
Main
Immune monitoring in both humans and animal models predominantly relies on analysing peripheral blood. However, this approach faces limitations, especially for detecting antigen-specific CD8+ T cells, which play crucial protective roles in infection, vaccination, cancer and autoimmune disease1,2. Such T cells are often present at extremely low frequencies in the peripheral blood (for example, ~0.05% in vaccinated hepatocellular carcinoma patients3 and SARS-CoV-2 mRNA-vaccinated children4), even following antigenic stimulation5,6. This low frequency combined with limited blood volumes means that biologically relevant T cell responses often go undetected by traditional methods, even using sensitive assays such as IFNγ ELISPOT, and can only be detected by ex vivo expansion7,8,9,10. Thus, new approaches to detect and characterize antigen-specific lymphocytes are of great interest.
Microneedle (MN) patches provide a minimally invasive means to sample immune cells and soluble factors from the skin. These patches, consisting of microscopic projections that penetrate the stratum corneum and epidermis11, have been extensively explored for drug and vaccine delivery12,13,14,15,16,17,18,19,20,21 and, more recently, for sampling interstitial fluid (ISF)22,23,24,25. We previously developed a hydrogel-coated MN patch capable of sampling both soluble factors in ISF and immune cells from skin26. While less invasive than biopsies or suction blisters27, this earlier approach yielded few cells unless antigen or adjuvant was included in the hydrogel26, and primarily recovered tissue-resident memory T cells (TRMs). Thus, while very useful for probing tissue-specific immunity, this approach would not necessarily provide a holistic window on the systemic immune response. We were inspired by the unique properties of TRMs to consider whether these skin-resident immune cells could be leveraged to provide enhanced sampling of both the local and systemic antigen-specific immune response in individuals.
TRMs are one subset of an array of immune cells that are strategically positioned in barrier sites, such as the skin, lungs and intestines, and act as a localized rapid defence at portals of entry for pathogens28,29,30. In the skin and female reproductive tract, TRM cells actively patrol tissues locally without recirculating through blood or other organs. Upon re-encounter with cognate antigen, TRMs can exhibit immediate effector functions and alert the tissue to potential threats, including infections and autoimmune responses31,32. One of their most important roles is to ‘sound the alarm’ in response to antigen encounter, via the rapid production of chemokines and cytokines that recruit immune cells from the blood33,34. We hypothesized that this alarm function of TRM cells could be exploited to concentrate antigen-specific T cells from the circulation at a selected site in the skin, providing a means to efficiently sample even rare antigen-specific T cells circulating in the peripheral blood using MN patches applied to a site of TRM stimulation. We envisioned that TRM could be leveraged in two distinct ways: a first approach would be to intentionally establish a TRM population at a selected site in the skin via intradermal inoculation of small quantities of an antigen/adjuvant in immune animals, followed by subsequent recall of these TRM cells through re-administration of antigen in the skin. This recall step would lead to the recruitment and accumulation of antigen-specific T cells that could be sampled via MN patch application at the stimulation site. A second scenario is to exploit pre-existing TRM established in treatment or disease conditions, such as patients with inflammatory or autoimmune conditions in the skin. In this situation, restimulation of disease-associated TRM through antigen inoculation would similarly recruit circulating immune cells, again facilitating recovery through local MN patch application.
Here we tested these ideas in mouse and human models of vaccination and skin inflammation, respectively, using MN patches carrying a hydrogel cell- and fluid-sampling coating optimized for maximal cell recovery from the skin without the inclusion of antigens and adjuvants within the sampling hydrogel layer itself. In mice, we show mechanistically that induction and recall of TRM cells in the skin enable greatly amplified cell sampling with MN patches, especially antigen-specific CD8+ T cells. TRM recall enabled both tissue-resident and circulating cells to be captured from the blood. We demonstrate that hydrogel-coated MN application induces minimal adverse reactions in human volunteers, with the patches being well tolerated for up to 24 h without any adverse reactions, and present a case study of a human participant with allergic contact dermatitis, suggesting that reactivation of TRM cells in the skin during antigen recall attracts CD4+ and CD8+ T cells to the skin in humans. MN patches applied to antigen recall sites in this patient showed superior sampling efficiency compared with the established research method of suction blister skin sampling, allowing recovery of T cells, macrophages, natural killer cells, B cells and monocytes without aberrant irritation. Microneedle sampling together with TRM stimulation may thus enable collection of rare antigen-specific immune cell populations that would be challenging to detect by conventional means in the setting of vaccination, immunotherapy or disease monitoring.
Results
Hydrogel coatings maximizing cell migration enhance cell sampling by MN patches
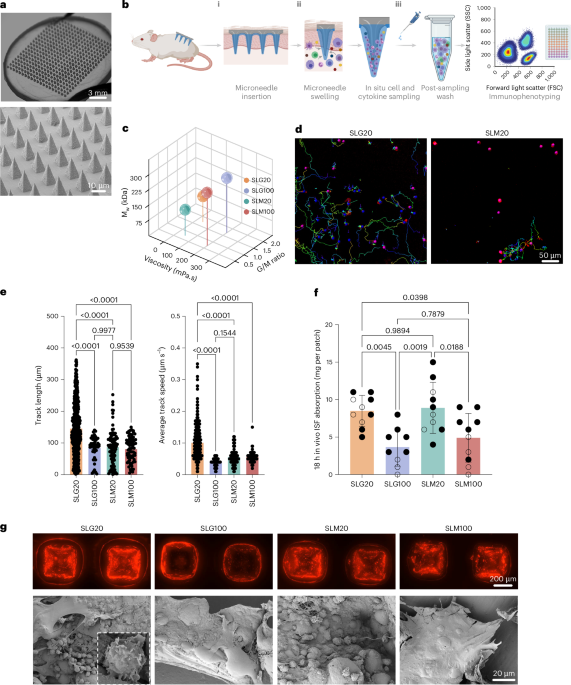
We previously developed minimally invasive MN patches capable of sampling both ISF and cells from skin. These patches are fabricated by melt-moulding of polylactide, a biodegradable polymer similar to that used in resorbable sutures35,36, to form an array of conical MN projections 550 μm long and 250 μm wide at the base, extending from a solid polymer backing. For small-animal studies, we employed patches 2 cm2 in area containing 400 MN projections, which are readily applied to the flanks of mice (Fig. 1a). These designs were based on MN dimensions shown to be effective for epidermal sampling in humans and rodents37: the human epidermis is typically <150 μm thick, while the dermis extends at least 600 μm, allowing our 550 μm microneedles to reliably reach the dermis without contacting deeper structures38,39. Similarly, in mice, the dermis ranges from ~600–700 μm in thickness40, and microneedles in the 500–700 μm range have been shown to be optimal for skin immune cell recovery41,42,43. Thus, our selected microneedle dimensions enable consistent penetration into the upper dermis in both species while minimizing tissue damage. A coating of sucrose and ionically crosslinked alginate, a US Food and Drug Administration (FDA) generally-regarded-as-safe biocompatible polysaccharide that swells strongly in water44,45, is cast over the MN projections to serve as the cell/fluid sampling layer. Sampling is carried out by applying the patch to the skin with mild pressure, which causes mechanical penetration of the stratum corneum and entry of MN projections into the epidermis (Fig. 1b(i)). Sucrose in the cell sampling coating (included to augment the mechanical strength of the sampling layer during skin insertion) rapidly dissolves and the alginate layer swells, absorbing ISF and enabling migration of cells into the gel coating (Fig. 1b(ii)). The coating is then dissolved with ethylenediaminetetraacetic acid (EDTA) to recover captured cells and fluid for analysis (Fig. 1b(iii)).

a, Photographs and scanning electron micrographs of the hydrogel-coated MN patch. b, Schematic view of the cell and interstitial fluid sampling process with MN array applied to the skin. c, Molecular weight, G/M ratio and viscosity of alginates tested as MN coatings. d, Still frames from time-lapse microscopy of activated T cells incubated in SLG20 and SLM20 hydrogels in vitro showing tracked individual cell paths as coloured lines. e, Quantification of T cell motility length and average track speed inside different hydrogels (n = 3 hydrogels per cell per group). f, Comparison of interstitial fluid sampling capacity between patches coated with different alginates (n = 10 animals per group). g, Optical and scanning electron micrographs of the patches after 18 h of in vivo sampling on the skin of OVA-immunized mice and sampled following the scheme of Fig. 2b (TRM recall), showing the retention of the alginate layer (labelled with Alexa647 for visualization) and collected cells on the patch (inset: high-magnification view of lymphocytes captured by the MN patch). Data shown are mean ± s.e.m. P values were determined using one-way ANOVA followed by Tukey’s multiple comparisons test (e,f). Panel created in BioRender: b, Jalili, S. https://biorender.com/b3buou9 (2026).
Although our previously reported prototype patch design26 was functional, we first sought to test whether the composition of the sampling layer could be further optimized to maximize cell recovery from the skin. The sampling layer is formed by dropcasting an alginate and sucrose solution over the MN patch, drying to a solid coating, then crosslinking the alginate with calcium chloride solution, followed by a second drying step to achieve a uniform alginate coating over the MN projections (Extended Data Fig. 1a,b). We first tested the influence of the alginate’s molecular weight, guluronic to mannuronic acid ratio (G/M ratio), and viscosity on in vitro and in vivo fluid absorption, as well as cellular interactions with the hydrogel layer. Four distinct ultra-pure alginates, SLG20, SLG100, SLM20 and SLM100, each with varied physical properties, were tested (Fig. 1c). Modifying the G/M ratio and molecular weight of alginate has been demonstrated to alter its physicomechanical properties, including porosity and stiffness44,46. In vitro bulk swelling of sampling layers prepared with each of these alginates was essentially identical (Extended Data Fig. 1c). However, we hypothesized that alginate properties could also affect the ability of cells to migrate within the sampling layer gel, and thereby impact cell collection in vivo. To test this, we encapsulated primary mouse T cells in gels prepared from each of the alginates (crosslinked under the same conditions used for sampling layer formation) and tracked cell migration over time by time-lapse microscopy. Strikingly, we observed substantially higher motility and cell migration within SLG20 gels compared with the other alginates, with ~2-fold greater mean track lengths and ~2.5-fold greater mean cell speeds than observed for lymphocytes in the other gels (Fig. 1d,e).
