
Introduction
Mosquitoes are considered the world’s deadliest animals due to their capacity to transmit an array of pathogens that cause severe infectious diseases1. Vector-borne diseases account for more than 17% of all infectious diseases globally and are responsible for more than 700,000 deaths annually. For instance, malaria alone leads to approximately 249 million cases and over 608,000 deaths each year2. Dengue, the most prevalent mosquito-borne viral infection, threatens more than 3.9 billion people in 139 countries and is associated with 96 million symptomatic cases and an estimated 40,000 deaths annually2. An important vector species of this disease is the Asian tiger mosquito Aedes albopictus. It is capable of transmitting up to 34 arboviruses and can enhance nematode infection3,4,5,6,7,8. These mosquito-borne diseases are often regarded as tropical, but due to human-mediated dispersal and climate change, the range of Aedes albopictus now extends beyond the tropics into temperate regions9.
As hematophagous Diptera, adult female mosquitoes inject a complex cocktail of pharmacologically relevant bioactive components into their hosts to facilitate blood meals10,11,12. These factors not only assist in overcoming host defenses but also interact synergistically with pathogens, having direct repercussions on their virulence13. Traditionally, the secretions injected during blood meal are considered salivary secretions, and the totality of its chemical arsenal is referred to as “sialome”14,15. Conceptually, the blood meal-facilitating secretions of mosquitoes and other hematophagous Diptera also align with the formal definition of venom sensu Fry et al. 2019: “ …a secretion, produced in a specialized gland in one animal, and delivered to a target animal through the infliction of a wound (regardless of how tiny it) a venom must further contain molecules that disrupt normal physiological or biochemical processes so as to facilitate feeding or defense by the producing animal”14, as well as others16,17,18. This perspective emphasizes the impact such secretions can have on physiological processes and recognizes the potentially greater diversity of bioactive compounds. Therefore, modern works acknowledge the venomous nature of mosquitoes and consider their chemical venom arsenal to be an important yet neglected subject18,19. For the purpose of this study, we hereafter refer to the salivary secretions of mosquitoes as venom. The venom of mosquitoes is a decisive factor in the complex triad of vector, pathogen, and host interactions where it serves as a mediating agent. On one hand, it is the active principle that facilitates blood meals and, partially, influences pathogen virulence. On the other hand, it contains several potent biomolecules that target the host immune and cardiovascular systems20. The injected venom can increase vascular permeability and manipulate immune cell recruitment. Within the salivary glands it creates an environment promoting pathogen survival and replication13. Hence, improving our understanding of mosquito venom composition, bioactivity, and molecular mechanisms can offer valuable insights for developing vaccines, novel insecticides and exploring potential vector control strategies, such as gene silencing via RNA interference13,21,22. For example, a recent study identified a labrum-interacting protein in Ae. albopictus venom that promotes the penetration of host skin by activating mouthpart movements and salivation. These proteins represent promising targets for disrupting blood feeding and thus reducing disease transmission23. Likewise, other previously identified mosquito venom components, such as peptides from the cecropin family, have shown antimicrobial activity24,25. These molecules may hold therapeutic potential or serve as lead compounds for novel antimicrobial agents26,27.
The methodological restraints of analytical and preparative chemistry have prevented the efficient analysis of venoms and their components from small invertebrates, such as mosquitoes28,29,30. Only recently, through the application of modern systems biology methods paired with biotechnology approaches, the targeted and efficient disentanglement of such venom systems has become feasible and has given rise to a new research field referred to as modern venomics29. Although these methods have paved the way for theoretical analysis of venoms from the smallest venomous lineages, including various insects, arachnids, and crustaceans, only a handful of these species have been investigated thus far19,30. Much remains to be learned about the chemical arsenals of the many remaining lineages, which include mosquitoes18. With the importance of providing a better understanding of vector biology and venom biodiscovery, an in-depth analysis of mosquito venoms via modern venomics appears particularly promising. In this study, we aimed to broaden the understanding of the molecular composition, function, and translational potential of mosquito venoms by studying the venom system of the invasive Asian tiger mosquito Ae. albopictus. This approach allows us to elucidate mosquito venom from different perspectives, as provided by traditional sialotranscriptomic methods. We offer new insights into the molecular mechanisms of blood feeding and vector competence and open up new translational avenues for mosquito venom studies.
Results
A transcriptomic perspective on the Ae. albopictus venom system
For a first assessment of the Ae. albopictus venom system, we analyzed the salivary gland transcriptome. Therefore, we dissected salivary glands from the thorax of 60 adult female specimens. The salivary gland samples were pooled, and mRNA was extracted for subsequent transcriptome sequencing. Sequencing revealed 19,201,370 paired-end reads (total of 5.8 Gbp, GC content 43.9%, Q30 88.0%), 13,278,893 of which remained after quality control. The concatenated transcriptome assembly revealed 346,630 contigs, to which our trimmed reads were mapped at a rate of 95.89%. A total of 4,555,524 open reading frames (ORFs) were identified and extracted. Following additional processing, we determined 2374 ORFs with an assigned InterPro ID and a predicted signal peptide, accounting for a total of 2,699,363 TPM. This ORF list served as an input database for our proteome analysis. The assigned ORFs most likely represent precursors of secreted polypeptides, a pivotal feature of venom components, and were thus further analyzed. The molecular weights ranged from 2.3 kDa to 369.3 kDa, with sequence lengths of 21–3344 amino acids (Supplementary data).
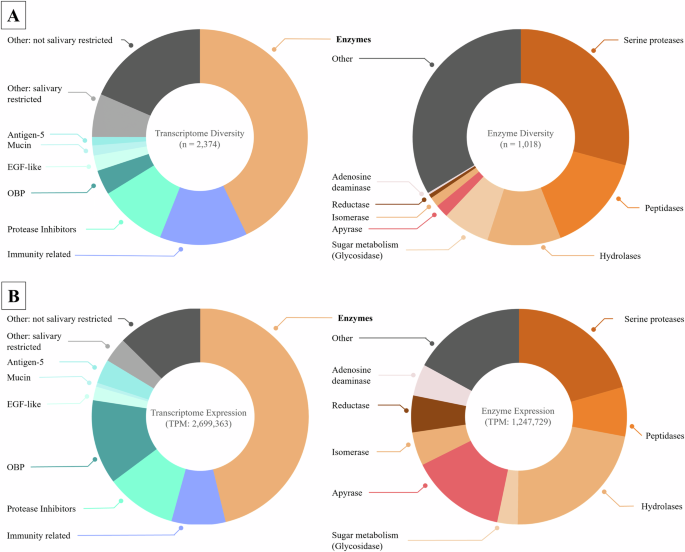
First, we explored the diversity of the whole transcriptome (Fig. 1A). Compounds annotated as enzymes appeared to be the most diverse class, representing 43% of all transcripts. Immunity-related peptides were the second most diverse class with 13%. This group contained several components that were similar to defensins, attacins, and cecropins. Next, 10% of the transcripts were protease inhibitors, and odorant-binding proteins (OBP), mainly D7 family, accounted for 4% of the transcripts. Mucins and epidermal growth factor (EGF)-like transcripts were detected with only 2% each, while Antigen-5 transcripts (CAP family) were represented in only 1%. A total of 6% of all transcripts could not be defined to specific classes and are here classified as “Other: salivary restricted”; these transcripts may feature protein families with roles for their venom or the salivary gland. The remaining 18% of the transcripts were similar to those of various housekeeping proteins and were classified as “Other: not salivary restricted”. As putative enzymes dominate the venom composition of Ae. albopictus, we investigated this class in more detail (Fig. 1A, right panel). Within the enzyme class, serine proteases are the most diverse and feature 29% of all enzymes identified. Other peptidases represented 15% of the enzymes. Hydrolases were found to represent 11% of the enzyme transcripts. Various Glycosidases and transcripts potentially relevant for sugar metabolism were detected with 7%. Apyrases display 2% and Isomerases 1% of enzyme diversity. Reductases only display 1% and Adenosine deaminases (ADA) > 1%. When considering only the total number of unique precursors, 34% of the precursors were associated with other enzymes, including phosphatases, metalloproteases or putative housekeeping enzymes, which are each represented in small amounts.

A Total number of unique transcripts for each protein family. The left panel displays the whole transcriptome diversity consisting of 2374 transcripts. The right panel focuses on enzymes and their subfamilies, with a total of 1018 transcripts. B Relative expression levels (TPM scores) summed for each protein family. The left panel shows the whole transcriptome expression profile, which included the expression of seven families and others, for a total of 2,699,363 TPM. The enzyme expression data are given in the right panel, considering eight enzyme families and others, for a total of 1,247,729 TPM.
While exploring transcript diversity has already provided a good overview of putatively contained venom compound classes, a quantitative metric such as the expression level could indicate which transcripts may be more relevant in biological processes (Fig. 1B). Enzymes comprised 46% of the summed-up TPMs. OBPs (D7 proteins) accounted for 13% of the transcript abundancy while protease inhibitors accounted for 11%, and immunity-related proteins for 8%. The Antigen-5 family claimed, 4% and EGF-like proteins 2% of TPMs. Mucins remained only slightly represented with 1% of the TPMs. Multiple other protein families that may have functions in the blood feeding process were detected in the transcriptome and are referred to as “Other: salivary restricted”. However, they together account for only ca. 4% of TPMs. This group contains, for example, 23.4 kDa salivary proteins, which are known to be secreted in saliva and upregulated in blood-fed individuals and have been reported in mosquitoes31,32. Lastly, 13% of the TPMs represented proteins likely employed for other functions, including housekeeping and basic cellular functions and are referred to as “Other: not salivary restricted”. When focusing only on enzyme transcripts, we observed that hydrolases were most abundant and corresponded to 22% of the TPMs. These were followed by serine proteases with 21%, apyrases with 17% and peptidases with 7%. Reductases regard 6%, while isomerases and adenosine deaminases display 5% of abundancy each. Glycosidases constitute only 3% of the total enzyme expression. Various other enzyme classes, such as metalloproteases or chitinases, can be found and feature 17% of the total enzyme abundancy, while each individual annotated enzyme class within the “Other” group contains less than 3% of the TPMs.
Proteomic integration reveals a complex venom in Ae. albopictus
While transcriptomic analysis reveals the entirety of transcripts expressed in the salivary glands, proteomic profiling can confirm which components are actually deployed in the venom. With this approach, the active molecular arsenal of the mosquito venom can be characterized33. Hence, we supplemented our salivary gland transcriptomic data with a high-resolution venom proteome for Ae. albopictus that used the generated transcriptome as a species- and tissue-specific database for peptide searches (Supplementary Data).
