
Introduction
De novo peptide sequencing from tandem mass spectra imposes no prior restrictions on the space of possible amino acid sequences, thus allowing for the identification of peptides or proteins outside existing knowledge databases, e.g., neoantigens, noncanonical antigens, synthetic peptides, antibodies, venom proteins, metaproteomes, and proteins from unknown species or species without sequenced genomes1,2,3,4,5 Traditionally, de novo sequencing algorithms have predominantly used the graph representation of tandem mass spectra and dynamic programming to search for the optimal sequence, such as PEAKS6, PepNovo7, pNovo8. Later, the Novor9 algorithm introduced a machine learning approach to predict amino acids. In recent years, with rapid advancements in deep learning, neural networks have been intensively applied to de novo sequencing, with DeepNovo10,11 as the pioneer, and dozens of followers such as pNovo312, PointNovo13, Casanovo14,15 PepNet16, GraphNovo17, Denovo-GCN18 and (pi)-PrimeNovo19. These algorithms show great capabilities in learning the hidden features of spectra and improving the accuracy of de novo sequencing.
However, existing de novo sequencing methods are still facing many challenges. Firstly, incomplete peptide fragmentation results in poor coverage of fragment ions for many peptides, leading to inaccurate or incomplete sequencing results20. Secondly, the digestion efficiency of commonly used proteases, e.g., trypsin, is often insufficient, resulting in missed cleavages of proteins, so that undigestible peptides cannot be sequenced. Thirdly, although recent deep learning-based algorithms, like GraphNovo17, can fill in missing ions to some extent through a trained network, these predicted ions still lack experimental evidence, making it difficult to confirm the correctness of the predicted sequences. Furthermore, the evaluation of de novo sequencing algorithms heavily depends on constructing benchmark datasets from peptide-spectrum matches (PSMs) obtained by database search, which impedes the direct performance comparison between the two approaches to peptide identification.
Mirror protease technology is a powerful way to improve fragment ion coverage of tandem mass spectra21. It typically uses two proteases to cleave proteins at the C- and N-termini of the same specific amino acid(s), such as K/R for trypsin and LysargiNase (Ac-LysargiNase), producing mirror peptides that share identical intermediate sequences but have the specific amino acid at the C- and N-termini, respectively. Such mirror peptides exhibit complementarity in their mass spectra, known as mirror spectra. Fragment ions absent in one spectrum can be present in the other spectrum22. For example, in higher-energy collisional dissociation (HCD) mass spectra, trypsin-digested peptides usually have rich y-type fragment ions, while LysargiNase-digested peptides have rich b-type fragment ions. Although de novo sequencing methods based on mirror proteases, such as pNovoM23 and Lys-Sequencer24, have been developed, they still suffer from several serious flaws. Firstly, there is still a lack of available software to support the entire workflow of mirror-spectra recognition and peptide sequencing. For instance, Lys-Sequencer has not been made public at all. Meanwhile, pNovoM lacks the program of mirror-spectra recognition, a crucial prerequisite for mirror peptide sequencing, in its publicly available package. Secondly, existing methods use only one pair of mirror proteases, which leaves the incomplete-digestion problem unsettled, leading to many sequences being undetectable. Thirdly, some key algorithmic problems have not been adequately addressed. For example, recognizing which spectral pairs are mirror spectra is the first step in using mirror spectra for de novo sequencing. In pNovoM, the recognition of mirror spectra relies on pre-sequencing of peptides from separate spectra obtained by single-protease digestion, which is both time-consuming and sensitive to spectrum quality, and the results of recognition lack an effective quality control standard. Moreover, deep learning techniques have not yet been explored for de novo sequencing of mirror peptides. All these issues hinder the practical application of the mirror-protease strategy in proteomics.