To assess the behaviour of these different alginate coatings in vivo, we applied patches to the skin of C57Bl/6 mice for 18 h. In contrast to the in vitro swelling measurements, in vivo patch sampling revealed distinct quantities of ISF recovered by patches bearing sampling layers prepared with different alginates; sampling layers prepared with SLG20 or SLM20 recovered approximately twice the interstitial fluid of patches prepared with the other two alginates (Fig. 1f). Optical and scanning electron microscopy (SEM) imaging revealed distinct differences in the microneedle patches after skin application. Patches coated with SLG100 or SLM100 alginate showed numerous microneedles that had lost their alginate coating following application to skin, resulting in fewer sampled cells accumulating on them (Fig. 1g). In contrast, patches coated with SLG20 retained their alginate layer on all microneedle projections and many more cells could be observed associated with the hydrogel coating (Fig. 1g). We thus focused further studies on SLG20 as the sampling layer polymer.
We next assessed effects of crosslinking density, sucrose content and coating. Crosslinking with less than 20 mM CaCl2 produced fragile gels, while higher calcium concentrations reduced swelling, which would reduce ISF recovery (Extended Data Fig. 1d). Sucrose concentration modestly affected fluid uptake, and thicker coatings increased swelling but not cell yield, probably due to limitations in migration depth of cells into the patch in the application time (Extended Data Fig. 1e,f). Altogether, these studies led us to focus on cell/fluid sampling patches prepared with sampling layers cast from 0.6 wt% SLG20 alginate/2.5 wt% sucrose solutions, ~10 µm thick in their final dried state.
Stimulation of local tissue-resident memory T cells augments skin patch MN cell sampling
In the absence of specific stimuli, MN patches applied to the skin recover few cells and provide little insight into vaccine- or disease-specific immune responses. We hypothesized that the efficiency of sampling antigen-specific lymphocyte populations could be amplified by inducing a population of tissue-resident memory T cells (TRM) in the skin, which would serve to actively recruit immune cells from the circulation before patch sampling. This concept is schematically outlined in Fig. 2a: To establish a TRM population, animals with an antigen-specific immune population of interest, for example, animals that have been vaccinated with a selected antigen(Fig. 2a(i)) are inoculated intradermally (i.d.) with a small dose of the antigen combined with an adjuvant to recruit and establish antigen-specific tissue-resident memory T cells at a selected site on the skin (Fig. 2a(ii)). Once established, this antigen-specific TRM population is recalled by i.d. injection of a small dose of the antigen and adjuvant at the same site, which triggers the ‘alarm’ function of TRM, leading to rapid production of chemokines that draw a large population of antigen-specific cells to the local skin site30,47,48,49 (Fig. 2a(iii)). MN patch sampling at the site is then used to non-invasively recover and analyse the makeup of the antigen-specific immune response (Fig. 2a(iv)).

a, Schematic view of TRM establishment, recall and recruitment of antigen-specific tissue-resident memory T cells for MN patch sampling at a selected site on the skin. b, Study design for establishing TRM populations in the skin and subsequent TRM recall in mice immunized with OVA protein (10 μg per dose) and Lipo-CpG (1.24 nmol per dose). Blood and MN samples were collected 7 days after TRM recall. c–e, Enumeration of recovered total live leucocytes, CD3+ T cells, TRM cells and antigen-specific CD8+ T cells recovered by MN patches under different sampling regimens. f, Representative flow cytometry plots showing SIINFEKL peptide–MHC tetramer staining to identify antigen-specific CD8+ T cells collected using MN patches 7 days after TRM recall. g, Enumeration of recovered antigen-specific CD8+ T cells in the MN patches at TRM establishment and recall steps in comparison with No TRM. h, study design for longitudinal cytokine sampling post TRM recall step. i, Expression of inflammatory cytokines and chemokines induced in the skin measured using multiplexed ELISA analysis of interstitial fluid samples recovered by MN patches following TRM recall. n = 10 animals per group (a–g) and n = 5 animals per group (i). Data shown are mean ± s.e.m. P values were determined using one-way ANOVA followed by Tukey’s multiple comparisons test (c–e,g,i). Panel created in BioRender: a, Jalili, S. https://biorender.com/awru57q (2026); panel elements created with BioRender: h, Jalili, S. https://biorender.com/8arfbyn (2026).
We first tested whether TRM stimulation would facilitate enhanced MN sampling of antigen-specific T cells in a vaccination model. Mice were primed and boosted with ovalbumin (OVA) + CpG adjuvant, then intradermally inoculated with OVA + CpG to establish TRMs at a defined skin site. One week later, a recall dose of OVA + CpG was administered at the same site, and local immune infiltration was analysed 6 days later (‘TRM recall’ in Fig. 2b). Controls included skin with TRM establishment but no recall (‘no recall’) and skin sites without TRM establishment or recall (Fig. 2b, ‘no TRM’). TRM recall increased total live and CD11b+ myeloid cells recruited to the site by ~3-fold compared to controls (Extended Data Fig. 2a,b). More strikingly, TRM induction led to 12-fold and 5-fold increases in CD4+ and CD8+ T cells, respectively, and a 35-fold increase in OVA-specific CD8+ T cells in the skin relative to the ‘no TRM’ group (Extended Data Fig. 2c,d). This recruitment depended on TRM restimulation as T cell infiltration was much lower in the ‘no recall’ condition (Extended Data Fig. 2c,d), and was probably mediated by recruitment of T cells from the circulation, as only a minority of recovered cells expressed the marker of tissue residency, CD103 (Extended Data Fig. 2e,f). Importantly, both antigen and adjuvant were required for robust TRM establishment, as neither alone induced significant T cell accumulation (Extended Data Fig. 2g).
Next, we tested cell recovery using MN patches under the same 3 treatment conditions (Fig. 2b). In the absence of TRM, few live cells were recovered from the MN patches (Fig. 2c). When TRMs were induced in the skin, a trend towards increased cell recovery at the sampled skin site was observed, but this did not reach statistical significance. However, live cell recovery increased 20-fold over the ‘no TRM’ case when the TRM population was recalled with antigen/adjuvant before MN sampling (Fig. 2c). In addition to absolute numbers, we also quantified the percentage of live leucocytes recovered (Fig. 2c); MN sampling enabled cell recovery with high viability. When we examined the cell types recovered by MN patches, we found that myeloid cell recovery was increased ~5-fold for both the TRM recall and no-recall groups vs ‘no TRM’ sampling (Fig. 2d). By contrast, TRM recall sampling led to 6.5-fold and 8-fold greater CD4+ and CD8+ T cells recovered, respectively, compared with the no-recall sampling conditions (Fig. 2e). This pattern of enhanced T cell recovery by sampling TRM-stimulated skin was even more pronounced when we examined the recovery of antigen-specific OVA tetramer+ T cells recovered by the MN patches, with TRM recall leading to 18-fold greater numbers of antigen-specific cells recovered compared with the no-recall condition (Fig. 2f,g). Notably, the majority of CD8+ T cells recovered in the patches using TRM recall were antigen specific (Fig. 2g). In addition to T cells and myeloid cells, we also observed a substantial CD11b−CD3− population among the recovered cells. This non-myeloid, non-T cell fraction probably includes other immune populations such as B cells, natural killer (NK) cells and innate lymphoid cells (ILCs) (Extended Data Fig. 2h,i). Furthermore, ~30% of the recovered cells were CD45−, probably non-immune skin-resident cells such as fibroblasts or keratinocytes (Extended Data Fig. 2h,i). These subsets were not further characterized here, as our primary emphasis was on T cells and myeloid cells. Consistent with our initial patch optimization findings, when MN patches with different alginate coatings were employed to sample cells from OVA-immunized mice skin, SLG20 demonstrated superior performance, collecting more live CD45+ cells, myeloid cells, CD4+ and CD8+ T cells, as well as antigen-specific OVA tetramer+ TRM cells (Extended Data Fig. 3a).
Two important parameters in this sampling approach are (1) the interval between TRM recall and patch application and (2) the duration of patch application. We applied patches in the above sampling experiments 6 days after TRM recall to allow time for robust T cell recruitment to the site from circulation. To determine whether this recruitment period was important, we compared T cell recovery after applying patches at 1 day or 6 days after TRM recall. Total and antigen-specific T cell recovery was much lower when patches were applied at 1 day post TRM recall (Extended Data Fig. 3b). When different durations of MN patch application were compared, we observed that the number of live lymphocytes and T cells recovered peaked with an 18-h duration of patch application to the skin (Extended Data Fig. 3c). We thus used SLG20 MN patches, with patches applied for 18 h at 6 days post TRM recall, for all subsequent cell sampling studies.
To gain insight into the signalling milieu established following TRM recall, we analysed a panel of inflammatory cytokines and chemokines induced in the skin using multiplexed enzyme-linked immunosorbent assay (ELISA) analysis of interstitial fluid samples recovered by MN patches applied to treated skin at different time points following TRM recall (Fig. 2h). TRM recall induced a coordinated response consisting of induction of an early burst of IFNγ, IFNβ and IL-6 at 24 h, which rapidly returned to baseline (Fig. 2i). Other chemokines such as CCL5 and CXCL1 showed a trend towards more sustained expression following recall. Thus, ‘TRM recall’ was accompanied by rapid induction of inflammatory cytokine/chemokine induction in the skin and greatly augmented MN sampling of antigen-specific T cells.
Finally, to confirm that the antigen/adjuvant administration used to induce and recall TRMs does not itself affect the systemic immune response, we assayed circulating antigen-specific T cell levels following administration of different doses of OVA and adjuvant during the TRM establishment/recall steps. We intentionally administered the TRM recall dose with a 3-week gap after TRM establishment to observe whether the intradermal TRM recall injection would lead to a sudden increase in antigen-specific cells in the blood (Extended Data Fig. 4a). We observed that the number of antigen-specific CD8+ T cells accumulating in the local site in response to TRM recall treatment was dependent on the dose of antigen inoculated in the skin (Extended Data Fig. 4b,c) and was amplified only following the final TRM recall inoculation (Extended Data Fig. 4d); however, no significant increase was observed in antigen-specific T cells in the blood following the TRM establishment or TRM recall intradermal injections (Extended Data Fig. 4e).
