform–methanol solution (1:1, v/v) was employed to extract the fatty acids. After centrifugation at 2000 rpm for 5 min, the chloroform phase was isolated and dried to a constant weight using nitrogen blowing. The total fatty acids content was determined via gravimetric analysis. The lipid content was expressed as % of DCW.
The fatty acids in the total lipid of S. cerevisiae were converted into fatty acid methyl esters (FAMEs) and analyzed using an Agilent gas chromatography-four-stage mass spectrometry (GC–MS 7890A-5975C, Agilent Corporation, USA) [54].
Whole-genome resequencing
For the whole-genome resequencing of MU2R48, genomic DNA was extracted and quantified using the SDS method [55] and Qubit® 2.0 Fluorometer (Thermo Scientific). Sequencing libraries were generated using NEBNext® Ultra™ DNA Library Prep Kit for Illumina (NEB, USA). The DNA sample was fragmented by sonication to a size of 350bp, and then DNA fragments were end-polished, A-tailed, and ligated with the full-length adapter for Illumina sequencing with further PCR amplification. At last, PCR products were purified by the AMPure XP system and libraries were analyzed for size distribution by Agilent 2100 Bioanalyzer and quantified using real-time PCR. The whole genome of MU2R48 was sequenced using Illumina NovaSeq PE150 at the Beijing Novogene Bioinformatics Technology Co., Ltd, and the sequencing data were uploaded to the NCBI database (Accession number: PRJNA1162638). The reference sequence was the whole-genome sequence of S. cerevisiae SC018 (Accession number in NCBI database: CP159412-CP159429). Detection of insertions and deletions of individual SNPs and small fragments (less than 50 bp) was performed by SAMTOOLS, and analysis of SNP/InDel changes in functional regions of the genome was conducted.
Transcriptome with reference sequencing analysis
Total RNA was extracted by the TIANGEN RNA Extraction Kit. RNA integrity and total quantity were detected accurately by Agilent 2100 Bioanalyzer. The mRNA with polyA tail was enriched by Oligo (dT) beads from the total RNA, and the library was obtained by purifying the mRNA. Initial quantification was performed using a Qubit 2.0 Fluorometer. The libraries were subsequently assayed using an Agilent 2100 Bioanalyzer and the effective library concentrations were accurately quantified by qRT-PCR to ensure library quality. Qualified library was sequenced by the Illumina and 150 bp paired-end reads were generated to obtain sequence information of the fragment to be detected. The sequencing raw data were uploaded to the NCBI database (Accession number: PRJNA1162648). High-quality clean reads were generated by removing reads containing adapters, poly-N (indicating unidentifiable base information) and inferior quality reads from the raw data. Reference genome and gene annotation files were obtained from whole-genome sequencing of SC018. FeatureCounts v1.5.0-p3 was used to count the number of reads mapped to each gene and then calculate the FPKM for each gene. Two differentially expressed groups (three biological replicates per condition) were analyzed using the DESeq2 R package (1.20.0), and genes with an adjusted p-value ≤ 0.05 were designated as differentially expressed.
Targeted metabolome analysis
For the analysis of central carbon metabolites, samples were ground in liquid nitrogen and diluted 100-fold with Milli-Q water, followed by vortexing. Subsequently, 100 μL of each diluted sample was amalgamated with 500 μL of a methanol/water (8:2) solution, which contained a blend of internal standards, using vortexing for homogenization. The mixture was then refrigerated on ice for 30 min and subjected to centrifugation at 12,000 rpm for 10 min. The resultant supernatant was subsequently injected into the LC–MS/MS system for analysis. For the detection of amino acids, 250 μL of precipitant containing a mixed internal standard (acetonitrile:methanol = 1:1) was added to the organisms. For the detection of medium and long chain fatty acids, 300 μL of precipitant containing a mixed internal standard (acetonitrile:isopropanol = 1:1) was added to the samples. Following this, liquid nitrogen quenching was performed and the samples were placed on ice for 30 min. Afterward, they were centrifuged at 12,000 rpm for 10 min. The supernatant was finally injected into the LC–MS/MS system, an ultra-high-performance liquid chromatography coupled to tandem mass spectrometry (UHPLC-MS/MS) system (ExionLC™ AD UHPLC-QTRAP® 6500 + , AB SCIEX Corp., Boston, MA, USA), for further analysis.
Supporting information
The supporting information is as follows.