Matching the proteomic data with the transcriptome database revealed 119 precursors with proteome coverage classified to various putative protein families. A majority of the proteins identified were enzymes, which constituted 31% of all the proteins identified (Fig. 2A). OBPs, comprising 28% of all proteins, are also very prominent and consist of short and long D7 proteins. The third most diverse group are immunity-related proteins, occupying 17% of the proteome. These were annotated as members of three different families: C-type lectins, Fibrinogen-Related Domain (FReD)/ficolin domain-containing proteins, and glycoside hydrolases (lysozymes). Protease inhibitors display 15% of the proteome, consisting of serpins, cystatins and kazal-type inhibitors. Proteins of the antigen-5 family represent 8% of the proteome. Finally, 2.8% of the proteins found in the proteome could be annotated to the SG1/62 kDa superfamily. A more in-depth look at the identified enzymes revealed four main classes. First, hydrolases, which constitute 49% of all identified enzymes, are divided into three groups: glycosyl hydrolases (22%), alpha-amylases (19%) and adenosine deaminases (8%). Second, apyrases accounted for 22% of the enzymes. One potential 5’-nucleotidase was detected, corresponding to 3%. Angiotensin-converting enzymes (peptidase M2) account for 24% of all enzymes. Finally, the histidine acid phosphatase family/inositol polyphosphate phosphatase 1 was recovered with 3% of the total enzyme amount.

A depicts diversity. The left panel highlights the whole proteome of 119 proteins in five main classes. The right panel shows the largest group, the enzymes, with a total of 37 proteins in seven main classes. B The whole proteome based on expression levels from the transcriptomic data for each protein group. The left panel focuses on the whole proteome, with a total of 854,796 TPMs and the right panel displays only the enzymes, with 321,898 TPMs.
In addition to proteome diversity, we analyzed how prominently each protein is expressed based on TPMs (Fig. 2B) to further identify key proteins that may drive venom biology. The 119 different proteins related to a total of 854,796 TPM of our transcriptome. Enzymes represent 37% of the TPMs; OBPs (D7 proteins) represented 30%; and Protease inhibitors, 16%. Antigen-5 proteins were related to 8% of all TPMs, while immunity-related proteins correspond only to 7% of the TPMs. The remaining 3% were related to the SG1/62 kDa superfamily. When focusing only on enzymes, hydrolases are presented by a total of 46% of TPMs split into three groups: alpha-amylases (23%), adenosine deaminases (18%) and glycosyl hydrolases (5%). Apyrases represent 45% of TPMs, and 5% are classified as 5’-nucleosidases. Only 7% of TPMs are Angiotensin-converting enzymes (peptidase M2). Histidine acid phosphatase family/inositol polyphosphate phosphatase 1 are found only as 1% of the enzyme transcripts.
Discovery of Ae. albopictus venom cecropins and their calculated properties
From a biodiscovery perspective, short linear peptides and immunity-related compounds are of high interest due to several aspects34. In addition to their roles in innate immunity, they could also play roles in modulating the host response and the microbiome and may affect the blood feeding process, vector competence and disease transmission35,36,37.
Within the transcriptomic diversity of Ae. albopictus salivary glands, various immunity-related compounds were identified (Figs. 2 and 3), some of which could be classified as members of known AMPs, for example, defensins or attacins. Within these our analysis indicated the presence of six peptide precursors that appear to represent members of the cecropin family. Reminiscent to other recent works that have reported cecropins from Ae. albopictus transcriptomes and cell lines25,28,38. To explore their putative functional space, we analyzed their physicochemical properties based on their primary structure to infer further details about their potential function and translational exploitability. The precursor sequences Cec_Aealb_B1, Cec_Aealb_A4 and Cec_Aealb_A3 showed more than 90% sequence similarity to previously reported mosquito cecropins from other Culicidae species (e.g., Anopheles arabiensis and Aedes aegypti), supporting our initial classification. Sequence analysis and comparative alignments revealed that the herein discovered cecropin precursors can be divided into two families. The A family (Cec_Aealb_A1, Cec_Aealb_A1, Cec_Aealb_A3) and the B family (Cec_Aealb_B1, Cec_Aealb_B2). Overall, both families exhibited high sequence similarity but higher degrees of sequence disparity at their C-terminus (Fig. 3).

Dark purple indicates 100%, purple 80%, and light purple 60% similarity. The noncolored amino acids indicate less than 60% similarity.
For in silico analysis, we focused on putative mature peptides and their potential modifications inferred from the precursor sequence data (Table 1, Fig. 3). Different in silico tools provided insights into mature peptide properties and sequence-structure relationships. Antimicrobial Peptide Scanner vr.2 indicated that all six have a very high probability of displaying AMPs and that all could form amphipathic α-helices (Table S1). The mature peptides ranged from 28 to 36 amino acids in length, with theoretical masses ranging from 3.5 kDa to 3.9 kDa. All the Ae. albopictus cecropins are highly cationic according to the analysis of their primary structure: the mature peptides mCec_Aealb_A3_amide and mCec_Aealb_A3 abound with a net charge of +10. mCec_Aealb_B2_amide and mCec_Aealb_B2 had a total charge of +9, while mCec_Aealb_A1 and mCec_Aealb_A4 had a total charge of +7. The percentage of hydrophobic amino acids ranged from 42–47%, and the number of hydrophobic amino acids constructing the helices hydrophobic surface was 9 for mCec_Aealb_B2_amide and mCec_Aealb_B2, 10 only for mCec_Aealb_A4, and 12 for the others (Table 2).
Structural dynamics of cecropins
Our structural analysis further supports that the cecropins fold into α-helices. All showed a hinged C-terminal end at amino acid positions 23 to 26. In most peptides, these regions contain proline and glycine. The distribution of hydrophobic amino acids occurs mainly on one side of the helices (Fig. S1). Interestingly, all the peptides developed a positively charged surface towards the N-terminus. A negative charge can be found at the C-terminal region (Fig. 4).

The α-helical organization of the peptides is presented in gray. Additionally, the peptide surface is displayed with red coloration for negative charge, white for neutral charge and blue indicating a positive charge. Models were created with UCSF ChimeraX 1.10.
Ae. albopictus venom cecropins have anti-E. coli activity
The structural and physicochemical properties as well as the literature on insect-derived cecropins prompted us to test the biological activities of the homologs identified herein, starting with their antimicrobial traits. Therefore, putative mature peptides of the precursors were synthesized with their likely modifications inferred from the sequence data (Table 1). Two cecropins precursors occur as the same putative peptide with and without a C-terminal glycine, which serves as an amidation signal. This indicates that the resulting peptide may be the truncated amidated version, respectively. Hence, for the peptides precursors, for which we detected full-length and truncated versions, we synthesized the unprocessed and amidated mature peptides (precursors Cec_Aealb_A2/A3 for mature peptides mCec_Aealb_A3_amide and mCec_Aealb_A3; as well as, Cec_Aealb_B1/B2 for mCec_Aealb_A2_amide and mCec_Aealb_A2) (Tables 1 and 2). For the other peptides, we synthesized the putative peptides as indicated by theor sequence.
A first pre-screening of all six synthesized mature structures of cecropins against E. coli DSMZ 102053 showed strong inhibitions of its growth over the course of 48 h at a concentration of 200 µM (Fig. S2, Table S2). This led us to further investigate the antimicrobial effects of these compounds against a larger panel of microorganisms and to calculate their MIC. The tested bacterial strains included E. coli ATCC25922, M. smegmatis ATCC607, the methicillin-resistant S. aureus strain ATCC33592 and the yeast C. albicans FH2173. None of the tested peptides had MICs lower than 50 µM against S. aureus or C. albicans, hence indicating little to no activity. For M. smegmatis, only mCec_Aealb_B2_amide seems to be active, with a MIC ranging from 25 to 12.5 µM. In contrast, strong activities were found against E. coli, with the exception of mCec_Aealb_A1 with a MIC of 50 µM. mCec_Aealb_A4 exhibited activity at concentrations ranging from 6.25 to 3.125 µM. The three cecropins mCec_Aealb_B2_amide, mCec_Aealb_B2 and mCec_Aealb_A3 had very low MICs of 0.4 µM, while mCec_Aealb_A3_amide had inhibitory effects at 0.2 µM (Table 3).
These strong effects against E. coli led us to further investigate the activity of selected peptides in presence of LPS. mCec_Aealb_A1 was excluded from this test because it lacked antibacterial effects. When tested in the presence of LPS, the activity of all tested cecropins decreased 15 to 30-fold. mCec_Aealb_A3_amide exhibited activity at 3.125 µM in the presence of LPS, while mCec_Aealb_B2_amide, mCec_Aealb_A4 and mCec_Aealb_A3 exhibited an inhibition range reduced to 6.25 µM, and that of mCec_Aealb_B2 was reduced to 12.5 µM.
Ae. albopictus venom cecropins show weak effects on mammalian cells
Given the antimicrobial properties observed, we next investigated whether the peptides exert cytotoxic or hemolytic activity. This will provide further insights into the biology of the peptides and highlight limits of potential therapeutic applicability.
First, we assessed the activity of the peptides against horse blood erythrocytes. The tested cecropins had no hemolytic effect on the tested blood cells at a concentration of 200 µM. We further tested the effects of cecropins on two vertebrate cell lines, MDCKII and Calu-3. In both cell lines, the peptides mCec_Aealb_A1, mCec_Aealb_A3_amide, mCec_Aealb_A4 and mCec_Aealb_A3 showed no noteworthy decline in cell viability. In MDCKII cells, mCec_Aealb_B2 displayed a reduction of cell viability of ~30% compared to the untreated control. mCec_Aealb_B2_amide reduced the viability of MDCKII and Calu-3 cells by ~70% (Fig. 5).

A Hemolytic activity of cecropins against horse blood erythrocytes at a concentration of 200 µM. The bar chart displays hemolytic activity for each cecropin after 1 h of incubation with erythrocytes. The data were normalized to the hemolytic activity of Triton X-100 as 100% of activity and to that of the solvent DMSO in 1:5 dilution as 0%. B The left bar chart represents Madin-Darby canine kidney cells (MDCKII) (blue), and the right bar chart represents human-derived airway epithelial cells (Calu-3) (purple). The bars show cell viability after 48 h of treatment with the cecropins. All experiments were carried out in triplicate, and the data is displayed as means with standard deviation. Charts were created using GraphPad Prism 10.0.2.