To directly address whether immune cell capture by MN patches can occur at later time points after sensitization alone, without TRM recall, we compared multiple experimental groups with and without recall stimulation. Our results demonstrate that waiting longer than 1 week after TRM establishment to carry out MN sampling, without TRM recall, did not yield increases in the number of antigen-specific CD8+tetramer+ T cells in the skin, even 3 weeks after TRM establishment (Supplementary Fig. 1). In contrast, groups that underwent both TRM establishment and recall showed a robust accumulation of antigen-specific T cells at the skin site. These findings indicate that TRM recall is mechanistically necessary to ‘alarm’ the skin and recruit circulating antigen-specific T cells, and that sensitization alone, even over an extended time, is insufficient to recruit additional immune cells to the tissue.
T cells recovered from TRM-stimulated skin reflect the circulating antigen-specific immune response
We hypothesized that TRM recall would lead to recruitment of antigen-specific T cells from the circulation, and thus provide a window into the systemic T cell response. However, stimulated T cells can also proliferate locally50, hence it was important to evaluate the role of systemic T cell recruitment vs local TRM expansion on the makeup of cells recovered by patch sampling. To monitor the migration of antigen-specific T cells to the TRM recall site, we employed photoconvertible KikGR mice51 to distinguish sampled cells that were recruited from the blood vs tissue-resident cells (Fig. 3a). Cells in these mice express the photoreactive KikGR protein in their cytoplasm, which exhibits green fluorescence unless photoactivated by UV light, whereupon a portion of the protein photoconverts to stable red fluorescence (Fig. 3b). To employ this tracer for tracking cell recruitment in the skin, OVA-vaccinated mice were inoculated with OVA + CpG adjuvant in the skin to establish TRM, and 1 week later, the sampling site was exposed to UV light to photoconvert skin-resident cells (Fig. 3a). Subsequently, TRM were recalled by inoculation of OVA and CpG adjuvant at the same site. Analysis of the skin immediately after photoconversion (0 h) confirmed the near-complete (94%) conversion of these cells from the default green fluorescence of the KikGR protein (‘KikGR Green+’) to the altered red fluorescence profile (‘KikGR Red+Green+’, Fig. 3c). We next vaccinated mice, established TRM by i.d. antigen/adjuvant inoculation (or not), photoconverted the TRM site 1 day before TRM recall on day 28, and sampled the infiltrated cells into the photoconverted area on day 34 via MN patches (Fig. 3a). When we analysed the KikGR reporter expression of recovered cells (Fig. 3d,e), we found that in the ‘no TRM’ case, the majority of cells sampled, including myeloid cells, CD4+ T cells and CD8+ T cells, were KikGR red+green+ double positive, indicating that they were resident in the skin at the time of photoconversion and that very few recovered cells came from the circulation (Fig. 3e). By contrast, the vast majority of cells recovered by TRM recall sampling (including antigen-specific CD8+ T cells) were KikGR green single-positive (Fig. 3d,e). This indicates that either they were recruited from the circulation or they had proliferated extensively since the photoconversion timepoint.

a, Schematic representation of temporal labelling of the skin of the C57BL/6 KikGR mice, photoconverted immediately before the TRM recall dose (day 28) by violet light exposure on the skin site. b,c, Representative flow cytometry plots showing KikGR red and green gene expression in skin before and immediately after photoconversion. d, Representative flow cytometry plots showing KikGR red and green expression by skin-infiltrating myeloid cells, CD3, CD4, CD8 and antigen-specific T cells on Day 34. e, Quantitation of frequencies of KikGR red+green+ and green-only+ cells by subtype, recovered from MN patch sampling following the timeline in a. f–j, Enumeration of recovered live leucocytes (f), myeloid cells (g), CD4+ T cells (h), CD8+ T cells (i) and antigen-specific CD8+ T cells (j) 7 days after TRM recall dose. n = 5 animals per group (all panels). Data shown are mean ± s.e.m. P values were determined using two-way ANOVA (e) and one-way ANOVA (f–j) followed by Tukey’s multiple comparisons test. Panel elements created in BioRender: a, Jalili, S. https://biorender.com/oz2aoky (2026).
To distinguish these possibilities, we carried out sampling under a third condition, where we restimulated TRM in the skin in the presence of systemically administered blocking antibody against Lymphocyte function-associated antigen-1 (LFA-1), as this receptor is important for the homing of T cells from the blood to inflammatory sites52,53,54 (Fig. 3a). LFA-1 blockade reduced the total live cells recovered by patch sampling of TRM-stimulated skin by ~2-fold (Fig. 3f). While the recovery of myeloid cells showed a slight, non-significant reduction (Fig. 3g), LFA-1 blockade significantly reduced CD4+ and CD8+ T cell recovery by 3-fold and 4-fold, respectively, and decreased OVA-specific CD8+ T cell recovery by 2.5-fold, (Fig. 3h–j). Further, recovered CD8+ T cells and antigen-specific CD8+ T cells were ~50/50 KikGR red+green+ vs KikGR green+ (Fig. 3e). These data suggest that at least ~50% of the total antigen-specific T cells recovered from TRM recall sampling was derived from the circulation, which is a conservative estimate because some proportion of T cell trafficking into inflamed tissues is LFA-1 independent. These data collectively suggest that antigen-specific T cells recovered by TRM-recall patch sampling represent both local tissue-resident cells and cells recruited from the circulating blood pool.
Unveiling TRM-driven virus-specific T cell responses with sampling MNs
While TRM recall was very effective for recovering T cells primed against OVA, this model antigen is highly immunogenic55. We thus next sought to test whether TRM recall-based MN sampling could enhance the detection and recovery of antigen-specific T cells primed against a bona fide viral antigen. To this end, we synthesized nucleoside-modified mRNA encoding a set of 5 Simian immunodeficiency virus (SIV) T cell epitopes as a model vaccine. This vaccine construct was designed to carry T cell epitopes presented by macaque MHC alleles, but one of the peptides in the mRNA, an epitope termed CL9, can also be presented by mouse class I molecules. A single intramuscular (i.m.) vaccination with lipid nanoparticles carrying this SIV mRNA construct elicited systemic T cell responses recognizing CL9 at a frequency of ~0.06% among all cells, which could be detected in the spleen by sensitive IFNγ ELISPOT assays on day 21 (Fig. 4a). To evaluate microneedle sampling of this response, TRM cells were established 3 weeks post mRNA vaccination via i.d. challenge with CL9 peptide and CpG adjuvant, TRMs were recalled by i.d. re-administration of CL9 + CpG at day 28, and cells were sampled with MN patches at day 34 (Fig. 4b, ‘TRM recall’). We compared TRM recall sampling to a group that only received i.d. peptide/CpG challenge at day 28 post vaccination (Fig. 4b, ‘TRM, No recall’). This control condition was intended to establish TRM and provide the same inflammatory stimulus to the skin just before MN patch sampling as done in the ‘TRM recall’ group (but without recall stimulation), to account for non-specific cell recruitment effects of the peptide/adjuvant injection. Skin biopsies at day 34 revealed that the TRM establishment enriched the presence of CD8+CD69+ and CD8+CD103+ TRM cells (Fig. 4c,d). However, following the TRM recall, we observed an increased number of CD69−CD103− cells recruited from the circulation to the site (Fig. 4d).

a, ELISPOT analysis of antigen-specific IFNγ-producing T cells from spleens of mice immunized with mRNA encoding SIV epitopes on day 21 post immunization (n = 5 animals per group). b, Study timeline comparing MN patch sampling under TRM recall vs ‘TRM, no recall’ conditions. c, Representative flow cytometry plots showing expression of tissue-resident phenotypic markers for cells recovered from MN patches 7 days after TRM establishment. d, Enumeration of tissue-resident (CD103+CD69+, CD103+CD69−, CD103−CD69+) or non-tissue-resident recruited (CD103−CD69−) CD8+ T cells recovered from MN patches with or without TRM recall stimulation. e–i, Quantitation of total live leucocytes (e), total T cells (f), CD4+ T cells (g), CD8+ T cells (h) and antigen-specific CD8+ T cells (i) recovered under recall or no-recall conditions. j, Quantitative comparison of antigen-specific CD8+ T cells recovered via blood draw (100 μl blood draw), skin biopsy (6 mm punch biopsy) or MN patches. Each data point represents an individual mouse. n = 5 animals per group (all panels). Data shown are mean ± s.e.m. P values were determined using unpaired two-tailed Student’s t-test (d–i) and one-way ANOVA followed by Tukey’s multiple comparisons test (j).
MN sampling of animals from these two groups revealed that TRM recall increased the recovery of total live cells, CD3+ T cells, CD4+ T cells and CD8+ T cells (Fig. 4e–h). Most strikingly, CL9 tetramer+CD8+ T cell recovery was increased 7.5-fold in the TRM recall group vs the TRM no-recall case (Fig. 4i). Comparing to a standard mouse blood draw and standard clinical skin punch biopsy (6 mm)56,57, patch sampling with TRM recall enabled >2-fold greater recovery of live antigen-specific T cells (Fig. 4j). In this analysis, we compared 2 cm² MN patches applied to murine flanks vs a 6-mm punch biopsy, which is the standard size in clinical practice. As expected, when normalized to skin area sampled, the punch biopsy, where the entire tissue is collected, recovered more cells per unit area than the patches (Extended Data Fig. 4f). While larger or multiple punch biopsies could theoretically yield greater cell numbers, these procedures are substantially more invasive, less acceptable to patients and poorly suited for longitudinal sampling. In summary, in response to bona fide viral antigen vaccination, TRM recall sampling with MN patches allows for greater recovery of live antigen-specific T cells than traditional blood sampling, and unlike blood sampling, allows recovery of both circulating and tissue-resident T cells.
Tracking TRM cells and cytokines in human allergic contact dermatitis
The studies above demonstrate a strategy of inducing antigen-specific TRM populations at selected skin sites for sensitive sampling of rare circulating T cells. A complementary approach is to leverage pre-existing disease-induced TRMs to recruit antigen-specific T cells locally for MN patch sampling, a strategy directly compatible with current dermatologic clinical practices. For human skin sampling, we fabricated square patches with a larger surface area of 4 cm2, bearing 400 microneedle projections attached to an adhesive backing for stable skin contact (Fig. 5a). Under Institutional Review Board (IRB)-approved protocols, we tested patch application across male and female volunteers of varying ages and skin tones, with wear times from 20 min to 18 h (Fig. 5a–c). Patches were well tolerated, causing only mild redness that resolved within an hour and no bleeding, swelling or adverse reactions (Fig. 5a).