The linear relationship between the lipid content of S. cerevisiae cells and the fluorescence intensity detected by BODIPY505/515 (Fig. S1); the frequency distribution of TAG Raman intensities for mutant libraries (Fig. S1); the functional enrichment and classification of genes based on transcript levels of the MU2R48 (Fig. S3); the mutation sites of genes with significant expression level difference (Table S1); the relative amounts of MU2R48 target metabolites (Table S2).
Data availability
No datasets were generated or analyzed during the current study.
References
-
Calder PC, Grimble RF. Polyunsaturated fatty acids, inflammation and immunity. Eur J Clin Nutr. 2002;56(Suppl 3):S14–9.
-
Yashodhara BM, Umakanth S, Pappachan JM, et al. Omega-3 fatty acids: a comprehensive review of their role in health and disease. Postgrad Med J. 2009;85(1000):84–90.
-
Scheinman SB, Sugasini D, Zayed M, et al. LPC-DHA/EPA-enriched diets increase brain dha and modulate behavior in mice that express human APOE4. Front Neurosci. 2021;15: 690410.
-
Morin C, Cantin AM, Vézina F-A, et al. The efficacy of MAG-DHA for correcting AA/DHA imbalance of cystic fibrosis patients. Mar Drugs. 2018;16(6):184.
-
Wang C-C, Wang D, Zhang T-T, et al. A comparative study about EPA-PL and EPA-EE on ameliorating behavioral deficits in MPTP-induced mice with Parkinson’s disease by suppressing oxidative stress and apoptosis. J Funct Foods. 2018;50:8–17.
-
Ai Q, Li S, Mai K, et al. The effect of dietary arachidonic acid (ARA) on growth performance, fatty acid composition and expression of ARA metabolism-related genes in larval half-smooth tongue sole (Cynoglossus semilaevis). Br J Nutr. 2015;113(10):1518–30.
-
Weir NL, Steffen BT, Guan W, et al. Circulating omega-7 fatty acids are differentially related to metabolic dysfunction and incident type II diabetes: The Multi-Ethnic Study of Atherosclerosis (MESA). Diabetes Metab. 2020;46(4):319–25.
-
Wu Y, Li R, Hildebrand DF. Biosynthesis and metabolic engineering of palmitoleate production, an important contributor to human health and sustainable industry. Prog Lipid Res. 2012;51(4):340–9.
-
Chan KL, Pillon NJ, Sivaloganathan DM, et al. Palmitoleate reverses high fat-induced proinflammatory macrophage polarization via AMP-activated protein kinase (AMPK). J Biol Chem. 2015;290(27):16979–88.
-
Cao H, Gerhold K, Mayers JR, et al. Identification of a lipokine, a lipid hormone linking adipose tissue to systemic metabolism. Cell. 2008;134:933–44.
-
Matthan NR, Dillard A, Lecker JL, et al. Effects of dietary palmitoleic acid on plasma lipoprotein profile and aortic cholesterol accumulation are similar to those of other unsaturated fatty acids in the F1B golden Syrian hamster. J Nutr Elderly. 2009;139(2):215–21.
-
Yang B, Kallio HP. Fatty acid composition of lipids in Sea Buckthorn (Hippophaë rhamnoides L.) berries of different origins. J Agric Food Chem. 2001;49(4):1939–47.
-
Badami RC, Patil KB. Structure and occurrence of unusual fatty acids in minor seed oils. Prog Lipid Res. 1980;19(3):119–53.
-
Vickery JR. The fatty acid composition of the seed oils of proteaceae: a chemotaxonomic study. Phytochemistry. 1971;10(1):123–30.
-
Canavar Ö. The influence of storage time on fatty acid, tocopherol and seed quality of peanut. Qual Assur Saf Crop. 2015;7(2):165–74.
-
Kolouchová I, Sigler K, Schreiberová O, et al. New yeast-based approaches in production of palmitoleic acid. Bioresour Technol. 2015;192:726–34.
-
Watsuntorn W, Chuengcharoenphanich N, Niltaya P, et al. A novel oleaginous yeast Saccharomyces cerevisiae CU-TPD4 for lipid and biodiesel production. Chemosphere. 2021;280: 130782.
-
Nie X, Xing Y, Li Q, et al. ARTP mutagenesis promotes selenium accumulation in Saccharomyces boulardii. LWT. 2022;168: 113916.
-
Yamada R, Kashihara T, Ogino H. Improvement of lipid production by the oleaginous yeast Rhodosporidium toruloides through UV mutagenesis. World J Microbiol Biotechnol. 2017;33(5):99.