In this paper, we present DiNovo, a comprehensive software system that supports the full workflow of de novo peptide sequencing from tandem mass spectra generated by mirror proteases. The features of DiNovo include support for multiple pairs of mirror proteases, a novel mirror-spectra recognition algorithm independent of pre-sequencing and with quality control, two optional de novo sequencing algorithms based on deep learning and graph theory, respectively, and a target-decoy approach for confidence evaluation of de novo sequencing results in the presence of a sequence database but free of prior peptide identification. We evaluate the performance of DiNovo using two pairs of mirror proteases, including trypsin/LysargiNase and Lys-C/Lys-N, which were used to digest proteins from E. coli and yeast proteomes. By taking full advantage of the complementarity of mirror spectra, DiNovo achieves much higher sequence coverage and confidence than state-of-the-art single-protease de novo sequencing algorithms. Compared with the trypsin protease used alone, DiNovo using two pairs of mirror proteases, leads to two to three times high-confidence amino acids sequenced. Furthermore, DiNovo identifies a comparable number of proteins to database search at the same false discovery rate (FDR) level, showing great potential as a powerful complement or even an alternative to database search for protein identification. DiNovo is designed to dramatically increase the coverage and confidence of de novo peptide sequencing and to facilitate mirror-protease proteomics.
Results
DiNovo workflow
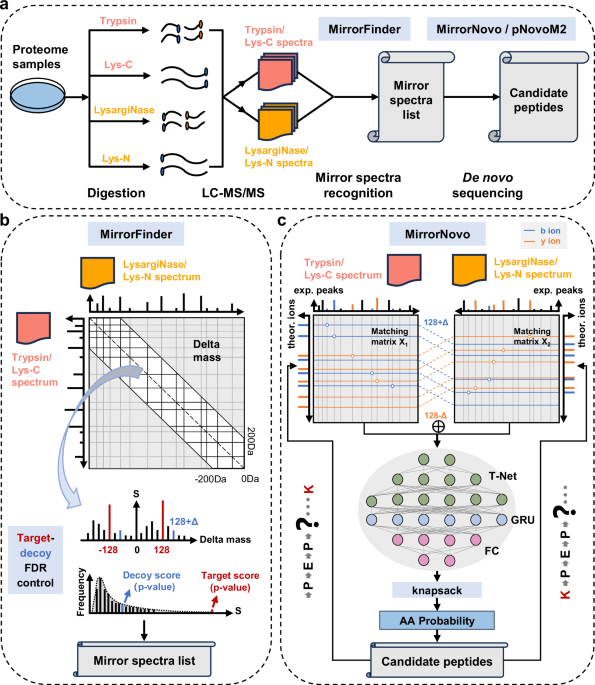
Figure 1 outlines the workflow and modules of DiNovo. In the first step, protein samples are digested by one or more pairs of mirror proteases, and the resulting peptides are analyzed using liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS). In this work, two pairs of mirror proteases are used, including trypsin/LysargiNase and Lys-C/Lys-N. To be specific, trypsin/LysargiNase cleaves C-/N-terminal to lysine and arginine, while Lys-C/Lys-N cleaves C-/N-terminal to lysine alone, producing longer peptides.

a The workflow of DiNovo software. Proteome samples are first digested using multiple mirror proteases, followed by LC-MS/MS analysis to obtain tandem mass spectra. Then, mirror spectral pairs are recognized by the MirrorFinder algorithm, and peptides are sequenced by the MirrorNovo or pNovoM2 algorithm. b MirrorFinder, the mirror-spectra recognition module of DiNovo, which leverages the distribution of fragment ion mass differences (delta masses) between two spectra to recognize mirror spectra and employs a target-decoy strategy for recognition error control. c MirrorNovo, the prime de novo sequencing module of DiNovo, which makes use of a designed neural network architecture to capture the peak features of mirror spectra and predict the probabilities of amino acids.