a, Representative photographs of pre- and post-MN patch application on the forearm of human volunteers. b,c. Tolerability of MN sampling was assessed on a cohort of volunteers (n = 45). Shown is the breakdown of volunteer gender and age (b) and skin areas tested (c). d, Human patient (n = 1) undergoes SADBE-induced allergic contact dermatitis with sites previously exposed (14 days before re-exposure) to SADBE. Suction blister and MN samples were collected at 2 and 4 days after SADBE treatment. A non-SADBE exposed site was selected as non-lesional skin. e, Representative flow cytometry plots showing expression of immune cell markers in ISF collected from MN patches and suction blisters. f, Enumeration of recovered total live, CD45, CD3, CD4, CD8, CD4+ TRM and CD8+ T cells as recovered by MN patches or suction blister sampling on day 4 of the ‘TRM recall’ condition. g, Comparison of cell yields from MN patches applied at 2 vs 4 days post TRM recall, alongside non-lesional skin and no-recall control groups. h, Olink proteomics data showing temporal changes in skin-associated proteins collected via MN patches. Panel elements created with BioRender: d, Jalili, S. https://biorender.com/ca1wx1s (2026).
Tissue-resident memory T cells play an important role in skin allergy, accumulating during initial allergen sensitization and driving inflammation upon re-exposure58,59,60,61. In a proof-of-concept human study, we applied MN patches following sensitization with the potent contact allergen squaric acid dibutyl ester (SADBE), which is absent in the natural environment and has been safely used in the clinic as an allergen and immunotherapeutic agent62,63. This initial exposure represents a ‘no recall’ condition, as there is no chance of previous exposure, and SADBE is safe for such studies due to the minimal risk of accidental re-exposure in the future. We then compared the no-recall case to cell recovery after a TRM recall response, induced by re-exposing the same subject to SADBE 2 weeks later. As an addition control, cell sampling was carried out on a distal non-allergen-exposed skin site (non-lesional, Fig. 5d). We also compared MN sampling to suction blister sampling64 at 2 and 4 days post resensitization to examine recall kinetics. Flow cytometry was used to identify recruited CD3+ T cells and assess markers of tissue-resident cells (CD69, CD103) as well as B cells and various innate immune cell populations (Fig. 5e and Supplementary Fig. 2a,b). While suction blister sampling recovered a larger number of live cells, almost none of these were CD45+ immune cells (probably, they are keratinocytes; Fig. 5f and Supplementary Fig. 2c). By contrast, MN patches captured primarily immune cells: CD4+, CD8+, NK, dendritic cells and monocytes (Fig. 5e,f). Strikingly, effective recovery of T cells required resensitization and increased markedly from day 2 to day 4 post recall (Fig. 5g). Although suction blistering yielded slightly higher viability, MN patches captured a greater fraction of CD45+ immune cells (Supplementary Fig. 2c).
Olink proteomic analysis of ISF from MN patches was used to track longitudinal immune responses, revealing that TRM recall was associated with an increase in cytokines linked to T cells in homing and activation (CXCL8, CXCL9, CXCL10, CXCL11, CXCL13, IFNγ, IL-17a, IL-13, IL-4, IL-15, TNF; Fig. 5h). Interestingly, MN patch protein profiles correlated well with those obtained via suction blistering (Supplementary Fig. 2d). However, blistering carries several drawbacks such as long procedure times, pain, hyperpigmentation, keratinocyte contamination and limited suitability for sensitive populations. Furthermore, the negative pressure and elevated temperature used in suction blistering can cause keratinocyte dissociation, potentially affecting the accuracy of results by introducing high numbers of non-immune cells65,66,67. MN patch sampling not only overcomes these limitations but also enables live cell recovery, enabling deeper insights into immune reactions occurring in the skin.
Discussion
We combined MN patch technology with the biology of TRMs to enable efficient sampling of both interstitial fluid and live immune cells from the skin in mice and humans. This approach captures both tissue-resident and circulating antigen-specific T cells and allows for longitudinal immune monitoring. Leveraging TRM cells can dramatically amplify the recruitment of antigen-specific T cells from the circulation to sites where TRM cells are established, enhancing cell recovery over blood sampling. The ability of TRM to orchestrate immune responses that span innate and adaptive immunity is one of many examples by which the immune system shares information between cell types. Comparison with invasive or complex sampling methods, such as skin biopsy and suction blisters, revealed similar or superior results with microneedle patches in both preclinical animal models and in a human case study.
Previous MN technologies primarily targeted soluble biomarkers in ISF. Some innovative designs incorporate paper reservoirs41 or integrate biosensors to perform in situ fluoroimmunoassay on-patch68, enhancing their analytical capabilities. However, many existing MN designs incorporate non-FDA approved biological components, limiting clinical translation. In addition, some methods rely on external vacuum devices that lack precise control over sampling depth and prevent remote or at-home sampling69. This reliance can lead to complications similar to those seen with suction blistering, including the risk of blood contamination in ISF samples24. Furthermore, most of these technologies were developed solely for sampling soluble factors in ISF. While there have been reports of cell sampling using hydrogel MNs, such as those based on hyaluronic acid, the yield of recovered cells remains limited70. Addressing these challenges is crucial for advancing MN sampling technology and enhancing its applicability in clinical settings.
To optimize cell recovery with sampling patches, we systematically screened a panel of hydrogel coatings with varied physicochemical properties. In our previous work, we compared MNs coated with two alginate hydrogels, SLM20 (molecular weight (MW) 75–150 kDa, G/M ratio < 1.0) and SLG100 (MW 150–250 kDa, G/M ratio > 1.5), and found no significant difference in immune cell recovery in vivo26. Here, expanding this analysis, we identified SLG20, characterized by a higher G/M ratio and lower molecular weight, as providing superior cell recovery in vivo. These polymer attributes, known to impact the physical, chemical and biological performance of alginate biomaterials44,46,71,72,73,74, proved critical for patch efficacy. SLG20’s high G/M ratio produces a stiffer, more porous alginate network due to the predominance of G-blocks forming ‘egg-box’ structures with Ca2+ ions. Combined with its lower molecular weight, this results in larger pores and reduced chain entanglement44,75. These features probably enhance the gel swelling capacity and cell migration, consistent with published findings showing increased cell mobility and nutrient transport in high-G-content, low-MW alginate hydrogels76.
To sample immune cell populations of interest, we previously mimicked the classical Mantoux test, where vaccinated animals were inoculated intradermally with small amounts of antigen (with or without adjuvant) to recruit antigen-specific T cells into the skin followed by MN sampling26. In those studies, most recovered cells were TRMs, as the platform was optimized for sampling this population. In contrast, the present study demonstrates that TRM recall can be harnessed not only to recover tissue-resident cells but also to efficiently recruit and sample circulating antigen-specific T cells at peripheral skin sites, enabling interrogation of the systemic T cell pool. A second major distinction is that our previous platform required incorporating antigen and adjuvant into the patch itself, a strategy that, while effective, would necessitate bespoke cGMP formulations for each antigen, posing a translational barrier. In contrast, the TRM recall strategy reported here is antigen agnostic and requires no antigen or adjuvant loading, allowing broad applicability across scenarios ranging from vaccine studies to sampling sites of skin pathology. In vaccination settings, TRM induction and recall could be achieved by the vaccine itself administered intradermally, followed by MN sampling, providing a simpler pathway for clinical translation.
TRM cells in peripheral tissues serve multiple functions. For instance, virus-specific TRMs in the vaginal mucosa enhance immunity against genital HSV-2 infection77, and reactivation of memory CD8+ T cells in the female reproductive tract triggers strong chemokine expression that recruits circulating memory T cells47. Further, CD8+ TRMs can also transiently increase vascular permeability, promoting antibody extravasation and local virus neutralization78. Both mechanisms may contribute to the MN sampling process established here. Although the precise mode of T cell and TRM recruitment into MN coatings remains unclear, murine and human TRMs are known to actively patrol the skin through dynamic migration, particularly in response to local tissue perturbations79,80. We anticipate that this migratory behaviour will facilitate their entry into the hydrogel layer upon patch application. Our data demonstrate that while skin-resident TRM can persist after antigen priming and boosting, their presence alone does not lead to substantial recovery of antigen-specific T cells by MN patches in the absence of a recall stimulus. Instead, efficient recruitment and capture of circulating antigen-specific T cells required TRM reactivation, which triggered a robust influx into the skin. These findings underscore the mechanistic importance of TRM recall in enabling reliable immune monitoring and highlight why recall-based models are more effective than relying on sensitization alone.
In a proof-of-concept study focusing on allergic dermatitis, where a patient’s skin was resensitized with a potent allergen, we observed an increase in the abundance of T cells and TRM cells in resensitized patients compared to those exposed to the allergen once and non-lesional skin sites. These data reaffirm the role of TRM cells in skin allergy, contributing to the recurrence of lesions by potentially recruiting CD4 and CD8 T cells to the skin58,61,81. Proteomics analysis of the interstitial fluid recovered from MN patches in this experiment revealed a longitudinal increase in cytokines responsible for homing and activation of T cells, including IL-17a, IFNγ, CXCL8, CXCL10, CXCL11 and CCL20 (refs. 82,83,84). Our findings demonstrate that sampling MN patches can facilitate longitudinal, non-invasive monitoring of humoral and cellular responses in human patients, comparable to the suction blister method but without its potential limitations. Blistering, for instance, can elevate skin temperatures, potentially affecting cytokines/chemokines, and may lead to lasting hyperpigmentation. Ethical concerns also restrict the use of blistering approaches, particularly in vulnerable populations such as infants or older adults.
While our murine studies clearly demonstrated recovery of antigen-specific CD8+ T cells using OVA and SIV tetramers, translating this analysis to the human allergic contact dermatitis model presents unique challenges, as no validated tetramers or canonical epitopes for the allergen SADBE have yet been defined. Consequently, hapten-specific T cell responses in humans have primarily been inferred indirectly. Previous reports85 showed that SADBE-induced dermatitis leads to oligoclonal TCR expansions in lesional skin distinct from peripheral blood repertoires, suggesting, but not definitively proving, hapten specificity. Unlike viral or model antigens, where tetramers can provide direct evidence of antigen specificity, hapten-driven responses remain technically difficult to resolve. While adapting MN patch sampling to systems with defined antigen specificity (for example, viral antigens or tumour neoantigens) is an important goal for future studies, our primary objective here was to establish proof of concept that viable lymphocytes can be recovered from human skin using MN patches. We also acknowledge the need for larger, more controlled cohorts to further define TRM-mediated recruitment. Future work will extend beyond immunophenotyping and proteomics to single-cell RNA sequencing to explore underlying mechanisms and T cell repertoires in greater depth.