-
Zhang X, Zhang X, Xu G, et al. Integration of ARTP mutagenesis with biosensor-mediated high-throughput screening to improve l-serine yield in Corynebacterium glutamicum. Appl Microbiol Biotechnol. 2018;102(14):5939–51.
-
Wang K, Liu Y, Wu Z, et al. Investigating formate tolerance mechanisms in Saccharomyces cerevisiae and its application. Green Carbon. 2023;1(1):65–74.
-
Guo X, Zhang M, Gao Y, et al. Repair characteristics and time-dependent effects in response to heavy-ion beam irradiation in Saccharomyces cerevisiae: a comparison with X-ray irradiation. Appl Microbiol Biotechnol. 2020;104(9):4043–57.
-
Su Y, Shao W, Zhang A, et al. Improving isobutanol tolerance and titers through EMS mutagenesis in Saccharomyces cerevisiae. FEMS Yeast Res. 2021;21(2):foab012.
-
Ul-Haq I, Ali S, Aslam A, et al. Characterization of a Saccharomyces cerevisiae mutant with enhanced production of β-d-fructofuranosidase. Bioresour Technol. 2008;99(1):7–12.
-
Baeshin NA, Abou-Zeid A-ZA, Baghlaf AO. The formation of oxytetracycline in a date medium by mutants of Streptomyces rimosus induced by chemical mutagens. Bioresour Technol. 1992;42(3):177–81.
-
Zheng DQ, Wang YT, Zhu YX, et al. Uncovering Bleomycin-induced genomic alterations and underlying mechanisms in the yeast Saccharomyces cerevisiae. Appl Environ Microbiol. 2022;88(2): e0170321.
-
Gu L-S, Tan M-Z, Li S-H, et al. ARTP/EMS-combined multiple mutagenesis efficiently improved production of raw starch-degrading enzymes in Penicillium oxalicum and characterization of the enzyme-hyperproducing mutant. Biotechnol Biofuels. 2020;13(1):187.
-
Zhang C, Shen H, Zhang X, et al. Combined mutagenesis of Rhodosporidium toruloides for improved production of carotenoids and lipids. Biotechnol Lett. 2016;38(10):1733–8.
-
Terashima M, Freeman ES, Jinkerson RE, et al. A fluorescence-activated cell sorting-based strategy for rapid isolation of high-lipid Chlamydomonas mutants. Plant J. 2015;81(1):147–59.
-
Cabanelas ITD, van der Zwart M, Kleinegris DMM, et al. Sorting cells of the microalga Chlorococcum littorale with increased triacylglycerol productivity. Biotechnol Biofuels. 2016;9(1):183.
-
Doan TTY, Obbard JP. Enhanced intracellular lipid in Nannochloropsis sp. via random mutagenesis and flow cytometric cell sorting. Algal Res. 2012;1(1):17–21.
-
Ji X, Chen L, Yang G, et al. Mutagenesis and fluorescence-activated cell sorting of oleaginous Saccharomyces cerevisiae and the multi-omics analysis of its high lipid accumulation mechanisms. Bioresour Technol. 2024;406: 131062.
-
Smalley T, Fields FJ, Berndt AJE, et al. Improving biomass and lipid yields of Desmodesmus armatus and Chlorella vulgaris through mutagenesis and high-throughput screening. Biomass Bioenergy. 2020;142: 105755.
-
He Y, Wang X, Ma B, et al. Ramanome technology platform for label-free screening and sorting of microbial cell factories at single-cell resolution. Biotechnol Adv. 2019;37(6): 107388.
-
Wu H, Volponi JV, Oliver AE, et al. In vivo lipidomics using single-cell Raman spectroscopy. PNAS. 2011;108(9):3809–14.
-
Wang T, Ji Y, Wang Y, et al. Quantitative dynamics of triacylglycerol accumulation in microalgae populations at single-cell resolution revealed by Raman microspectroscopy. Biotechnol Biofuels. 2014;7:58.
-
He Y, Zhang P, Huang S, et al. Label-free, simultaneous quantification of starch, protein and triacylglycerol in single microalgal cells. Biotechnol Biofuels. 2017;10:275.
-
Lee TH, Chang JS, Wang HY. Rapid and in vivo quantification of cellular lipids in Chlorella vulgaris using near-infrared Raman spectrometry. Anal Chem. 2013;85(4):2155–60.
-
Wang X, Ren L, Su Y, et al. Raman-Activated Droplet Sorting (RADS) for label-free high-throughput screening of microalgal single-cells. Anal Chem. 2017;89(22):12569–77.