Discussion
Given the critical role of mosquitoes as vectors, a precise understanding of the biochemical complexity of their venom is essential. To achieve this, a proteotranscriptomic-guided modern venomics experiment was employed to disentangle the molecular arsenal present in the venom of the Asian tiger mosquito (Ae. albopictus). We report another perspective to mitigate potential biases inherent in conventional sialotranscriptomic approaches. This strategy allowed us to explore the components of mosquito venom, which may play key roles in blood feeding, pathogen transmission, and host manipulation while also serving as potential targets for vector control and as sources of novel therapeutic agents. Our proteo-transcriptomic investigation of the Ae. albopictus venom system was guided by the sialome nomenclature established by Ribeiro et al. (2010) and VectorBase39 paired with the venom databases VenomZone (available online: https://venomzone.expasy.org/) and Tox-Prot40. Overall, our findings align with previous pioneering works of Ribeiro, Arca and colleagues, which paved the foundations of sialomic studies in hematophagous dipterans15,32. Considering the totality of the so far existing data, Mosquitoes appears to abound with a chemically and functionally surprisingly complex venom, with several shared protein families that may be of importance32,41. This includes families, such as D7, antigen-5, apyrase, and ADA, which are repeatedly found in various Mosquito venoms and deserve additional scientific attention32,42,43,44. However, our study refines the current understanding of the molecular arsenal of the Ae. albopictus venom system and is followed by a discussion of the particularities and potential roles of the most abundantly represented components of our analysis.
Proteomic analysis revealed the diversity and functional space of Aedes albopictus venom enzymes. Enzymes constitute a major portion of mosquito venom. The largest enzymatic group comprises hydrolases, a diverse class known to participate in processes such as feeding, digestion, and reproduction in arthropods45. Alpha-amylases and glycosyl hydrolases were found in high abundance and were present across all blood-feeding Nematocera. They may display the evolutionary link to the conserved ancestral trait of sugar feeding and the independent evolution of hematophagy between members of Culicomorpha and Psychodidae42. Another prominent enzyme group comprises ADA, which is proposed to facilitate blood feeding via anti-hemostatic and anti-inflammatory activities and to act as immunomodulatory agents42,46. Notably, ADA has also been linked to enhanced dengue virus replication in Ae. albopictus47. Apyrases and 5′-nucleotidases were identified which hydrolyze ATP and ADP, which are key components of the blood coagulation cascade and thereby may facilitate blood uptake by perturbing blood clotting20,42. We also detected a high diversity of ACE homologs, which are likewise considered as blood-feeding facilitators based on their role in vertebrate blood pressure regulation48,49,50, raising the intriguing possibility that mosquitoes might exploit this function to maintain blood flow during feeding, although this hypothesis has yet to be experimentally validated. Another identified group of feeding-related enzymes are phosphatases, primarily histidine acid phosphatases51. To our knowledge, this is the first report of phosphatases in mosquito venom. Their role may involve blood feeding by hydrolyzing dinucleotides, thereby suppressing platelet-derived inflammatory mediators, which remains to be further investigated52,53. In sandflies, they have been shown to inhibit host coagulation proteins, and similarly, in cattle ticks, elevated phosphatase activity has been observed during blood feeding54,55. Overall, our analysis revealed Ae. albopictus venom as a rich source of enzymes, similar to the venoms of other neglected arthropods as suggested by recent studies56,57,58,59. Considering their biological function, many of those are associated with blood feeding and thus serve a primarily trophic function. Additionally, multiple of the enzyme families proposed herein could be targeted for vector control to inhibit blood feeding efficiency to disturb other physiological processes within mosquitoes or as biomarkers60,61.
In addition to the array of enzymatic components, we detected several components from various non-enzymatic protein families across our generated Ae. albopictus venom proteome. Henceforth, we discuss these components in light of their known biological activities and their potential role in the venom system. One major protein group is OBPs, which are primarily represented by D7 salivary proteins. These are well characterized in mosquito venom and are present in other blood-feeding Diptera10. D7 proteins are grouped into long and short forms, containing one or two OBP-like domains with six α-helices and three disulfide bonds each. OBPs can be found in the antennae, where they may facilitate olfactory functions10. The function of the sialomic analogs (D7 proteins) function was first described by James et al. (1991) in Ae. aegypti salivary glands as supporting the feeding capabilities of female mosquitoes43. Since then, they have been reported and described regularly within mosquitoes62. The first specific D7 protein was described by Valenzuela et al. (2003) in Anopheles stephensi and was suggested to play roles in both sugar and blood feeding by inhibiting factor XII63. Isawa et al. (2002) characterized Hamadarin, a D7 protein with anti-hemostatic and anti-inflammatory properties, as a promising candidate for anti-inflammatory and analgesic drug development62,64,65,66. D7 proteins likely contribute to host detection, but they also exert direct effects during blood feeding. The C-terminal domains of these proteins can bind biogenic amines and host mediators such as serotonin, histamine, and norepinephrine, which are molecules that induce vasoconstriction, platelet aggregation, itching, and pain. Inhibiting these mediators promotes painless and efficient blood feeding64. The N-terminal regions may be involved in suppressing vasoconstriction or interfering with the coagulation cascade44. D7 proteins affect pathogen survival and transmission. They bind dengue virions, inhibiting viral replication and infection both in vivo and in vitro67,68. This dual role presents them as a two-edged sword: while they may protect mosquitoes from infection, they can simultaneously suppress host immune responses, potentially increasing disease transmission risk. These contrasting effects make D7 proteins compelling targets for further investigation in medical and vector control contexts. Another well-known mosquito venom component, mucin, has surprisingly been detected in only small amounts within the transcriptome, even though it is usually recalled as common in the sialotranscriptomes of mosquitoes and other hematophagous insects63,69,70. These proteins are proposed to act as lubricants in the food canal32,70. Their absence from the proteome and low expression in the transcriptome may suggest rapid degradation, low translational activity or a less important role for the venom, as previously expected.
Several immunity-related protein families were identified in the venom proteome. C-type lectins (CTLs) may have paradoxically two converse effects: (1) protection against microbial infection during feeding as components of the mosquito’s innate immune system71 and (2) enhancement of arbovirus infection and replication, as shown in Ae. aegypti, by binding to viral envelope proteins72. Other immunity-related peptides include FReD proteins and ficolins described from the salivary glands and hemolymph of several arthropods. These molecules are upregulated following exposure to bacteria or arboviruses and may function in pathogen recognition73. Additionally, cecropins are known antimicrobial peptides involved in mosquito immune responses and possibly in venom function. Another group are protease inhibitors, which show high diversity in the mosquito venom. Proteases such as thrombin and trypsin are essential for vertebrate hemostasis and immune responses74, and their inhibition is a key element to support blood feeding. Common protease inhibitors in our dataset included Kazal-type inhibitors, serpins, cathepsins and cystatins. Among these, cathepsins stand out as multifunctional protease inhibitors that may facilitate blood digestion and amino acid release for egg development75,76. Furthermore, in An. gambiae, cathepsins have been shown to exert negative effects on Plasmodium parasites, and in Aedes species, correlations with dengue virus titers have been reported77,78. Another family found in the proteome is the CAP superfamily, particularly antigen-5 peptides. Although these peptides are widely distributed among blood-feeding insect venoms, no functional characterization of CAP from mosquito venom has been carried out thus far. However, in hematophagous triatomine bugs, CAP peptides are thought to inhibit collagen-induced platelet aggregation and ATP release44,79. A similar role could be speculated for Ae. albopictus derived CAPs, but this hypothesis requires more extensive research and confirmation. Additionally, a minor protein group identified in the proteome was the SG1/62 kDa superfamily, which included the secreted salivary protein SGS1. Besides being a protein group identified only as a minor fraction, we were able to confirm its presence at the protein level. The literature describes the 62 kDa family as exclusive to Aedes species32, and our data validates them on protein level in the Ae. albopictus venom system. They are of particular interest due to their proposed role in pathogen transmission80. Strikingly, we did not find members of the 34 kDa protein family, which are known to be of pivotal importance during the blood feeding process of mosquitoes23,32. This may be because we analyzed 5-day-old mosquitoes, while previous studies reporting these proteins relied on older specimens23. Taken together, these findings suggest that non-enzymatic proteins are rich sources of biomolecules that play roles in pathogen transmission or blood feeding. The non-enzymatic may further serve as biomarkers for mosquito exposure. For instance, the 62 kDa superfamily could be a biomarker candidate because its members show unique sequence patterns in metazoans and they are quite restricted to the Aedes venom system32. A thorough understanding of mosquito venom and its non-enzymatic elements may thus form the basis of future pharmacological innovations or devices15,32.
We focused primarily on six components that were classified into the AMP family of cecropins, one of the largest and most studied families of insect AMPs81. Typically, by adopting an α-helical structure, these compounds are known for their potent activity against both gram-negative and gram-positive bacteria while exhibiting low toxicity and even anti-inflammatory effects in mammalian systems37,82,83. In addition to antibacterial activity, cecropins have shown efficacy against fungi, protozoa, and viruses84,85,86,87. Even antimalarial effects of cecropins were reported in Anopheles mosquitoes88, and their role in microbial clearance during pupation has been highlighted24. In addition, cecropins possess anti-inflammatory properties which they exert by binding to bacterial membranes. Various insect-derived cecropins have been investigated as potential candidates for therapeutic applications81,89.
Structural predictions and bioinformatic analysis of the cecropins identified in this study revealed the presence of α-helical conformations, a high net positive charge, and a distinctly amphipathic nature with hydrophobic amino acids predominantly localized on one side of the helix. These features are consistent with typical membrane-active linear cytolytic AMPs and suggest that these peptides may disrupt microbial membranes through pore formation or lipid bilayer destabilization90,91. A particularly interesting structural element is the helix breaking proline after the N-terminal amphipathic helix region, leading to a hinged hydrophobic and negatively charged C-terminal domain. This organization is common for cecropins and might be crucial for their interaction with bacterial membranes81,92,93,94. This architecture points to a well-described mechanism of action referred to as “type II channel formation”. According to this model, the positively charged C-terminal region anchors the peptide to the negatively charged outer membrane surface, primarily via interactions with LPS, while the hydrophobic hinge region inserts into the membrane. As multiple peptides aggregate, they form stable transmembrane pores that compromise membrane integrity and ultimately lead to cell lysis90,91. The structural predictions suggest that the cecropins in Ae. albopictus venom likely conform to this same type II channel mechanism. Their predicted amphipathic and electrostatic profiles support membrane interactions, and the presence of a defined hinge region further points toward potential pore formation, as documented for other insect-derived cecropins (Fig. 4)94. Based on these structural cues, we investigated the activity of these compounds via functional screenings against several microbial strains. The antimicrobial screenings of Ae. albopictus cecropins revealed that these compounds strongly inhibited the gram-negative bacterium E. coli, with MICs as low as 0.2 µM. This confirmed our expectations, as their bacterial membranes structurally differ from those of gram-positive bacteria, where no activity was detected. Interestingly, mCec_Aealb_A1 had weaker antibacterial effects than the other cecropins while still yielding high charge and hydrophobicity. This difference may be explained by the unique sequence features of this peptide compared to those of its relatives. This includes the lysine at position 2 or the histidine at position 16, among others. In venom components and AMPs, single amino acid exchanges can exert tremendous functional effects on peptides95,96. Therefore, carrying out alanine scanning on the various cecropin A family members would be an interesting avenue for future research to infer the functional and structural importance of specific residues along the peptides primary structures.