The interventions required for TRM recall could be readily incorporated into existing clinical visits to minimize additional procedures. In vaccination or immunotherapy settings, TRM establishment could coincide with standard booster doses, with recall occurring during follow-up visits. Moreover, many immune-mediated skin diseases already involve repeat intradermal or intralesional interventions, making TRM recall highly compatible with existing clinical routines. In vitiligo, lesions persist chronically due to autoreactive TRM activity, and patients often undergo repeated treatments and monitoring86. Similarly, in alopecia areata, patients frequently receive multidose intralesional corticosteroid or platelet-rich plasma injections every 2–3 weeks87. Incorporating TRM recall into such established regimens could enable longitudinal immune sampling without added burden, underscoring the clinical feasibility of our approach in scenarios where repeat skin-directed interventions are already standard care.
While the integration of MN patch technology with TRM biology enables minimally invasive immune sampling, several considerations are important for interpretation and further development. Effective sampling depends on the presence and functional responsiveness of TRMs, which may vary across anatomical sites, disease states or patient populations, including immunosuppressed individuals or aged skin. Further, TRM recall may represent an active immune perturbation, and repeated stimulation could influence local immune tone or tissue homeostasis, necessitating careful optimization for longitudinal studies. From a technical perspective, the current rigid MN patch geometry may be suboptimal for curved or mechanically dynamic skin surfaces. Future efforts will therefore focus on engineering advances to further enhance platform performance and clinical utility, including incorporation of point-of-care or in situ sensing modalities for real-time biomarker readouts, development of next-generation hydrogel layers to further promote immune cell migration and selective enrichment, and fabrication of flexible or conformal microneedle arrays for anatomically complex sites. Together, these advances will help transition MN-based immune sampling from a powerful experimental approach to a broadly deployable technology for clinical immune monitoring.
Beyond vaccines and skin allergies, this technology offers broad applications for monitoring immune responses to infections, predicting autoimmune flares or therapeutic responses and assessing tissue health in transplantation. MN patches could also sample injection site reactions often observed in clinical trials of new vaccines, including recent HIV trials. Some skin reactions have been reported in individuals who previously received SARS-CoV-2 mRNA vaccines, although the responsible components—lipids, mRNA, polyethylene glycol (PEG) or anti-glycan reactions—remain unclear88. Given these considerations, MN patches provide a non-invasive method to sample and analyse local immune responses in such cases, enabling longitudinal monitoring without biopsies. They can also be adapted for mucosal sites or cutaneous tumours, offering a practical alternative to invasive procedures that require in-person visits. The scalability of MN sampling supports remote, large-scale studies, particularly valuable for sensitive populations such as infants or older adults with skin fragility.
Methods
Fabrication and characterization of sampling MNs
PDMS moulds (Sylgard 184, Dow-Corning) for fabrication of MN patches were prepared by laser micromachining (Blueacre Technology). Poly(L-lactide) (PLLA; RESOMER L 207 S, Evonik Industries) was melted over the moulds under vacuum (−20 mmHg, 200 °C, 120 min). Patches were treated with oxygen plasma (Diener Electronic, 2 min under 0.5 mbar pressure) and then a 0.58% w/v aqueous solution of alginate (PRONOVA SLG20, SLM20, SLG100 or SLM100, Novamatrix, IFF) containing 4.6% v/v sucrose (Teknova, S00572PK) was pipetted onto each patch and allowed to dry at 25 °C for at least 4 h. A crosslinking solution containing 20 mM CaCl2 (Sigma, 21115) was then pipetted onto the surface of MN patches and dried in a tissue culture hood overnight.
Microneedle patches were characterized by SEM using a Zeiss Crossbeam 540 SEM/FIB. For samples applied in vivo, SEM was performed after fixing the samples with paraformaldehyde (4%; Electron Microscopy Sciences, 157-4) and glutaraldehyde (2.5%; Sigma, G7776) for 2 h and incubation in osmium tetroxide (0.5%; Electron Microscopy Sciences, 19152) for 1 h before serial dehydration in ethanol. Samples were then dried overnight and imaged. The in vitro absorption capacity of the patches was assessed by immersing them in phosphate-buffered saline (PBS) and placing them in a 37 °C incubator for a duration ranging from 30 min to 18 h, followed by the measurement of the absorbed liquid mass. For in vivo swelling measurements, the patches were weighed both before and after 18 h insertion into the skin of mice.
Mice
Animal studies were approved by the Massachusetts Institute of Technology (MIT) Institutional Animal Care and Use Committee, and animals were cared for in the US Department of Agriculture-inspected MIT Animal Facility under federal, state, local and National Institutes of Health guidelines for animal care. Female, 6–8 week-old C57BL/6 mice (B6(Cg)-Tyrc−2J/J, JAX 000058) and KikGR transgenic mice (Tg(CAG-KikGR)33Hadj/J, JAX 013753)89, were obtained from the Jackson Laboratory, and colonies were maintained at the animal Koch Institute mouse facility at MIT. Photoconversion was performed as previously described51. The skin was exposed to 405-nm LED light from a fixed distance of 1 cm for 3 min at 50% intensity, with 5-s breaks every 20 s. Black cardboard and aluminum foil were used to shield the rest of the mouse during exposure.
In vitro T cell mobility measurements
Activated T cells were introduced to various alginate groups and then placed inside a chambered coverglass (ThermoFisher, 155360). Wells were incubated in Live Cell Imaging Solution (Invitrogen, A59688DJ) and images were acquired using a Leica SP8 laser-scanning confocal microscope equipped with a ×25 water lens. Five fields of view were imaged for 35 min for each condition under identical settings and subsequently processed using Imaris v.10 image analysis software. T cell movement was tracked using the Spots algorithm within the software. At least 50 tracks per condition were analysed.
Skin application of sampling MNs
Animals were anaesthetized using isofluorane, and MNs were placed on their back after shaving with an approved depilatory cream. Sampling MNs were laid out flat on 3M Nexcare waterproof tape and subsequently applied by pressing down vertically with the thumb or index finger while securing Nexcare tape around the MN to keep it securely in place. Another layer of waterproof tape was secured around the first layer to keep the MN application site dry during the application period.
Skin punch biopsy and tissue digestion
Punch biopsies (6-mm) were performed on murine trunk skin at sites previously sampled by MN patches. Following hair removal, biopsied tissue was dissected free of subcutaneous fat and processed into single-cell suspensions. Briefly, biopsies were minced with scissors and digested in RPMI medium containing collagenase XI (2 mg ml−1; Sigma, C9407), hyaluronidase (0.5 mg ml−1; Sigma, H3506) and DNase I (0.1 mg ml−1; Sigma, DN25) at 37 °C for 45 min with agitation. Digested tissue was resuspended in complete RPMI (C10 medium) to quench enzyme activity, filtered through a 100-µm strainer and washed before flow cytometry analysis. Cell suspensions were immediately kept on ice to preserve viability and minimize further enzymatic digestion.
Model protein immunizations
For model protein immunizations, 6–8-week-old mice were immunized via subcutaneous injection (at the tail base) of Ovalbumin (OVA, 2.5–10 μg, Worthington) along with a lipid-conjugated CpG90 (Lipo-CpG, 0.4–1.24 nmol, Oligo Factory), a TLR9 agonist, as an adjuvant. To induce a classic delayed-type hypersensitivity reaction or establish TRM populations on the skin, OVA-immunized mice were intradermally injected with 10 μl of OVA and adjuvant in the flank. For TRM cell recall, mice were intradermally injected with 10 μl of OVA and adjuvant at the same location where TRM had been initially established. To block the infiltration of T cells, mice were treated intravenously with anti-LFA-1 (200 μg per mouse, clone M17/4, eBioscience, 14-0111-82). Microneedle sampling, blood draws and skin biopsies were performed at different intervals. For all in vivo studies, mice were randomly assigned to groups, each consisting of 4 or more individuals per treatment.
mRNA synthesis
A set of T cell SIV sequences containing putative T cell epitopes91 (Supplementary Table 1) each separated by an AAY proteasomal cleavage motif were cloned into a DNA plasmid backbone using in-fusion cloning. The plasmid sequence was verified using zero-prep sequencing (Primordium Labs). The resultant plasmid DNA was linearized via HindIII-HF (New England Biolabs) endonuclease digestion and purified with PureLink PCR Purification columns (ThermoFishe) following manufacturer instructions. To synthesize mRNA, 20 μl in vitro transcription (IVT) reactions were performed using the HiScribe T7 High Yield RNA Synthesis kit (NEB, E2040S), Cleancap AG (TriLink BioTechnologies) and 1–2 μg of linear DNA template. N1-methylpseudouridine (TriLink BioTechnologies) was used for the IVT reaction in place of uridine. The IVT product was purified using PureLink RNA mini columns (ThermoFisher) following manufacturer instructions. The quality of the resulting mRNA was assessed using UV–Vis spectrophotometry and gel electrophoresis.
Lipid nanoparticle (LNP) formulations
LNPs were formulated using flash nanoprecipitation in a microfluidic mixer. Briefly, SM-102 ionizable lipid (BroadPharm), cholesterol (Avanti Polar Lipids), DSPC (Avanti Polar Lipids) and PEG-DMG (Avanti Polar Lipids) were dissolved in ethanol at a molar ratio of 50:38.5:10:1.5, respectively. mRNA was dissolved in 10 mM citrate buffer pH 3.0 (Alfa Aesar). The lipid and RNA solutions were mixed together at N/P ratio of 6:1 and final RNA concentration of 0.1 mg ml−1 using an Ignite NanoAssemblr (Precision NanoSystems) at a flow rate of 12 ml min−1 and a lipid/RNA flow ratio of 3:1. Formulated LNPs were dialysed in PBS for 2 h in 3500 MWCO Slide-A-Lyzer dialysis cassettes (ThermoFisher).
mRNA immunization
Mice were injected intramuscularly with mRNA–LNPs encoding T cell SIV epitopes. Three weeks after injection, spleens were excised and splenocytes were isolated using mechanical dissociation. ELISPOT was conducted on the splenocytes using the mouse IFNγ ELISPOT kit following manufacturer protocol. Briefly, well plates were coated with mouse IFNγ, and 106 splenocytes per well were seeded on coated wells. Cells were stimulated with SIV peptides at a dose of 2 μg ml−1 and incubated overnight at 37 °C. Plates were developed according to manufacturer protocol and imaged using the CTL-ImmunoSpot plate reader. Numbers of spots were calculated using the CTL-ImmunoSpot software. To establish and recall TRM cells, mice received intradermal injections on days 21 and 28, respectively, with subsequent collection of skin and microneedle samples for further analyses.