In the second step, mirror spectral pairs are recognized using a novel algorithm, called MirrorFinder. This algorithm does not rely on pre-sequencing of peptides from separate spectra, making it less sensitive to spectrum quality and more efficient. It directly utilizes the internal information of spectra to match spectral pairs, and employs a matching score to measure the possibility of a pair of spectra being mirror spectra (Fig. 1b). The internal information includes precursor mass, fragment ion mass, and fragment ion intensity, which possess specific patterns for mirror spectral pairs. Furthermore, a target-decoy strategy is used to estimate and control the FDR of mirror spectral pairs. More details about the MirrorFinder algorithm are given in “Methods”.
In the third step, peptides are sequenced from the recognized mirror spectra using two developed algorithms. The first algorithm, called MirrorNovo, is based on a deep neural network (DNN), as shown in Fig. 1c. Specifically, a T-Net is used to extract peak features from the feature matrix for matching peaks and theoretical fragment ions. Then, a Gated Recurrent Unit (GRU) layer is employed to capture the relationships between adjacent peak features. Finally, a fully connected (FC) layer outputs the probabilities of amino acids. In addition, a knapsack algorithm is applied at each step to constrain the search space. The second sequencing algorithm is pNovoM2 (Supplementary Fig. 1), an improved version of pNovoM23, which is based on the graph representation of spectra and dynamic programming. While MirrorNovo is more accurate than pNovoM2, it requires GPU(s) for acceptable running speed. On the other hand, pNovoM2 runs on a CPU and is much faster than MirrorNovo when GPU(s) are unavailable. We recommend using MirrorNovo if GPU(s) are available, and pNovoM2 otherwise. In addition to mirror spectra, all separate spectra are also subjected to de novo sequencing for as high sequence coverage as possible. Further details on the sequencing algorithms can be found in “Methods”.
Performance evaluation
To evaluate the performance of DiNovo, we constructed eight datasets using E. coli and yeast proteome samples, which were digested by two pairs of mirror proteases, i.e., trypsin/LysargiNase and Lys-C/Lys-N (see “Methods” for details on sample preparation and mass spectrometry experiments). In addition, a public antibody dataset23 was also used to test the performance of DiNovo. We compared our mirror-protease strategy with the conventional single-protease strategy for de novo sequencing. We also compared DiNovo with previous single-protease de novo sequencing algorithms as well as the commonly used database search approach for peptide identification. Additionally, we evaluated the accuracy and speed of the two built-in sequencing algorithms in DiNovo, i.e., MirrorNovo and pNovoM2.
The traditional approach to evaluating de novo sequencing results primarily relies on constructing a benchmark dataset from peptide identifications obtained by protein sequence database search. However, this method cannot guarantee data quality due to the inherent error rate in database search results. Moreover, it directly prevents the horizontal performance comparison between de novo sequencing and database search. Another approach is to map de novo peptides to protein sequences in the database, and evaluate the number of peptide sequences mapped16. However, this approach lacks error control, as incorrect sequences can randomly match database proteins.
In this paper, we propose target-decoy (TD) mapping to evaluate de novo sequencing results, which is free of database search and allows for error control (Fig. 2a). Specifically, we map all de novo peptides to a target-decoy protein database and count the number of target or decoy matches for FDR estimation, just as in the TD database search approach25,26 The rationale for the TD mapping algorithm is detailed in “Methods”.

a The schematic diagram of the TD mapping method. The decoy database is generated by reversing or shuffling the sequences in the original (target) database. Sequenced peptides whose masses are above a certain threshold are mapped to the protein sequences in both the target and decoy databases, and the FDR is subsequently estimated based on the mapping results. b FDR estimated by TD mapping as a function of peptide mass threshold on the datasets of E. coli (first two) and yeast (last two) for various sequencing algorithms. Try and Lys are abbreviations of trypsin and LysargiNase, respectively. Source data are provided as a Source Data file.