To gather further insights into the mode of action of the cecropins, we explored their interaction with LPS, a major component of the outer membrane of gram-negative bacteria. Membrane-disrupting AMPs generally bind to LPS in bacterial membranes as an initial step in their disassembling mode of action97. The screening of antimicrobial growth inhibition in the presence of free LPS highlighted that the antimicrobial effect of the tested cecropins against E. coli was reduced by a factor of 15 to 30. This finding suggested that the cecropins bind to free LPS, reducing the amount of cecropin available to bind bacterial membranes. These results support the hypothesis drawn from structural analysis that these peptides act via a membrane targeting mechanism. To further characterize the biological function of the cecropins and to evaluate the pharmaceutical potential of these peptides, we explored their activity on various mammalian cells. Specifically, we tested two mammalian epithelial cell lines and erythrocytes. In particular, the latter are a common first-line system estimate for potential side effects caused by AMPs. In our hemolytic activity screening, none of the tested cecropins displayed hemolytic effects. Cytotoxicity screening revealed that only mCec_Aealb_B2_amide exhibited cytotoxic effects on epithelial cells. mCec_Aealb_B2 exhibited toxic effects on Calu-3 cells. The cytotoxic effects of the Cec_Aealb_B-family members may be exerted by their distinct N-terminal organization, which makes them unique from the A-family members. The other compounds showed no noteworthy cytotoxicity. Given the potent anti-E. coli activity of these cecropins and the absence of cytotoxic or hemolytic effects, we identified these peptides as promising for further exploration of their pharmaceutical potential. Taken together, the tested cecropins appear to function as membrane-active, potentially pore-forming AMPs with strong activity against gram-negative bacteria such as E. coli but not against other microbes or mammalian cells. Since some cecropins have shown to act synergistically, future studies should explore combinations of cecropins to provide a more conclusive picture of their function and bacterial targeting ability.
Nonetheless, our study is affected by some limitations that need to be taken into account when interpreting our findings. First, the taxonomic coverage of studied Mosquito venom systems, into which our work is embedded, represents an intrinsic limitation towards interpretation of our findings. Hematophagous dipterans, as a whole, but particularly mosquitoes, are a diverse group of insects that form multiple evolutionary lineages and ecotypes98. Many are important vectors of pathogens, and their venom systems are key elements in the transmission process. That said, only a small fraction of the ca. 3500 species within Culicidae (Mosquitoes and their allies) has been studied in this regard99. In our work, we shed light on the biochemical profile of the Ae. albopictus venom system and found similarities to the few other previously analyzed venoms, which revealed, e.g., Hydrolases, Apyrases, and OBP(D7-family) as commonly expressed major components across various taxa32. However, before any general conclusions about the relevance of specific components in mosquito venoms can be drawn, the number of Culicidae species studied from a venomics perspective needs to increase. Else, overinterpretation of the available small fraction could give rise to a form of taxonomic bias, as commonly discussed in arachnological toxinology100. A second limitation lies in the sampling process. We analyzed non-blood-fed specimen aged 5 days, while some studies partially analyzed specimens at later stages, others at earlier stages, and some used blood fed animals, which limits the comparability between studies23,101,102. In many venomous lineages, ontogenetic venom variation (variation in venom composition over time and life history stages) is well-established103,104, but whether this process manifests in Mosquitoes is currently unclear, and understanding this would be an important future task. Conceptually, it is reasonable to assume that if venom variation occurs in Mosquitoes, divergent venom profiles will be created within only a few days considering the short life span of most adults and their prioritized sugar feeding in the first days of emergence105. This could explain some of the discrepancies observed. Another potential impediment is the milking process. Venom collection from small invertebrates is extremely challenging and often fails completely17,30, rendering transcriptomics the most reliable source of information for many arthropod venom systems30. Here, we successfully milked crude venom by using osmotic pressure from mineral oil and subsequent isolation of the hydrophilic venom components. Nevertheless, at the time of writing, it is unclear whether our isolation procedure is absolute and equally effective for every component, and some may be retained in the oil fraction and hence remain absent from the proteomics experiment. In light of that, our proteomic analysis should be understood as a conservative perspective. The full Ae. albopictus venomic arsenal may be even greater than that reported herein. A final caveat revolves around the identification of cecropins, which we have identified at the mRNA level and characterized in more detail in light of the families well-known antimicrobial potential. It is important to note that we have not yet verified their existence on protein level. This stems, on the one hand, from their low abundance and diversity, which renders them difficult to detect. Second, we applied a bottom-up approach in which polypeptides are digested into smaller fractions and, in case of short AMPs, often leads to fragments that are poorly captured by mass spectrometric devices. This obstacle is often encountered in venom biodiscovery from small arthropods and often leads to the compound-inference from transcriptomic data and other sequence-based sources30,106,107. It would be important to employ one of the rapidly developing mass spectrometry technologies of Modern Venomics or to chemically isolate the peptides to verify their existence and their modifications on protein level108,109,110.
In conclusion, we can state that mosquitoes, especially Ae. albopictus, are vector insects that rely on venom to obtain a blood meal, which also facilitates the transmission of pathogens. Their venoms lie at the functional interface between host, pathogen and vector and hence plays a key regulatory role. Furthermore, considering their coagulo-modulatory and immune-related roles, mosquito venoms represent a rich source for novel biomedical leads. Nevertheless, comparatively little is known about their venom profile. Here, we fill this important research gap by carrying out an in-depth modern venomics profiling of Ae. albopictus venom. Our analysis revealed a chemically complex venom composed of at least 119 proteins from six protein families, that were verified at the protein level. Based on the functional annotation of the venom compounds, Ae. albopictus venom was found to be rich in enzymes (1018 transcripts, 37 components), but non-enzymatic families were also abundant (1356 transcripts, 82 components). With respect to compounds directly affecting blood feeding, the identified putative hydrolases, in particular, have homologs known to target the cardiovascular system of vertebrates (e.g., ACE and D7-salivary proteins). Likewise, the enzymatic families might interfere with the coagulation cascade (e.g., Apyrases, 5′-nucleotidases, phosphatases and peptidases). Our analysis revealed a plethora of biomacromolecules in Ae. albopictus venom that likely facilitate blood feeding and therefore add to our understanding of the biochemical basis of mosquito feeding and, consequently, their vector transmission process. We further identified a range of components with potential immune function (C-type lectins, FReD proteins and ficolins), including novel cecropin peptides. Structural in silico analysis revealed these peptides to be linear cytolytic peptides. In vitro screenings for bioactivity highlighted these peptides as potent AMPs that target gram-negative bacteria but no other pathogens nor mammalian cells. This work demonstrates the power of modern venomics, which enables the discovery of bioactive molecules that may be underrepresented or undetected by classical approaches of sialome research. This study thereby provides novel insights into venom complexity and expands the repertoire of candidates for therapeutical applications and potential targets for vector control. Future works exploring synergistic combinations and bioengineering strategies may further enhance the efficacy and application potential of these bioresources.
Methods
Mosquito rearing
Ae. albopictus mosquitoes (Rimini strain, kindly provided as eggs by Dr. Hanano Yamada) were reared at 28 ± 3 °C and 80 ± 10% relative humidity with a 12 h photoperiod. The animals were fed on 8% D(-)-fructose (Carl Roth) ad libitum. For rearing, the females were provided weekly with defibrinated sheep blood (Thermo Fisher Scientific) for 1 h using the Hemotek system (Hemotek Ltd) set at 37 °C. After blood meal, an oviposition site was prepared from a black plastic pot with humid filter paper and put into the rearing cage. The egg-containing filter papers were carefully dried over 10 to 14 days. Eggs were hatched in jars containing deoxygenated water. After hatching, the larvae were transferred to plastic trays containing two liters of tap water supplemented with two Pleco Tablets (Tetra) as larval feed. Over the next 5 days, new feed was provided daily. Pupae were collected in a jar and placed in a rearing cage for adult emergence (Fig. 6A).

A Mosquitoes in the rearing cages prior to processing. B Immobilized mosquitoes with their mouthparts inserted into a pipet tip containing mineral oil to induce salivation and collect saliva, yellow arrow highlights the position of mosquitoes, while (C) is zoomed in on a single mosquito. D A magnified view on the pipet tip with the mosquito saliva visible in the oil. E A mosquito after successful dissection process. In (F), a magnified view of a salivary gland pair (g) connected via the salivary ducts (d) is displayed. Organization of a gland pair into three lobes is recognizable (l). Photographs are original and created by Ludwig Dersch.
Salivary gland dissection
Dissections were performed on non-blood-fed 5-day-old female mosquitoes. Prior to dissections, the sugar solutions were removed from the rearing cages and animals were starved for 3 h. Individual female mosquitoes were collected from the cages and kept separated in Ø 29 × 95 mm vials with Ø 30 × 30 mm foam stoppers (Nerbe Plus). Animals were immobilized by CO2 and passed through ethanol to disinfect the animal surface and neutralize charges that may complicate the dissection process and then placed on glass slides with 50 µL droplets of PBS. All dissection steps were performed in PBS and under a Stemi 508 Stereomicroscope (Zeiss) at 40× magnification. For the dissection process, wings and legs were removed with extra fine straight forceps No. 5 (neoLab), then the head was carefully pulled off the thorax. Afterwards, gland pairs were precisely removed and washed by passing them through PBS (Fig. 6E, F). Gland pairs were stored and pooled in 500 µL RNA-later (Thermo Fisher Scientific) at −20 °C until further processing. In total, 60 salivary gland pairs were dissected and used for transcriptomics.