Cytokine analysis and flow cytometry
For cytokine sampling, MN patches were placed on the skin for 20 min, and for cell sampling, MN patches were placed for 6, 18 or 24 h. After retrieving the MN patches from skin, they were immersed in 200 μl of PBS containing 2% bovine serum albumin and 100 mM EDTA, and incubated at room temperature on a shaker at 150 r.p.m. for 20 min. The supernatant was collected and centrifuged to pellet cells. For cytokine analysis, MN patches were washed in the absence of FBS. The levels of interstitial fluid cytokines and chemokines were determined by a bead-based multiplex assay using the LEGENDplex mouse cytokine response panel kit (740622, BioLegend) following manufacturer protocol, and samples were subsequently subjected to flow cytometry analysis on either an LSR Fortessa or Symphony flow cytometer (BD Biosciences). The concentration of the different cytokines was calculated using the software LEGENDplex v.8.0 supplied by BioLegend. For surface staining, cells were incubated with anti-Fc receptor antibody (clone 2.4G2) and stained with antibodies in PBS and 2% fetal calf serum for 30 min on ice. All flow cytometry was conducted on either an LSR Fortessa or Symphony (BD Biosciences), and data were collected in Diva (BD Biosciences) and analysed using FlowJo (BD Biosciences). Antibodies used in these studies for mice experiments include: CD45 (BUV395, 564279, 2259805, BD Biosciences), CD3ɛ (AF488, 100321, B366226, BioLegend), CD3ɛ (PerCP Cy5.5, 100218, B365335, Biolegend), CD4 (BUV496, 612952, 1328397, BD Biosciences), CD8 (BV421, 100738, B358297, BioLegend), CD11b (BV786, 101243, B361001, Biolegend), CD103 (BUV 805, 741948, 3033478, BD-Optibuild), CD69 (PE cy7, 104512, B320104/B253212, Biolegend) and SIINFEKL/H-2Kb peptide–MHC (major histocompatibility complex) tetramer (iTAg Tetramer/PE–H-2Kb OVA (SIINFEKL) from MBL). Antibodies used for the human study include: CD45 (PerCP, 368506, Biolegend), CD3 (Brilliant Violet 510, 344828, Biolegend), CD4 (Alexa Fluor 700, 317426, Biolegend), CD8 (Brilliant Violet 605, 344742, Biolegend), CD69 (Brilliant Violet 650, 310934, Biolegend), CD103 (BUV 805, 748501, BD Biosciences), CD19 (Spark NIR 685, 302270, Biolegend), CD11b (Brilliant Violet 785, 101243, Biolegend), CD56 (BUV737, 612766, BD Biosciences), CD16 (Brilliant Violet 570, 302036, Biolegend), CD14 (Spark Blue 550, 367148, Biolegend), HLA-DR (PE/Fire 810, 307683, Biolegend) and CD11c (eFluor 450, 48011642, eBioscience). Viability was assessed by staining with fixable Live/Dead Zombie NIR stain (BioLegend).
Human study design
A healthy individual was recruited at the University of Massachusetts Chan Medical School under IRB-approved protocols (STUDY 00000321 and H00021295). The volunteer underwent sensitization with SADBE in acetone base (Boulevard Compounding Pharmacy). In brief, allergic skin reactions were induced by occluding SADBE on the skin using Finn Chamber AQUA patch test chambers (SmartPractice Canada). The following sites were induced and samples were collected at specific time points: (1) a site naive to SADBE, sampled 2 days later; (2) a site previously exposed to SADBE, sampled 2 days later; (3) a site naive to SADBE, sampled 4 days later; and (4) a site previously exposed to SADBE, sampled 4 days later. In addition, a non-SADBE-exposed site was selected as (5) non-lesional skin for comparison.
Human MN sampling
For each site, two sets of MN were firmly secured to the skin using hypoallergenic Scanpor tape (SmartPractice Canada). The first set of MN patches was applied overnight for cell sampling. After removal from the skin, these patches were immersed in 500 μl of PBS with 100 mM EDTA and 2% fetal bovine serum, and then subjected to agitation on a plate shaker for 20 min at 350 r.p.m. The fluid was subsequently centrifuged at 4 °C at 300 g for 10 min to pellet the cells. After discarding the supernatant, the pelleted cells were processed for flow cytometry analysis. The second set of MN patches was applied for 20 min to collect extracellular fluid. After removal from the skin, these patches were placed in 200 μl of PBS with 100 mM EDTA and subjected to agitation on a plate shaker for 20 min at 350 r.p.m. The fluid was then frozen at −20 °C and stored for subsequent proteomic analysis.
Human suction blister skin sampling
Suction blisters were induced on each site using the Negative Pressure Instrument Model NP-4 (Electronic Diversities), with a negative pressure set between 178–255 mmHg until blisters formed. Once formed, blister fluid was aspirated using a 1.0-ml insulin syringe, carefully transferred to a collection tube, and then centrifuged at 4 °C at 300 g for 10 min to pellet the cells. The supernatant of the blister fluid was subsequently frozen at −20 °C and stored for further proteomic analysis. Pelleted cells from the blister fluid were processed for flow cytometry analysis.
Proteomics analysis
Suction blister and MN patch fluids were analysed using the Olink Flex platform with a custom panel of targets (see Supplementary Table 2). The resulting Olink Normalized Protein Expression (NPX) data were converted to pg ml−1 values and plotted using Prism.
Statistical analysis
Datasets were analysed using the unpaired Student’s t-test or one-way analysis of variance (ANOVA), followed by Tukey’s HSD test for multiple comparisons in GraphPad Prism. P values less than 0.05 were considered statistically significant. All values are reported as means ± s.e.m.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The main data supporting the results in this study are available within the paper and its Supplementary Information. The raw and analysed datasets generated during the study are available for research purposes from the corresponding authors on reasonable request.
References
-
Sun, L., Su, Y., Jiao, A., Wang, X. & Zhang, B. T cells in health and disease. Signal Transduct. Target. Ther. 8, 235 (2023).
-
Koh, C.-H., Lee, S., Kwak, M., Kim, B.-S. & Chung, Y. CD8 T-cell subsets: heterogeneity, functions, and therapeutic potential. Exp. Mol. Med. 55, 2287–2299 (2023).
-
Mizukoshi, E. et al. Peptide vaccine-treated, long-term surviving cancer patients harbor self-renewing tumor-specific CD8+ T cells. Nat. Commun. 13, 3123 (2022).
-
Philpott, J. D. et al. Antigen-specific T cell responses in SARS-CoV-2 mRNA-vaccinated children. Cell Rep. Med. 4, 101298 (2023).
-
Bacher, P. & Scheffold, A. Flow-cytometric analysis of rare antigen-specific T cells. Cytometry A 83A, 692–701 (2013).
-
Anikeeva, N., Grosso, D., Flomenberg, N. & Sykulev, Y. Evaluating frequency and quality of pathogen-specific T cells. Nat. Commun. 7, 13264 (2016).
-
Tippalagama, R. et al. Antigen-specificity measurements are the key to understanding T cell responses. Front. Immunol. 14, 1127470 (2023).
-
Bercovici, N., Duffour, M.-T., Agrawal, S., Salcedo, M. & Abastado, J.-P. New methods for assessing T-cell responses. Clin. Diagn. Lab. Immunol. 7, 859–864 (2000).
-
Yang, J. et al. Antigen-specific T cell analysis reveals that active immune responses to β cell antigens are focused on a unique set of epitopes. J. Immunol. 199, 91–96 (2017).
-
Oelke, M. et al. Ex vivo induction and expansion of antigen-specific cytotoxic T cells by HLA-Ig–coated artificial antigen-presenting cells. Nat. Med. 9, 619–625 (2003).
-
Himawan, A. et al. Where microneedle meets biomarkers: futuristic application for diagnosing and monitoring localized external organ diseases. Adv. Healthc. Mater. 12, 2202066 (2023).
-
DeMuth, P. C. et al. Polymer multilayer tattooing for enhanced DNA vaccination. Nat. Mater. 12, 367–376 (2013).
-
Backlund, C., Jalili-Firoozinezhad, S., Kim, B. & Irvine, D. J. Biomaterials-mediated engineering of the immune system. Annu. Rev. Immunol. 41, 153–179 (2023).
-
vander Straeten, A. et al. A microneedle vaccine printer for thermostable COVID-19 mRNA vaccines. Nat. Biotechnol. 42, 510–517 (2024).
-
Rouphael, N. G. et al. Immunologic mechanisms of seasonal influenza vaccination administered by microneedle patch from a randomized phase I trial. npj Vaccines 6, 89 (2021).
-
Garg, N. et al. Phase 1, randomized, rater and participant blinded placebo-controlled study of the safety, reactogenicity, tolerability and immunogenicity of H1N1 influenza vaccine delivered by VX-103 (a MIMIX microneedle patch [MAP] system) in healthy adults. PLoS ONE 19, e0303450 (2024).
-
Caudill, C. et al. Transdermal vaccination via 3D-printed microneedles induces potent humoral and cellular immunity. Proc. Natl Acad. Sci. USA 118, e2102595118 (2021).
-
Ng, H.-I. et al. Microprojection arrays applied to skin generate mechanical stress, induce an inflammatory transcriptome and cell death, and improve vaccine-induced immune responses. npj Vaccines 4, 41 (2019).
-
Li, W. et al. Rapidly separable microneedle patch for the sustained release of a contraceptive. Nat. Biomed. Eng. 3, 220–229 (2019).
-
Sullivan, S. P. et al. Dissolving polymer microneedle patches for influenza vaccination. Nat. Med. 16, 915–920 (2010).