-
Zhang P, Ren L, Zhang X, et al. Raman-Activated Cell Sorting based on dielectrophoretic single-cell trap and release. Anal Chem. 2015;87(4):2282–9.
-
Wang X, Ren L, Diao Z, et al. Robust spontaneous raman flow cytometry for single-cell metabolic phenome profiling via pDEP-DLD-RFC. Adv Sci. 2023;10(16):2207497.
-
Wang X, Xin Y, Ren L, et al. Positive dielectrophoresis–based Raman-activated droplet sorting for culture-free and label-free screening of enzyme function in vivo. Sci Adv. 2020;6(32):eabb3521.
-
Czamara K, Majzner K, Pacia MZ, et al. Raman spectroscopy of lipids: a review. J Raman Spectrosc. 2015;46(1):4–20.
-
Tang X, Lee J, Chen WN. Engineering the fatty acid metabolic pathway in Saccharomyces cerevisiae for advanced biofuel production. Metab Eng Commun. 2015;2:58–66.
-
Chen Y, Siewers V, Nielsen J. Profiling of cytosolic and peroxisomal acetyl-CoA metabolism in Saccharomyces cerevisiae. PLoS ONE. 2012;7(8): e42475.
-
de Jong BW, Shi S, Siewers V, et al. Improved production of fatty acid ethyl esters in Saccharomyces cerevisiae through up-regulation of the ethanol degradation pathway and expression of the heterologous phosphoketolase pathway. Microb Cell Fact. 2014;13(1):39.
-
Ratledge C, Cohen Z. Microbial and algal oils: do they have a future for biodiesel or as commodity oils? Lipid Technol. 2008;20(7):155–60.
-
Nakamura MT, Nara TY. Structure, function, and dietary regulation of Δ6, Δ5, and Δ9 desaturases. Annu Rev Nutr. 2004;24:345–76.
-
Kerkhoven EJ, Pomraning KR, Baker SE, et al. Regulation of amino-acid metabolism controls flux to lipid accumulation in Yarrowia lipolytica. NPJ Syst Biol Appl. 2016;2(1):16005.
-
Thepnok P, Ratanakhanokchai K, Soontorngun N. The novel zinc cluster regulator Tog1 plays important roles in oleate utilization and oxidative stress response in Saccharomyces cerevisiae. Biochem Biophys Res Commun. 2014;450(4):1276–82.
-
Turcotte B, Liang XB, Robert F, et al. Transcriptional regulation of nonfermentable carbon utilization in budding yeast. FEMS Yeast Res. 2009;10(1):2–13.
-
Wang Y, Ji X, Chen L, et al. Conversion of saturated fatty acid to unsaturated one: Whole-cell catalysis of Saccharomyces cerevisiae. Biochem Eng J. 2023;196: 108960.
-
Cooper MS, Hardin WR, Petersen TW, et al. Visualizing “green oil” in live algal cells. J Biosci Bioeng. 2010;109(2):198–201.
-
Zhou W, Wang H, Chen L, et al. Heterotrophy of filamentous oleaginous microalgae Tribonema minus for potential production of lipid and palmitoleic acid. Bioresour Technol. 2017;239:250–7.
-
Lim HJ, Lee E-H, Yoon Y, et al. Portable lysis apparatus for rapid single-step DNA extraction of Bacillus subtilis. Appl Microbiol Biot. 2016;120(2):379–87.
Funding
This work was funded by the National Key Research and Development Program of China (Grant no. 2021YFA0909702), the National Natural Science Foundation of China (Nos. 31972932, 32170103, and 32470087), Chinese Academy of Sciences Project for Young Scientists in Basic Research (No.YSBR-111). Natural Science Foundation of Shandong Province (No. 2021ZDSYS29 and ZR2024Y0048), the Youth Innovation Promotion Association CAS, Shandong Taishan Scholarship, and the Science and Technology Benefit for the People Demonstration Special Project of Qingdao (No. 23–2-8-smjk-3-nsh).
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All the authors listed have approved the manuscript, agreed to authorship, and submission of the manuscript for publication.
Competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ji, X., Wang, X., Zhou, W. et al. Label-free isolation of lipid-rich Saccharomyces cerevisiae mutant by high-throughput flow-mode Raman-activated cell sorting and multi-omics analysis for uncovering the mechanism of enhanced lipid accumulation. Biotechnol. Biofuels Bioprod. 18, 75 (2025). https://doi.org/10.1186/s13068-025-02677-8
-
Received:
-
Accepted:
-
Published:
-
DOI: https://doi.org/10.1186/s13068-025-02677-8