Venom collection
For venom collection, non-blood-fed 5-day-old female mosquitoes were used. Therefore, groups of 5 animals were collected from rearing cages, kept in Ø 29 × 95 mm vials with Ø 30 × 30 mm foam stoppers (Nerbe Plus) and starved for 3 h. Before venom collection, a plate with a stripe of common double-sided adhesive tape for mosquito immobilization and modeling clay for pipet tip stabilization was prepared (Fig. 6B). Mosquitoes were cold-immobilized by placing the vials on ice for 3 min. Wings and legs were removed by forceps and the body of the mosquito was affixed on the double-sided adhesive tape. The mouthparts were carefully inserted in pipet tips filled with 2 µL of mineral oil (Carl Roth). Plates prepared with mosquitoes were placed in a climate chamber at 28 ± 3 °C and 80 ± 10% relative humidity for up to 1 h to further support passive salivation. Afterwards, droplets of salivary secretion were visible within the oil in the pipet tips (Fig. 6C, D). Pipet tips were carefully removed and all containing liquid was acquired by centrifuging for 2 min at 2000 RCF into an Eppendorf tube. For centrifugation tubes containing 20 µL of ddH2O and a custom-made pipet tip spin-down adapter were used. Finally, samples from 250 mosquitoes were pooled in a single tube. The tube was centrifuged at 4 °C and 20,000 RCF for 10 min to separate the venom from the oil. The aqueous phase containing water and venom was recovered and lyophilized. The sample was then stored at −80 °C until further proteomic processing.
Proteotranscriptomics
For a detailed venomic assessment of Ae. albobictus, we utilized our in-house modern venomics platform established previously111,112. Total RNA extraction, library preparation, and sequencing was outsourced to Macrogen. SMART-Seq v4 Ultra Low Input RNA Kit for Sequencing and TruSeq RNA Library Preparation v2 Kit was used for library preparation and sequencing. The kit includes SMART (Switching Mechanism at 5′ end of RNA Template) technology to select mRNA. Sequencing was performed on an Illumina platform to produce 151 bp paired-end reads. The raw sequencing data are available under project PRJEB96266 at the European Nucleotide Archive (ENA, https://www.ebi.ac.uk/ena/). Crude venom was analyzed by bottom-up mass spectrometry. Our proteo-transcriptomic investigation of the Ae. albopictus venom system was partly guided by the sialome nomenclature established by Ribeiro et al. (2010) and VectorBase39 paired with venom databases VenomZone (available online: https://venomzone.expasy.org/) and Tox-Prot40.
Transcriptomics
Sequencing data were quality controlled with FastQC v0.11.9113. Sequencing adapters were removed and reads quality trimmed with cutadapt v4.2114 with a quality cutoff of 28 and a minimum length of 30 bp. We used a multiple assembler strategy including Trinity v2.13.2115 with a minimum contig size of 30 bp and maximum read normalization of 50, rnaSPAdes v3.15.5116 with and without reads corrected with Rcorrector v1.0.5117. Both rnaSPAdes assemblies were built with k-mer sizes of 21, 33 and 55. All three resulting assemblies were concatenated for further analysis as one assembly. We mapped all trimmed reads with HISAT2 v2.2.1118 against the assembly for quality control and used StringTie v2.2.1119 to calculate the transcripts per million (TPM) value for each contig. We also used samtools v1.16.1120 for data transformation. TransDecoder v5.5.0 (RRID:SCR_017647) was performed to extract and identify open reading frames (ORFs) with a minimum length of 10 amino acids. All ORFs without a predicted stop codon were removed. Resulting ORFs with identical length and amino acid sequence were combined with fastanrdb from the Exonerate suite v2.4.0121 and handled as single ORFs with the corresponding TPMs summarized and kept for the united ORF. We screened all remaining ORFs with InterProScanv5.61-93.0122 including all databases available. ORFs without an associated InterPro entry nor a predicted signal peptide by SignalP v6.0 g123 in slow-sequential mode for eukarya were removed from our dataset. These data served as database for our proteome analysis.
For the annotation step DIAMOND v2.0.15124 searches against the public available databases were performed: VenomZone (de Castro, E.; Jungo, F. VenomZone. Available online: https://venomzone.expasy.org/) (downloaded on 19 January 2023), UniProtKB/Swiss-Prot Tox-Prot40 (downloaded on 10 January 2023), UniProtKB/Swiss-Prot v2022_05, UniProtKB/TrEMBL v2022_05125 (both downloaded on 20 January 2023), and annotated protein sequences from VectorBase Release 6539,126 (downloaded on 12 October 2023) assigned to Arthropoda. The E-value was set to a maximum of 1 × 10−3 in ultra-sensitive mode with all target sequences reported (–max-target-seqs 0). We then calculated the coverage of query and subject, and the similarity with the BLOSUM62127 matrix using BioPython v1.81128 for each hit. Sorting by similarity, query and subject coverage for each toxin candidate led to the resulting hit for the final analysis. With these annotations and the InterPro entries, we assigned corresponding peptide families for each putative toxin (Table S3).
Proteomics
For proteomics we utilized a bottom-up approach proven to be effective for venomic analyses as described earlier128,129,130. Briefly, 10 μg of lyophilized venom from 250 female individuals was dissolved in 25 mM ammonium bicarbonate with 0.6 nM ProteasMaxTM (Promega). For disulfide reduction we added 5 mM DTT for 30 min at 50 °C, followed by alkylation of free thiols via 10 mM iodacetamide for 30 min at 24 °C. After quenching the reaction by excess cysteine, trypsin was added at a 50:1 ratio and digested the venom for 16 h at 37 °C. After reaction stoppage by adding trifluoroacetic acid to a concentration of 1%, the sample was purified with C18-ZipTip (Millipore), dried them under a vacuum and redissolved the material in 10 μl of aqueous 0.1% trifluoroacetic acid.
Prior to mass spectrometry, fractionation of the venom peptides was achieved by a directly coupled chromatographic separation on a Thermo Fisher Scientific UltiMate 3000RSLCnano device. For this purpose, 1 µg of the sample material was injected into a 50 cm μPAC C18 column (Pharma Fluidics) in 0.1% formic acid at 35 °C. Peptide elution was performed using a linear gradient of acetonitrile increasing from 3% to 44% over 240 min at a flow rate of 300 nl/min. Finally, the column was washed with 72% acetonitrile. MS of the peptides was carried out on an Orbitrap Eclipse Tribrid MS (Thermo Fisher Scientific). Positive ionization with spray was established by an Advion TriVersa NanoMate (Advion BioSciences) with spray voltage set to 1.7 kV and source temperature set to 275 °C. MS scans were performed in data-independent acquisition mode with the following settings: Scanning time 3 s, mass range of m/z 375–1500 with resolution of 120,000. Auto-gain control was set to standard with a maximal injection time of 50 ms. The most intense ions occurring at each cycle with a threshold ion count of over 50,000 and charge states of 2–7 were selected with an isolation window of 1.6 m/z for higher-energy collisional dissociation (normalized collision energy 30%). MSMS spectra were acquired in the linear ion trap with rapid scan rate and normal mass range. The maximum injection time was set to 100 ms and selected precursor ions were excluded for 15 s following fragmentation.
Mass spectrometry proteomics data have been deposited in the Zenodo repository (https://zenodo.org) under the project identifier 16942242 [10.5281/zenodo.16942242].
Matching proteomic and transcriptomic data
To identify Ae. albopictus venom compounds with proteomic coverage, the fragment spectra generated for the venom sample were matched against a database comprising a six-frame translation of the salivary gland transcriptome using the software PEAKS v. 12.5 (BSI). The analysis was performed with a parent error mass tolerance of 15 ppm and a fragment mass error tolerance of 0.1 Da. The enzyme mode was set to ‘Trypsin’ with ‘semi specific’ as digest mode. Carbamidomethylation was set as fixed post-translational modification (PTM), whereas pyro-glu from Q, amidation, half of a disulfide bridge, and carboxymethyl were considered as variable PTMs.
Peptide identification, physicochemical properties and similarities
Physicochemical properties of salivary gland transcriptome derived cecropins were calculated using in silico tools. Briefly, we employed the antimicrobial peptide (AMP) database APD3 with its AMP calculator and predictor131 to find antimicrobial characteristics and similarities to known AMPs. We further employed PepCalc (Innovagen AB) to recognize basic physicochemical properties, HeliQuest132 to better understand the alpha-helical organization. Additionally, we used the AMP scanner vr.2133 to extrapolate the probability of functioning as AMP for a given sequence. Raw data from all used tools can be found in the supplementary material Table S1. Multiple sequence alignments were performed with Geneious 11.1.5 (Dormatics). Specifically, we employed ClustalW algorithm134 with the BLOSUM62 cost matrix127. Open gap costs were set to 12 and gap extension costs to 3. Further, a structural prediction with AlphaFold3135 was conducted to gather structural insights on the investigated peptides. ChimeraX v1.10136 was used for 3D structure analysis and visualizations.
Peptide synthesis
Peptides have been synthesized by a commercial supplier of custom-made peptides (GenScript Biotech) via solid phase synthesis as described previously137 for further bio activity screens.
Antimicrobial activity
For a pre-screening of antimicrobial activities, the peptides were tested against Escherichia coli DSMZ 102053 at a final concentration of 200 µM. Bacteria from cryo-conserved vials were transferred to tryptone soy broth (TSB) agar plates and incubated for 24 h at 37 °C. Then, single colonies were picked, transferred into 3 mL liquid TSB medium and cultivated overnight at 37 °C and 180 RPM. Subcultures were prepared in 3 mL fresh TSB medium, by adding 40 µL of overnight culture and grown for 4 h at 37 °C before testing. Optical density of each culture was measured at λ = 600 nm (OD600) using a DR2800 spectral photometer (Hach Lange) and then diluted to the start inoculum (Table S3). Peptides were available as 0.5 mg lyophilized powder, to ensure solubility stocks of 10 mmol/L in 100% DMSO (9,75 to 10,60 µL, depending on peptide molecular weight) were prepared for each peptide. They have been further diluted with TSB medium to concentrations of 400 μmol/L. As negative control 100% DMSO in a final 1:5 ratio with TSB medium was used. The 400 µmol/L peptide solution and bacteria were transferred to 96-multiwellplates in volumes of 50 µL per well each. Resulting in final volumes of 100 µL with 200 µmol/L peptide concentrations per well. Gentamicin in a final concentration of 10 µg/mL (Sigma-Aldrich) was used as positive controls. We prepared growth curves over a 48 h period at 37 °C by taking OD600 measurements every 30 min using a BioTek Eon microplate reader with short intervals of double-orbital shaking prior to each measurement. Data were analyzed and visualized using GraphPad Prism v10.0.2 (GraphPad Software) (Fig. S2).