-
Tran, K. T. M. et al. Transdermal microneedles for the programmable burst release of multiple vaccine payloads. Nat. Biomed. Eng. 5, 998–1007 (2020).
-
Steinbach, S. et al. Temporal dynamics of intradermal cytokine response to tuberculin in Mycobacterium bovis BCG-vaccinated cattle using sampling microneedles. Sci. Rep. 11, 7074 (2021).
-
Dahis, D. et al. Monitoring melanoma responses to STING agonism and focused ultrasound thermal ablation using microneedles and ultrasensitive single molecule arrays. Adv. Funct. Mater. 33, 2301659 (2023).
-
Samant, P. P. et al. Sampling interstitial fluid from human skin using a microneedle patch. Sci. Transl. Med. 12, eaaw0285 (2020).
-
Kolluru, C., Williams, M., Chae, J. & Prausnitz, M. R. Recruitment and collection of dermal interstitial fluid using a microneedle patch. Adv. Healthc. Mater. 8, e1801262 (2019).
-
Mandal, A. et al. Cell and fluid sampling microneedle patches for monitoring skin-resident immunity. Sci. Transl. Med. 10, eaar2227 (2018).
-
Wiig, H. & Swartz, M. A. Interstitial fluid and lymph formation and transport: physiological regulation and roles in inflammation and cancer. Physiol. Rev. 92, 1005–1060 (2012).
-
Szabo, P. A., Miron, M. & Farber, D. L. Location, location, location: tissue resident memory T cells in mice and humans. Sci. Immunol. 4, eaas9673 (2019).
-
Lefrançois, L. & Marzo, A. L. The descent of memory T-cell subsets. Nat. Rev. Immunol. 6, 618–623 (2006).
-
Schenkel, J. M. & Masopust, D. Tissue-resident memory T cells. Immunity 41, 886–897 (2014).
-
Khalil, S., Bardawil, T., Kurban, M. & Abbas, O. Tissue-resident memory T cells in the skin. Inflamm. Res. 69, 245–254 (2020).
-
Farber, D. L. Tissues, not blood, are where immune cells function. Nature 593, 506–509 (2021).
-
Boutet, M. et al. Memory CD8+ T cells mediate early pathogen-specific protection via localized delivery of chemokines and IFNγ to clusters of monocytes. Sci. Adv. 7, eabf9975 (2021).
-
Moser, B. T-cell memory: the importance of chemokine-mediated cell attraction. Curr. Biol. 16, R504–R507 (2006).
-
Lopes, M. S., Jardini, A. L. & Filho, R. M. Poly (lactic acid) production for tissue engineering applications. Procedia Eng. 42, 1402–1413 (2012).
-
Narayanan, G., Vernekar, V. N., Kuyinu, E. L. & Laurencin, C. T. Poly (lactic acid)-based biomaterials for orthopaedic regenerative engineering. Adv. Drug Deliv. Rev. 107, 247–276 (2016).
-
Rouphael, N. G. et al. The safety, immunogenicity, and acceptability of inactivated influenza vaccine delivered by microneedle patch (TIV-MNP 2015): a randomised, partly blinded, placebo-controlled, phase 1 trial. Lancet 390, 649–658 (2017).
-
Lintzeri, D. A., Karimian, N., Blume-Peytavi, U. & Kottner, J. Epidermal thickness in healthy humans: a systematic review and meta-analysis. J. Eur. Acad. Dermatol. Venereol. 36, 1191–1200 (2022).
-
Chopra, K. et al. A comprehensive examination of topographic thickness of skin in the human face. Aesthet. Surg. J. 35, 1007–1013 (2015).
-
Hansen, L. S., Coggle, J. E., Wells, J. & Charles, M. W. The influence of the hair cycle on the thickness of mouse skin. Anat. Rec. 210, 569–573 (1984).
-
Samant, P. P. & Prausnitz, M. R. Mechanisms of sampling interstitial fluid from skin using a microneedle patch. Proc. Natl Acad. Sci. USA 115, 4583–4588 (2018).
-
Al Sulaiman, D. et al. Hydrogel-coated microneedle arrays for minimally invasive sampling and sensing of specific circulating nucleic acids from skin interstitial fluid. ACS Nano 13, 9620–9628 (2019).
-
Boopathy, A. V. et al. Enhancing humoral immunity via sustained-release implantable microneedle patch vaccination. Proc. Natl Acad. Sci. USA 116, 16473–16478 (2019).
-
Lee, K. Y. & Mooney, D. J. Alginate: properties and biomedical applications. Prog. Polym. Sci. 37, 106–126 (2012).
-
Kuo, C. K. & Ma, P. X. Ionically crosslinked alginate hydrogels as scaffolds for tissue engineering: part 1. Structure, gelation rate and mechanical properties. Biomaterials 22, 511–521 (2001).
-
Majedi, F. S. et al. T-cell activation is modulated by the 3D mechanical microenvironment. Biomaterials 252, 120058 (2020).
-
Schenkel, J. M., Fraser, K. A., Vezys, V. & Masopust, D. Sensing and alarm function of resident memory CD8+ T cells. Nat. Immunol. 14, 509–513 (2013).
-
Masopust, D. & Soerens, A. G. Tissue-resident T cells and other resident leukocytes. Annu. Rev. Immunol. 37, 521–546 (2019).
-
Rosato, P. C., Beura, L. K. & Masopust, D. Tissue resident memory T cells and viral immunity. Curr. Opin.Virol. 22, 44–50 (2017).
-
Strobl, J. et al. Long-term skin-resident memory T cells proliferate in situ and are involved in human graft-versus-host disease. Sci. Transl. Med. 12, eabb7028 (2020).
-
Li, Z. et al. In vivo labeling reveals continuous trafficking of TCF-1+ T cells between tumor and lymphoid tissue. J. Exp. Med. 219, e20210749 (2022).
-
Becker, M. D., Garman, K., Whitcup, S. M., Planck, S. R. & Rosenbaum, J. T. Inhibition of leukocyte sticking and infiltration, but not rolling, by antibodies to ICAM-1 and LFA-1 in murine endotoxin-induced uveitis. Invest. Ophthalmol. Vis. Sci. 42, 2563–2566 (2001).
-
Walling, B. L. & Kim, M. LFA-1 in T cell migration and differentiation. Front. Immunol. 9, 952 (2018).
-
Thatte, J., Dabak, V., Williams, M. B., Braciale, T. J. & Ley, K. LFA-1 is required for retention of effector CD8 T cells in mouse lungs. Blood 101, 4916–4922 (2003).
-
Nishikawa, M. et al. Induction of tumor-specific immune response by gene transfer of Hsp70-cell-penetrating peptide fusion protein to tumors in mice. Mol. Ther. 18, 421–428 (2010).
-
Stratman, E. J., Elston, D. M. & Miller, S. J. Skin biopsy. J. Am. Acad. Dermatol. 74, 19–25 (2016).
-
Alguire, P. C. & Mathes, B. M. Skin biopsy techniques for the internist. J. Gen. Intern. Med. 13, 46–54 (1998).
-
Chen, L. & Shen, Z. Tissue-resident memory T cells and their biological characteristics in the recurrence of inflammatory skin disorders. Cell. Mol. Immunol. 17, 64–75 (2020).
-
Akdis, M. et al. T helper (Th) 2 predominance in atopic diseases is due to preferential apoptosis of circulating memory/effector Th1 cells. FASEB J. 17, 1026–1035 (2003).
-
Sans-de San Nicolàs, L. et al. Allergen sensitization stratifies IL-31 production by memory T cells in atopic dermatitis patients. Front. Immunol. 14, 1124018 (2023).
-
Lefevre, M.-A., Vocanson, M. & Nosbaum, A. Role of tissue-resident memory T cells in the pathophysiology of allergic contact dermatitis. Curr. Opin. Allergy Clin. Immunol. 21, 355–360 (2021).
-
Happle, R. et al. Contact allergy as a therapeutic tool for alopecia areata: application of squaric acid dibutylester. Dermatology 161, 289–297 (1980).
-
Harris, J. E., Seykora, J. T. & Lee, R. A. Renbök phenomenon and contact sensitization in a patient with alopecia universalis. Arch. Dermatol. 146, 422–425 (2010).
-
Strassner, J. P., Rashighi, M., Ahmed Refat, M., Richmond, J. M. & Harris, J. E. Suction blistering the lesional skin of vitiligo patients reveals useful biomarkers of disease activity. J. Am. Acad. Dermatol. 76, 847–855.e5 (2017).
-
Gao, P.-R. et al. A comparative study of suction blister epidermal grafting and automated blister epidermal micrograft in stable vitiligo. Sci. Rep. 12, 393 (2022).
-
Friedel, M. et al. Opportunities and challenges in the diagnostic utility of dermal interstitial fluid. Nat. Biomed. Eng. 7, 1541–1555 (2023).
-
Saifullah, K. M. & Faraji Rad, Z. Sampling dermal interstitial fluid using microneedles: a review of recent developments in sampling methods and microneedle-based biosensors. Adv. Mater. Interfaces 10, 2201763 (2023).
-
Wang, Z. et al. Microneedle patch for the ultrasensitive quantification of protein biomarkers in interstitial fluid. Nat. Biomed. Eng. 5, 64–76 (2021).
-
Jiang, X., Wilkirson, E. C., Bailey, A. O., Russell, W. K. & Lillehoj, P. B. Microneedle-based sampling of dermal interstitial fluid using a vacuum-assisted skin patch. Cell Rep. Phys. Sci. 5, 101975 (2024).
-
Dosta, P. et al. Polymeric microneedles enable simultaneous delivery of cancer immunomodulatory drugs and detection of skin biomarkers. Theranostics 13, 1–15 (2023).
-
Neves, M. I., Moroni, L. & Barrias, C. C. Modulating alginate hydrogels for improved biological performance as cellular 3D microenvironments. Front. Bioeng. Biotechnol. 8, 665 (2020).
-
Rosiak, P., Latanska, I., Paul, P., Sujka, W. & Kolesinska, B. Modification of alginates to modulate their physic-chemical properties and obtain biomaterials with different functional properties. Molecules 26, 7264 (2021).
-
Enobakhare, B., Bader, D. L. & Lee, D. A. Concentration and M/G ratio influence the physiochemical and mechanical properties of alginate constructs for tissue engineering. J. Appl. Biomater. Biomech. 4, 87–96 (2006).
-
Martinsen, A., Skjåk-Bræk, G. & Smidsrød, O. Alginate as immobilization material: I. Correlation between chemical and physical properties of alginate gel beads. Biotechnol. Bioeng. 33, 79–89 (1989).
-
de Vos, P., Lazarjani, H. A., Poncelet, D. & Faas, M. M. Polymers in cell encapsulation from an enveloped cell perspective. Adv. Drug Deliv. Rev. 67–68, 15–34 (2014).
-
Rowley, J. A., Madlambayan, G. & Mooney, D. J. Alginate hydrogels as synthetic extracellular matrix materials. Biomaterials 20, 45–53 (1999).
-
Shin, H. & Iwasaki, A. A vaccine strategy that protects against genital herpes by establishing local memory T cells. Nature 491, 463–467 (2012).
-
Rosato, P. C. et al. Tissue-resident memory T cells trigger rapid exudation and local antibody accumulation. Mucosal Immunol. 16, 17–26 (2023).
-
Dijkgraaf, F. E. et al. Tissue patrol by resident memory CD8+ T cells in human skin. Nat. Immunol. 20, 756–764 (2019).
-
Ariotti, S. et al. Tissue-resident memory CD8+ T cells continuously patrol skin epithelia to quickly recognize local antigen. Proc. Natl Acad. Sci. USA 109, 19739–19744 (2012).
-
Migayron, L., Merhi, R., Seneschal, J. & Boniface, K. Resident memory T cells in nonlesional skin and healed lesions of patients with chronic inflammatory diseases: appearances can be deceptive. J. Allergy Clin. Immunol. 153, 606–614 (2024).
-
Schmidt, J. D. et al. Rapid allergen-induced interleukin-17 and interferon-γ secretion by skin-resident memory CD8+ T cells. Contact Dermatitis 76, 218–227 (2017).
-
Marchesini Tovar, G., Gallen, C. & Bergsbaken, T. CD8+ tissue-resident memory T cells: versatile guardians of the tissue. J. Immunol. 212, 361–368 (2024).
-
Kienzl, P. et al. The cytokine environment influence on human skin-derived T cells. FASEB J. 33, 6514–6525 (2019).
-
Gaide, O. et al. Common clonal origin of central and resident memory T cells following skin immunization. Nat. Med. 21, 647–653 (2015).
-
Richmond, J. M. et al. Antibody blockade of IL-15 signaling has the potential to durably reverse vitiligo. Sci. Transl. Med. 10, eaam7710 (2018).
-
Lintzeri, D. A. et al. Alopecia areata – current understanding and management. J. Dtsch. Dermatol. Ges. 20, 59–90 (2022).
-
Cohen, J. Puzzling skin side effects stymie advance of promising HIV vaccine. Science https://doi.org/10.1126/science.zwkry00 (2024).
-
Nowotschin, S. & Hadjantonakis, A.-K. Use of KikGR a photoconvertible green-to-red fluorescent protein for cell labeling and lineage analysis in ES cells and mouse embryos. BMC Dev. Biol. 9, 49 (2009).
-
Liu, H. et al. Structure-based programming of lymph-node targeting in molecular vaccines. Nature 507, 519–522 (2014).
-
Gaiha, G. D. et al. Structural topology defines protective CD8+ T cell epitopes in the HIV proteome. Science 364, 480–484 (2019).
Acknowledgements
We thank L. Santollani for guidance on the KikGR mice, C. Backlund and A. Hostetler for helping with immunoassays, and J. Dye for assisting with mRNA synthesis; D. Garafola for assisting with animal studies and S.-Y. Kang for helping with photography; M. Melo for valuable advice on in vitro and in vivo assays; and the Koch Institute Swanson Biotechnology Center for technical support, specifically the Flow Cytometry and Microscopy Core facilities. This work was supported by the NIH (award U01AI176310 to J.E.H., D.J.I. and S.J.), the Jackson Laboratory, the Ragon Institute of MGH, MIT and Harvard, and by the Koch Institute Support (core) Grant P30-CA14051from the National Cancer Institute. We thank the King Trust, Bank of America Private Bank, Co-Trustees Fellowship, for supporting K.A. D.J.I. is an investigator at the Howard Hughes Medical Institute. Panels created in BioRender: Fig. 1b, Jalili, S. https://biorender.com/b3buou9 (2026); Fig. 2a, Jalili, S. https://biorender.com/awru57q (2026); panel elements created with BioRender: Fig. 2h, Jalili, S. https://biorender.com/8arfbyn (2026); Fig. 3a, Jalili, S. https://biorender.com/oz2aoky (2026); Fig. 5d, Jalili, S. https://biorender.com/ca1wx1s (2026); Extended Data Fig. 1a, Jalili, S. https://biorender.com/b3buou9 (2026); authors own a full licence to publish the BioRender figures.
Ethics declarations
Competing interests
S.J., P.T.H. and D.J.I. have submitted a patent application (no. PCT/US25/43105) filed by MIT related to the data presented in this work. M.R. is principal or co-investigator of studies sponsored by Pfizer, Biogen, AbbVie, Incyte, LEO Pharma, Abeona Therapeutics, Dermavant and Target RWE; and M.R. provides consulting for Pfizer, Biogen, Incyte, Takeda, Inzen, ROME Therapeutics, Almirall, Medicxi, Related Sciences and VisualDx. The other authors declare no competing interests.
Peer review
Peer review information
Nature Biomedical Engineering thanks Scott Mueller and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Optimization and characterization of alginate-coated microneedle patches.
a, Schematic representation of the fabrication and alginate coating process of the MN patches. b, Scanning electron micrographs of uncoated (bare) and alginate coated MN patches. Electron micrographs of collected cells in SLG20 patch after insertion into the mouse skin. Cells were false-colored in purple to distinguish from the MN base and hydrogel. c, In vitro swelling properties of MN patches coated with different alginates (n = 5 hydrogels per group). d, e, In vitro PBS uptake by SLG20 MN patches as a function of crosslinking concentrations, alginate concentrations, and sucrose content (n = 3 hydrogels for panel e). f, Enumeration of CD3 + T cells collected in thin and thick SLG20 MN patches. Green and red dots represent two separate experiments (n = 9 animals for naïve and Alg-Thin groups, and n = 13 animals for Alg-Thick group). Data shown are means ± SEM. P values were determined by one-way ANOVA followed by Tukey’s multiple comparison test (f). Panel elements created with BioRender: a, Jalili, S. https://biorender.com/b3buou9 (2026).
Extended Data Fig. 2 Induction and recall of skin tissue-resident memory T cells (TRMs) enhance local immune recruitment.
a-d, Enumeration of recovered total live leukocytes, myeloid cells, T cells, and antigen-specific T cells sampled using skin biopsies (Naïve group animal number: n = 3 for panel 1 and n = 4 for panel b–d; n = 6 animal for No TRM, No recall, and TRM recall groups). e, Representative flow cytometry plots showing expression of CD3+CD103+ T cells and antigen-specific CD8+CD103+ TRM cells in the skin. f, Enumeration of recovered CD3+CD103+ T cells and antigen-specific CD8+CD103+ TRM cells in the skin at TRM establishment and recall steps and in comparison with No TRM. (n = 5 animals per group). g, Mice were immunized subcutaneously (prime and boost) with OVA (10 µg) plus CpG (1.24 nmol). For TRM establishment, animals then received intradermal administration of either OVA (10 µg) plus CpG (1.24 nmol), OVA alone (10 µg), CpG alone (1.24 nmol), or PBS control, followed by MN sampling to assess TRM establishment in the skin 7 days later. Quantification of cells collected per patch revealed that co-delivery of OVA and CpG significantly increased the number of total T cells (CD3+), CD8+ T cells, and antigen-specific CD8+ T cells compared to antigen or adjuvant alone. (n = 5 animals per group). h, Representative flow cytometry plots for identifying and quantifying immune cells in the MN patch. i, Enumeration of recovered CD45− and CD45+CD11b−CD3− cells in the MN patch at TRM establishment and recall steps and in comparison with No TRM. At least n = 3 animals per group were used for all panels (n = 10 animals per group). Data shown are means ± SEM. P values were determined by one-way ANOVA followed by Tukey’s multiple comparison test (a-d, f, g, i).
Extended Data Fig. 3 Key parameters influencing microneedle sampling yield.
a, Enumeration of recovered total live leukocytes, myeloid cells, T cells, TRM cells, and antigen-specific TRM cells in MN patches coated with different alginates (n = 5 animals per group). b, Temporal comparison of CD3+CD8+ T cells and antigen-specific T cells collection 1 day and 6 days post-TRM recall (n = 4 animals per group). c, Effect of MN patch insertion duration on the number of live cells, myeloid cells, and T cells collected by MN patches (n = 4 animals per group). Data shown are means ± SEM. P values were determined by one-way ANOVA followed by Tukey’s multiple comparison test.
Extended Data Fig. 4 Effect of antigen/adjuvant dose during TRM establishment and recall on local and systemic antigen-specific T cells.
a, study design to compare the effect of multiple amounts of OVA and adjuvant during the prime/boost and TRM establishment/recall steps on the number of antigen-specific lymphocytes systemically and locally. b, Representative flow cytometry plots of antigen-specific CD8+ T cells in the control group at 1 and 3 weeks post-prime/boost vaccination. c, d, Quantification of antigen-specific T cells in the skin at the injection site across different vaccination regimens and at various intervals after TRM establishment dose. e, Quantification of antigen-specific T cells in blood following different doses of TRM establishment and after TRM recall dose. f, Quantitative comparison of antigen-specific CD8 + T cells recovered via skin biopsy (6 mm punch biopsy) and MN patches normalized per 1 cm2 sampling area. n = 5 animals per group were used for all panels. Data shown are means ± SEM. P values were determined by one-way (c,d) and two-way (e) ANOVA followed by Tukey’s multiple comparison test, and unpaired two-tailed Student’s t-test (f).
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jalili, S., Hosn, R.R., Ko, WC. et al. Leveraging tissue-resident memory T cells for non-invasive immune monitoring via microneedle skin patches. Nat. Biomed. Eng (2026). https://doi.org/10.1038/s41551-026-01617-7
-
Received:
-
Accepted:
-
Published:
-
Version of record:
-
DOI: https://doi.org/10.1038/s41551-026-01617-7