After pre-screening, the antimicrobial effects of the synthesized peptides were further evaluated against a set of indicator strains. The used method for minimum inhibitory concentrations (MIC) determination is derived from the methodology proposed by the CLSI committee138. E. coli ATCC25922 and Staphylococcus aureus ATCC33592 (methicillin-resistant strain) were incubated overnight (37 °C, 180 rpm) and subsequently diluted to 5 × 105 cells/mL in cation adjusted Mueller Hinton II medium (Becton Dickinson). For E. coli ATCC25922 an additional cell suspension was prepared in cation adjusted Mueller Hinton II medium supplemented with 100 µg/mL lipopolysaccharide (LPS) from E. coli 0111:B4 (Sigma-Aldrich), to investigate the potential interaction between peptide and lipopolysaccharides. As positive controls, dilution series of ceftazidime (Biomol GmbH), gentamicin (Sigma-Aldrich) ranging from 64 to 0.03 μg/mL, and ciprofloxacin (Sigma-Aldrich) ranging from 0.5 to 0.0002 µg/mL were used, to show inhibitions of all tested bacterial strains, including gram-negative, gram-positive and multidrug-resistant strains. Antibiotics stocks have been prepared in ultra-pure water as 10 mg/mL stocks and further diluted in Mueller Hinton II medium (MHII) (Tables S5 and S6). Bacterial suspensions without peptide or antibiotic control were used as negative controls. After assay incubation (37 °C, 180 rpm, 85% relative humidity), cell growth was assessed by measuring the turbidity with a microplate spectrophotometer at 600 nm (LUMIstar® Omega BMG Labtech). Growth inhibition was calculated relative to the absorption of the controls. The preculture of Mycobacterium smegmatis ATCC607 was incubated in brain–heart infusion broth (Becton Dickinson) for 48 h, at 37 °C and 180 RPM before the cell concentration was adjusted in Mueller Hinton II medium. Isoniazid (Sigma-Aldrich) was used at 64–0.03 µg/mL instead of gentamicin while the other two controls were done as described above. Cell viability was evaluated after 48 h (37 °C, 180 RPM, 85% relative humidity) via ATP quantification (BacTiter-Glo, Promega) according to the manufacturer’s instructions. Candida albicans FH2173 was incubated for 48 h at 28 °C and 180 RPM before the preculture was diluted to 1 × 106 cells/mL in MHII. Tebuconazole ranging from 64 to 0.03 μg/mL and azoxystrobin at 4–0.002 µg/mL (both Cayman Chemical Company) were used as positive controls. Readout was performed via ATP quantification.
The antibacterial/antifungal MIC value is defined as the lowest concentration of an agent that inhibits the growth of a microorganism by 85% relative to the growth controls (high value). The medium background (low value) is subtracted from all measurements (AU = absorption units; luminescence for cell viability assays). The peptides stocks were dissolved in type I ultra-pure water (conductivity 0.055 µS/cm) and tested in a 12-step dilution series ranging from 50 to 0.02 μM. All concentrations were tested in triplicate. Relative growth inhibition was calculated according to:
$${text{Relative}},{text{growth}},{text{inhibition}},left[ % right]=left(1-frac{{{AU}}_{{sample}}-{{AU}}_{{low}}}{{{AU}}_{{high}}-{{AU}}_{{low}}}right)times 100$$
(1)
AU = absorption units, low = medium blank, high = untreated control.
Equation (1): Calculation of relative growth.
Hemolytic and cytotoxic activity
Erythrocytes derived from horse blood were used to detect hemolytic activities, as previously described111. Briefly, erythrocytes were purified by washing 300 µL of defibrinated horse blood (Thermo Fisher Scientific) with 1 mL of DPBS and centrifugation at 4 °C and 804 RCF for 5 min. Resulting supernatant was discarded and the erythrocyte containing pellet was resuspended in 1 mL of DPBS. This was repeated 3 times until the supernatant was clear. Stock solutions of cecropins were prepared at concentrations of 10 mmol/L in DMSO and mixed with the erythrocyte suspension in DPBS to gain a 1% erythrocyte suspension with 200 µmol/L of peptide. This suspension was transferred in a 96-multiwell v-bottom plate. Afterwards, the plate was incubated for 1 h at 37 °C and 130 RCF, then the plate was centrifuged at 4 °C and 804 RCF for 5 min. Finally, 50 μL of the supernatant was transferred to a flat bottom plate. Hemolytic activity was measured photometrically at λ = 405 nm (OD405) in a BioTek Eon microplate reader. We used 1% Triton X-100 as a positive control and the solvent DMSO in 1:5 ratio with DPBS as a negative control. Measurements were carried out in triplicates. Standard deviations, normalizations and visualizations were made with GraphPad Prism v10.0.2.
Cytotoxic activity of cecropins against mammalian cells was determined as previously described139. Briefly, Madin-Darby canine kidney II (MDCK II) cells and human-derived airway epithelial cells (Calu-3) were cultured in Dulbecco’s modified Eagle’s medium (DMEM GlutaMAX) and DMEM/F12, respectively. Both media were supplemented with 1% penicillin/streptomycin and 10% fetal calf serum (all reagents from Thermo Fisher Scientific) and the cell lines were maintained at 37 °C in a 5% CO2 atmosphere. Stock solutions of cecropins were prepared at concentrations of 10 mmol/L in DMSO. MDCKII cells were grown to 90% confluency in 96-well plates and treated with 200 µmol/L of the cecropins. The positive control was ionomycin (Cayman Chemicals) at 100 µM and untreated cells served as negative control. Cell viability was quantified after 48 h of incubation using the CellTiter-Glo Luminescent Cell Viability Assay (Promega). Luminescence was measured in black 96-well plates in a Synergy H4 microplate reader (Biotek). Relative light units were normalized to the untreated control set as 100% and ionomycin as 0% cell viability. Treatments with cell viability below 80% were considered toxic. Means and standard deviations were calculated from triplicate measurements using GraphPad Prism v10.0.2.
Data availability
Raw sequencing reads are available under the project PRJEB96266 at the European Nucleotide Archive (ENA, https://www.ebi.ac.uk/ena/). All data underlying the findings are fully available and included in the manuscript. Please see Supporting Information. Mass spectrometry proteomics data have been deposited in the Zenodo repository (https://zenodo.org) under the project identifier 16942242. The bio assay raw datasets generated for this study are published as supplementary files alongside this study.
References
-
Prudêncio, M. In fairness to mosquitoes. Trends Parasitol. 36, 876–877 (2020).
-
World Health Organization. Vector-borne diseases. World Health Organization, 26 Sept. 2024. https://www.who.int/news-room/fact-sheets/detail/vector-borne-diseases (2024).
-
Braack, L., Gouveia de Almeida, A. P., Cornel, A. J., Swanepoel, R. & de Jager, C. Mosquito-borne arboviruses of African origin: review of key viruses and vectors. Parasites Vectors 11, 29 (2018).
-
Grard, G. et al. Zika Virus in Gabon (Central Africa) – 2007: a new threat from Aedes albopictus? PLoS Negl. Trop. Dis. 8, e2681 (2014).
-
Kraemer, M. U. G. et al. Past and future spread of the arbovirus vectors Aedes aegypti and Aedes albopictus. Nat. Microbiol. 4, 854–863 (2019).
-
Paupy, C., Delatte, H., Bagny, L., Corbel, V. & Fontenille, D. Aedes albopictus, an arbovirus vector: from the darkness to the light. Microbes Infect. 11, 1177–1185 (2009).
-
Willems, W. R. et al. Semliki forest virus: cause of a fatal case of human encephalitis. Science 203, 1127–1129 (1979).
-
Wong, P.-S. J., Li, M. I., Chong, C.-S., Ng, L.-C. & Tan, C.-H. Aedes (Stegomyia) albopictus (Skuse): a potential vector of Zika virus in Singapore. PLoS Negl. Trop. Dis. 7, e2348 (2013).
-
European Centre for Disease Prevention and Control and European Food Safety Authority. Mosquito maps. ECDC, 2025. https://ecdc.europa.eu/en/disease-vectors/surveillance-and-disease-data/mosquito-maps (2025).
-
Andersen, J. F. Structure and mechanism in salivary proteins from blood-feeding arthropods. Toxicon 56, 1120–1129 (2010).
-
Krenn, H. W. & Aspöck, H. Form, function and evolution of the mouthparts of blood-feeding Arthropoda. Arthropod Struct. Dev. 41, 101–118 (2012).
-
Xu, X. et al. Toward an understanding of the molecular mechanism for successful blood feeding by coupling proteomics analysis with pharmacological testing of horsefly salivary glands. Mol. Cell. Proteom. 7, 582–590 (2008).
-
Guerrero, D., Cantaert, T. & Missé, D. Aedes mosquito salivary components and their effect on the immune response to arboviruses. Front. Cell. Infect. Microbiol. 10, 407 (2020).
-
Fry, B. G. et al. The toxicogenomic multiverse: convergent recruitment of proteins into animal venoms. Annu. Rev. Genomics Hum. Genet. 10, 483–511 (2009).
-
Ribeiro, J. M. C. & Francischetti, I. M. B. Role of arthropod saliva in blood feeding: sialome and post-sialome perspectives. Annu. Rev. Entomol. 48, 73–88 (2003).
-
Hayes, W. K. et al. It’s a small world after all: the remarkable but overlooked diversity of venomous organisms, with candidates among plants, fungi, protists, bacteria, and viruses. Toxins 17, 99 (2025).
-
von Reumont, B. M., Campbell, L. I. & Jenner, R. A. Quo vadis venomics? A roadmap to neglected venomous invertebrates. Toxins 6, 3488–3551 (2014).
-
Jenner, R. A., Casewell, N. R. & Undheim, E. A. B. What is animal venom? Rethinking a manipulative weapon. Trend. Ecol. Evol. https://doi.org/10.1016/j.tree.2025.05.009 (2025).
-
Walker, A. A. et al. Entomo-venomics: The evolution, biology and biochemistry of insect venoms. Toxicon 154, 15–27 (2018).
-
Ribeiro, J. M. C., Mans, B. J. & Arcà, B. An insight into the sialome of blood-feeding Nematocera. Insect Biochem. Mol. Biol. 40, 767–784 (2010).
-
Omokungbe, B. et al. Gene silencing in the aedine cell lines C6/36 and U4.4 using long double-stranded RNA. Parasites Vectors 17, 255 (2024).
-
Lopez, S. B. G. et al. RNAi-based bioinsecticide for Aedes mosquito control. Sci. Rep. 9, 4038 (2019).
-
Arnoldi, I. et al. A salivary factor binds a cuticular protein and modulates biting by inducing morphological changes in the mosquito labrum. Curr. Biol. 32, 3493–3504.e11 (2022).
-
Barreto, C., Cardoso-Jaime, V. & Dimopoulos, G. A novel broad-spectrum antibacterial and anti-malarial Anopheles gambiae Cecropin promotes microbial clearance during pupation. PLoS Pathog. 20, e1012652 (2024).
-
Lowenberger, C. et al. Antimicrobial activity spectrum, cDNA cloning, and mRNA expression of a newly isolated member of the cecropin family from the mosquito vector Aedes aegypti. J. Biol. Chem. 274, 20092–20097 (1999).
-
Santos de, E. A. et al. Phylogeny of Anopheles Darlingi (Diptera:Culicidae) based on the antimicrobial peptide genes cecropin and defensin. SSRN J. https://doi.org/10.2139/ssrn.3931611 (2021).
-
Zheng, Z. et al. Synergistic efficacy of Aedes aegypti antimicrobial peptide cecropin A2 and tetracycline against Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 61, e00686-17 (2017).
-
Sun, D., Eccleston, E. D. & Fallon, A. M. Cloning and expression of three cecropin cDNAs from a mosquito cell line. FEBS Lett. 454, 147–151 (1999).
-
von Reumont, B. M. et al. Modern venomics-Current insights, novel methods, and future perspectives in biological and applied animal venom research. Gigascience 11, giac048 (2022).
-
Von Reumont, B. M. Studying smaller and neglected organisms in modern evolutionary venomics implementing RNASeq (transcriptomics)—a critical guide. Toxins 10, 292 (2018).
-
Sharma, P. et al. Unraveling dual feeding associated molecular complexity of salivary glands in the mosquito Anopheles culicifacies. Biol. Open 4, 1002–1015 (2015).
-
Arcà, B. et al. An insight into the sialome of the adult female mosquito Aedes albopictus. Insect Biochem. Mol. Biol. 37, 107–127 (2007).
-
Smith, J. & Undheim, E. True lies: using proteomics to assess the accuracy of transcriptome-based venomics in centipedes uncovers false positives and reveals startling intraspecific variation in Scolopendra subspinipes. Toxins 10, 96 (2018).
-
da Costa, J. P., Cova, M., Ferreira, R. & Vitorino, R. Antimicrobial peptides: an alternative for innovative medicines? Appl Microbiol Biotechnol. 99, 2023–2040 (2015).
-
Godreuil, S. et al. Aedesin: structure and antimicrobial activity against multidrug resistant bacterial strains. PLoS ONE 9, e105441 (2014).
-
Manniello, M. D. et al. Insect antimicrobial peptides: potential weapons to counteract the antibiotic resistance. Cell. Mol. Life Sci. 78, 4259–4282 (2021).
-
Wei, L. et al. Anti-inflammatory activities of Aedes aegypti cecropins and their protection against murine endotoxin shock. Parasites Vectors 11, 470 (2018).
-
Sun, D., Eccleston, E. D. & Fallon, A. M. Peptide sequence of an antibiotic cecropin from the vector mosquito, Aedes albopictus. Biochem. Biophys. Res. Commun. 249, 410–415 (1998).
-
Giraldo-Calderón, G. I. et al. VectorBase.org updates: bioinformatic resources for invertebrate vectors of human pathogens and related organisms. Curr. Opin. Insect Sci. 50, 100860 (2022).
-
Jungo, F. & Bairoch, A. Tox-Prot, the toxin protein annotation program of the Swiss-Prot protein knowledgebase. Toxicon 45, 293–301 (2005).
-
Ribeiro, J. M. C. & Arcà, B. In Advances in Insect Physiology Vol. 37, 59–118 (Elsevier, 2009).
-
Ribeiro, J. M. et al. An annotated catalogue of salivary gland transcripts in the adult female mosquito, Ædes ægypti. BMC Genomics 8, 6 (2007).
-
James, A. A., Blackmer, K., Marinotti, O., Ghosn, C. R. & Racioppi, J. V. Isolation and characterization of the gene expressing the major salivary gland protein of the female mosquito, Aedes aegypti. Mol. Biochem. Parasitol. 44, 245–253 (1991).
-
Arcà, B., Lombardo, F., Struchiner, C. J. & Ribeiro, J. M. C. Anopheline salivary protein genes and gene families: an evolutionary overview after the whole genome sequence of sixteen Anopheles species. BMC Genomics 18, 153 (2017).
-
Argentine, J. A. & James, A. A. Characterization of a salivary gland-specific esterase in the vector mosquito, Aedes aegypti. Insect Biochem. Mol. Biol. 25, 621–630 (1995).
-
Ribeiro, J. M. C. & Modi, G. The salivary adenosine/AMP content of Phlebotomus argentipes annandale and brunetti, the main vector of human kala-azar. J. Parasitol. 87, 915–917 (2001).
-
Mu, X. et al. Aedes albopictus salivary adenosine deaminase is an immunomodulatory factor facilitating dengue virus replication. Sci. Rep. 13, 16660 (2023).
-
Burnham, S., Smith, J. A., Lee, A. J., Isaac, R. E. & Shirras, A. D. The angiotensin-converting enzyme (ACE) gene family of Anopheles gambiae. BMC Genomics 6, 172 (2005).
-
Ekbote, U., Coates, D. & Isaac, R. E. A. mosquito (Anopheles stephensi) angiotensin I-converting enzyme (ACE) is induced by a blood meal and accumulates in the developing ovary. FEBS Lett. 455, 219–222 (1999).
-
Khurana, V. & Goswami, B. Angiotensin converting enzyme (ACE). Clin. Chim. Acta 524, 113–122 (2022).
-
Funk, C. J. Alkaline phosphatase activity in whitefly salivary glands and saliva. Arch. Insect Biochem Physiol. 46, 165–174 (2001).
-
Koupenova, M., Clancy, L., Corkrey, H. A. & Freedman, J. E. Circulating platelets as mediators of immunity, inflammation, and thrombosis. Circ. Res. 122, 337–351 (2018).
-
Müller, F. et al. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell 139, 1143–1156 (2009).
-
Breijo, M. et al. Hematobin is a novel immunomodulatory protein from the saliva of the horn fly Haematobia irritans that inhibits the inflammatory response in murine macrophages. Parasites Vectors 11, 435 (2018).
-
Lu, S., Andersen, J. F., Bosio, C. F., Hinnebusch, B. J. & Ribeiro, J. M. Acid phosphatase-like proteins, a biogenic amine and leukotriene-binding salivary protein family from the flea Xenopsylla cheopis. Commun. Biol. 6, 1280 (2023).
-
Delgado-Prudencio, G. et al. The enzymatic core of scorpion venoms. Toxins 14, 248 (2022).
-
Dresler, J. et al. A roadmap to the enzymes from spider venom: biochemical ecology, molecular diversity, and value for the bioeconomy. Front. Arachn. Sci. 3, 1445500 (2024).
-
Dresler, J., Herzig, V., Vilcinskas, A. & Lüddecke, T. Enlightening the toxinological dark matter of spider venom enzymes. NPJ Biodivers. 3, 25 (2024).
-
Drukewitz, S. et al. A Dipteran’s novel sucker punch: evolution of arthropod atypical venom with a neurotoxic component in robber flies (Asilidae, Diptera). Toxins 10, 29 (2018).
-
Valenzuela, J. G., Garfield, M., Rowton, E. D. & Pham, V. M. Identification of the most abundant secreted proteins from the salivary glands of the sand fly Lutzomyia longipalpis, vector of Leishmania chagasi. J. Exp. Biol. 207, 3717–3729 (2004).
-
Lapidus, S. et al. Two mosquito salivary antigens demonstrate promise as biomarkers of recent exposure to Plasmodium falciparum-infected mosquito bites. J. Infect. Dis. 231, e570–e581 (2025).
-
Valenzuela, J. G. et al. The D7 family of salivary proteins in blood sucking diptera. Insect Mol. Biol. 11, 149–155 (2002).
-
Valenzuela, J. G., Francischetti, I. M. B., Pham, V. M., Garfield, M. K. & Ribeiro, J. M. C. Exploring the salivary gland transcriptome and proteome of the Anopheles stephensi mosquito. Insect Biochem. Mol. Biol. 33, 717–732 (2003).
-
Calvo, E., Mans, B. J., Andersen, J. F. & Ribeiro, J. M. C. Function and evolution of a mosquito salivary protein family. J. Biol. Chem. 281, 1935–1942 (2006).
-
Isawa, H., Yuda, M., Orito, Y. & Chinzei, Y. A Mosquito salivary protein inhibits activation of the plasma contact system by binding to factor XII and high molecular weight kininogen. J. Biol. Chem. 277, 27651–27658 (2002).
-
Mans, B. J., Calvo, E., Ribeiro, J. M. C. & Andersen, J. F. The crystal structure of D7r4, a salivary biogenic amine-binding protein from the malaria mosquito Anopheles gambiae. J. Biol. Chem. 282, 36626–36633 (2007).
-
Conway, M. J. et al. Aedes aegypti D7 saliva protein inhibits Dengue virus infection. PLoS Negl. Trop. Dis. 10, e0004941 (2016).
-
Olajiga, O. et al. Vertebrate responses against arthropod salivary proteins and their therapeutic potential. Vaccines 9, 347 (2021).
-
Rayms-Keller, A., McGaw, M., Oray, C., Carlson, J. O. & Beaty, B. J. Molecular cloning and characterization of a metal responsive Aedes aegypti intestinal mucin cDNA. Insect Mol. Biol. 9, 419–426 (2000).
-
Acosta-Serrano, A. The mucin-like glycoprotein super-family of Trypanosoma cruzi: structure and biological roles. Mol. Biochemical Parasitol. 114, 143–150 (2001).
-
Cheng, J. et al. Cloning and characterization of a mannose binding C-type lectin gene from salivary gland of Aedes albopictus. Parasites Vectors 7, 337 (2014).
-
Adelman, Z. N. & Myles, K. M. The C-type lectin domain gene family in Aedes aegypti and their role in arbovirus infection. Viruses 10, 367 (2018).
-
Hanington, P. C. & Zhang, S.-M. The primary role of fibrinogen-related proteins in invertebrates is defense, not coagulation. J. Innate Immun. 3, 17–27 (2011).
-
López-Otín, C. & Bond, J. S. Proteases: multifunctional enzymes in life and disease. J. Biol. Chem. 283, 30433–30437 (2008).
-
Cho, W.-L. et al. Mosquito cathepsin B-like protease involved in embryonic degradation of vitellin is produced as a latent extraovarian precursor. J. Biol. Chem. 274, 13311–13321 (1999).
-
Valzania, L., Mattee, M. T., Strand, M. R. & Brown, M. R. Blood feeding activates the vitellogenic stage of oogenesis in the mosquito Aedes aegypti through inhibition of glycogen synthase kinase 3 by the insulin and TOR pathways. Dev. Biol. 454, 85–95 (2019).
-
Santiago, P. B. et al. Proteases of haematophagous arthropod vectors are involved in blood-feeding, yolk formation and immunity—a review. Parasites Vectors 10, 79 (2017).
-
de Araújo, C. N. et al. The biotechnological potential of proteases from hematophagous arthropod vectors. Front. Cell. Infect. Microbiol. 13, 1287492 (2023).
-
Assumpção, T. C. F. et al. Salivary antigen-5/CAP family members are Cu2+-dependent antioxidant enzymes that scavenge O2⨪ and inhibit collagen-induced platelet aggregation and neutrophil oxidative burst. J. Biol. Chem. 288, 14341–14361 (2013).
-
Korochkina, S. et al. A mosquito-specific protein family includes candidate receptors for malaria sporozoite invasion of salivary glands. Cell Microbiol 8, 163–175 (2006).
-
Brady, D., Grapputo, A., Romoli, O. & Sandrelli, F. Insect cecropins, antimicrobial peptides with potential therapeutic applications. IJMS 20, 5862 (2019).
-
Kaushal, A., Gupta, K., Shah, R. & van Hoek, M. L. Antimicrobial activity of mosquito cecropin peptides against Francisella. Dev. Comp. Immunol. 63, 171–180 (2016).
-
Vizioli, J. et al. Cloning and analysis of a cecropin gene from the malaria vector mosquito, Anopheles gambiae. Insect Mol. Biol. 9, 75–84 (2000).
-
Chicharro, C., Granata, C., Lozano, R., Andreu, D. & Rivas, L. N-terminal fatty acid substitution increases the leishmanicidal activity of CA(1-7)M(2-9), a cecropin-melittin hybrid peptide. Antimicrob. Agents Chemother. 45, 2441–2449 (2001).
-
Jaynes, J. M. et al. In vitro cytocidal effect of novel lytic peptides on Plasmodium falciparum and Trypanosoma cruzi. FASEB J. 2, 2878–2883 (1988).
-
Liu, D. et al. A potential food biopreservative, CecXJ-37N, non-covalently intercalates into the nucleotides of bacterial genomic DNA beyond membrane attack. Food Chem. 217, 576–584 (2017).
-
Wang, J. et al. A novel cecropin B-derived peptide with antibacterial and potential anti-inflammatory properties. PeerJ 6, e5369 (2018).
-
Lou, J. et al. Antimalarial activity of cecropin antimicrobial peptides derived from Anopheles mosquitoes. Antimicrob. Agents Chemother. 68, e00311-24 (2024).
-
Yeaman, M. R. & Yount, N. Y. Mechanisms of antimicrobial peptide action and resistance. Pharm. Rev. 55, 27–55 (2003).
-
Christensen, B., Fink, J., Merrifield, R. B. & Mauzerall, D. Channel-forming properties of cecropins and related model compounds incorporated into planar lipid membranes. Proc. Natl. Acad. Sci. USA 85, 5072–5076 (1988).
-
Durell, S. R., Raghunathan, G. & Guy, H. R. Modeling the ion channel structure of cecropin. Biophys. J. 63, 1623–1631 (1992).
-
Oh, D. et al. Role of the hinge region and the tryptophan residue in the synthetic antimicrobial peptides, cecropin A(1−8)−magainin 2(1−12) and its analogues, on their antibiotic activities and structures. Biochemistry 39, 11855–11864 (2000).
-
Efimova, S. S., Medvedev, R. Y., Chulkov, E. G., Schagina, L. V. & Ostroumova, O. S. Regulation of the pore-forming activity of cecropin A by local anesthetics. Cell Tiss. Biol. 12, 331–341 (2018).
-
Efimova, S. S., Schlagina, L. V. & Ostroumova, O. S. Channel-forming activity of cecropins in lipid bilayers: effect of agents modifying the membrane dipole potential. Langmuir 30, 7884–7892 (2014).
-
Klubthawee, N., Adisakwattana, P., Hanpithakpong, W., Somsri, S. & Aunpad, R. A novel, rationally designed, hybrid antimicrobial peptide, inspired by cathelicidin and aurein, exhibits membrane-active mechanisms against Pseudomonas aeruginosa. Sci. Rep. 10, 9117 (2020).
-
Liu, S., Bao, J., Lao, X. & Zheng, H. Novel 3D structure based model for activity prediction and design of antimicrobial peptides. Sci. Rep. 8, 11189 (2018).
-
Jakubec, M. et al. Goldilocks dilemma: LPS works both as the initial target and a barrier for the antimicrobial action of cationic AMPs on E. coli. Biomolecules 13, 1155 (2023).
-
Ruzzante, L., Reijnders, M. J. M. F. & Waterhouse, R. M. Of genes and genomes: mosquito evolution and diversity. Trends Parasitol. 35, 32–51 (2019).
-
Arcà, B. & Ribeiro, J. M. Saliva of hematophagous insects: a multifaceted toolkit. Curr. Opin. Insect Sci. 29, 102–109 (2018).
-
Herzig, V., King, G. F. & Undheim, E. A. B. Can we resolve the taxonomic bias in spider venom research? Toxicon X 1, 100005 (2019).
-
Calvo, E. et al. An insight into the sialotranscriptome of the West Nile mosquito vector, Culex tarsalis. BMC Genomics 11, 51 (2010).
-
Gavor, E. et al. Identification of Aedes aegypti salivary gland proteins interacting with human immune receptor proteins. PLoS Negl. Trop. Dis. 16, e0010743 (2022).
-
Columbus-Shenkar, Y. Y. et al. Dynamics of venom composition across a complex life cycle. eLife 7, e35014 (2018).
-
Surm, J. M. & Moran, Y. Insights into how development and life-history dynamics shape the evolution of venom. EvoDevo 12, 1 (2021).
-
Becker, N. et al. Mosquitoes: Identification, Ecology and Control (Springer, 2020).
-
von Reumont, B. M. et al. The first venomous crustacean revealed by transcriptomics and functional morphology: remipede venom glands express a unique toxin cocktail dominated by enzymes and a neurotoxin. Mol. Biol. Evol. 31, 48–58 (2014).
-
Guan, C., Torres, M. D. T., Li, S. & de la Fuente-Nunez, C. Computational exploration of global venoms for antimicrobial discovery with Venomics artificial intelligence. Nat. Commun. 16, 6446 (2025).
-
Melani, R. D., Nogueira, F. C. S. & Domont, G. B. It is time for top-down venomics. J. Venom. Anim. Toxins Incl. Trop. Dis. 23, 44 (2017).
-
Damm, M., Vilcinskas, A. & Lüddecke, T. Mapping the architecture of animal toxin systems by mass spectrometry imaging. Biotechnol. Adv. 81, 108548 (2025).
-
Slagboom, J. et al. Analytical strategies in venomics. Microchem. J. 175, 107187 (2022).
-
Dersch, L. et al. Linear peptides from the venom of large marsh horsefly (Tabanus autumnalis) larvae facilitate prey capture and defense. Insect Biochem. Mol. Biol. 182, 104349 (2025).
-
Lüddecke, T. et al. Comparative venomics suggests an evolutionary adaption of spider venom from predation to defense. Commun. Biol. 8, 1496 (2025).
-
Andrews, S. FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc (2010).
-
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet j. 17, 10 (2011).
-
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512 (2013).
-
Bushmanova, E., Antipov, D., Lapidus, A. & Prjibelski, A. D. rnaSPAdes: a de novo transcriptome assembler and its application to RNA-Seq data. GigaScience 8, giz100 (2019).
-
Song, L. & Florea, L. Rcorrector: efficient and accurate error correction for Illumina RNA-seq reads. Gigascience 4, s13742-015-0089-y (2015).
-
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
-
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 20, 278 (2019).
-
Danecek, P. et al. Twelve years of SAMtools and BCFtools. GigaScience 10, giab008 (2021).
-
Slater, G. S. C. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinforma. 6, 31 (2005).
-
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
-
Teufel, F. et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 40, 1023–1025 (2022).
-
Buchfink, B., Reuter, K. & Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368 (2021).
-
The UniProt Consortium UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 47, D506–D515 (2019).
-
Alvarez-Jarreta, J. et al. VEuPathDB: the eukaryotic pathogen, vector and host bioinformatics resource center in 2023. Nucleic Acids Res. 52, D808–D816 (2024).
-
Henikoff, S. & Henikoff, J. G. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA 89, 10915–10919 (1992).
-
Cock, P. J. A. et al. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422–1423 (2009).
-
Avella, I. et al. Proteogenomics-guided functional venomics resolves the toxin arsenal and activity of Deinagkistrodon acutus venom. Int. J. Biol. Macromol. 278, 135041 (2024).
-
Schulte, L. et al. Venomics of the milos viper (Macrovipera schweizeri) unveils patterns of venom composition and exochemistry across blunt-nosed viper venoms. Front. Mol. Biosci. 10, 1254058 (2023).
-
Wang, G., Li, X. & Wang, Z. APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 44, D1087–D1093 (2016).
-
Gautier, R., Douguet, D., Antonny, B. & Drin, G. HELIQUEST: a web server to screen sequences with specific α-helical properties. Bioinformatics 24, 2101–2102 (2008).
-
Veltri, D., Kamath, U. & Shehu, A. Deep learning improves antimicrobial peptide recognition. Bioinformatics 34, 2740–2747 (2018).
-
Larkin, M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007).
-
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
-
Meng, E. C. et al. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. 32, e4792 (2023).
-
Lüddecke, T. et al. Functional profiling of the A-family of venom peptides from the wolf spider Lycosa shansia. Toxins 15, 303 (2023).
-
Erkoc, P. et al. Determining the pharmacological potential and biological role of linear pseudoscorpion toxins via functional profiling. iScience 27, 110209 (2024).
-
Hurka, S. et al. Bioactivity profiling of in silico predicted linear toxins from the ants Myrmica rubra and Myrmica ruginodis. Toxins 14, 846 (2022).
Acknowledgements
Aedes albopictus mosquitoes (Rimini strain, kindly provided as eggs by Dr. Hanano Yamada, Joint FAO/IAEA Center of Nuclear Techniques in Food and Agriculture, Insect Pest Control Sub-Program, Vienna, Austria). We acknowledge technical assistance of the Bioinformatics Core Facility at the professorship of Systems Biology at JLU Giessen and the provision of IT resources and general support by the NBI/ELIXIR-DE (W-de.NBI-010) funded by the Federal Ministry of Education and Research.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dersch, L., Krämer, J., Hurka, S. et al. Vector venom: venomics of Aedes albopictus reveals a large enzyme repertoire and novel cecropins with activity against E. coli. npj Drug Discov. 3, 7 (2026). https://doi.org/10.1038/s44386-026-00041-w
-
Received:
-
Accepted:
-
Published:
-
Version of record:
-
DOI: https://doi.org/10.1038/s44386-026-00041-w