Considering that short peptides are more likely to produce random matches, we filter the de novo peptides according to their precursor masses, which provide a more continuous measure than peptide lengths. Figure 2b shows how the estimated FDR changes with the precursor mass threshold for different de novo sequencing algorithms. It is evident that the FDR curve declines sharply as the mass threshold increases, and stabilizes at close to zero after reaching 700–800 Da. As the mass threshold increases, mismatched peptides are gradually excluded. We ultimately chose a mass threshold corresponding to 1% FDR for each dataset and algorithm, as detailed in Supplementary Table 1. Although two steps of quality control are sequentially performed for mirror-spectra recognition and peptide sequencing, respectively, our analysis shows that the false positives are not accumulated and the FDR of the final sequenced peptides can be effectively controlled (Supplementary Note 1).
Furthermore, we focus on high-confidence sequencing results. A de novo peptide is considered high-confidence if it achieves 100% ion coverage, meaning that every amino acid residue of the peptide is supported by at least one spectral peak in the mass spectrum (considering only the top 200 spectral peaks). These high-confidence peptides are backed by more complete experimental evidence and are more convincing in practical applications.
Near-complete fragment ion coverage in mirror spectra
The absence of some fragment ions presents a major challenge for de novo peptide sequencing. Our strategy in this paper is to improve the ion coverage using mirror proteases. To demonstrate the effectiveness of this strategy, we calculated the ion coverage of separate spectra and mirror spectra recognized by MirrorFinder. Figure 3 and Supplementary Fig. 2 show that mirror spectra significantly complement the missing ions in separate spectra on both E. coli and yeast datasets, which is highly beneficial for subsequent de novo peptide sequencing. Notably, a large portion of mirror spectra achieved near-complete fragment ion coverage, with average coverages of 98.4% for the E. coli datasets and 98% for the yeast datasets. In contrast, the average ion coverages for the separate spectra were only 90.2% and 89.7% for the E. coli and yeast datasets, respectively.

a Illustration of the mirror-protease strategy and the complementarity of mirror spectra. The mirror proteases trypsin and LysargiNase cleave proteins at the C- and N-termini of lysine and arginine residues, respectively. The complementary fragment ions are highlighted by the yellow bar. Fragment ions absent in one spectrum are mostly present in the other, which improves the coverage of fragment ions. b Fragment ion coverages on the datasets of E. coli (first two) and yeast (last two). The total number ((n)) of spectra analyzed was (n=3008625) (E. coli Try/Lys), (n=3236563) (E. coli LysC/LysN), (n=984524) (yeast Try/Lys), and (n=1053672) (yeast LysC/LysN). Data are presented as violin plots, with the central vertical mark indicating the median (values labeled). The y+ (b+) ion coverage is calculated as the proportion of theoretical y+ (b+) ions with matching spectral peaks. In mirror spectra, the spectral peak is matched once one of the mirror spectra is matched. The ‘all’ category represents the proportion of fragmentation sites identified. A fragmentation site is considered identified when any of the b+, b++ (if any), y+, y++ (if any), or a+ ions corresponding to that site are matched. Source data are provided as a Source Data file.
Comparison with single-protease strategy
To compare the mirror-protease strategy with the commonly used single-protease strategy for de novo peptide sequencing, in addition to DiNovo we also used GraphNovo (the built-in algorithm in PEAKS-online 12), to sequence the E. coli and yeast proteomes, which were digested by trypsin/LysargiNase and Lys-C/Lys-N. PEAKS is the most popularly used software for de novo sequencing. We evaluated the number of peptides sequenced at an estimated 1% FDR, as well as amino acid coverage (the proportion of sequenced amino acids in the database), and protein coverage (the proportion of identified proteins). Throughout this paper, MirrorNovo was used within DiNovo unless otherwise specified.
As shown by Fig. 4a, b, the mirror-protease strategy (DiNovo) significantly outperformed the single-protease strategy (GraphNovo) in terms of all three metrics. In most cases, using either pair of mirror proteases, Try + Lys or LysC + LysN, was superior to using any of the four proteases alone. Combined use of the two pairs of mirror proteases within DiNovo resulted in the highest sequencing coverage and confidence. On the combined results of four proteases (All) for the E. coli (yeast) datasets, DiNovo sequenced 133.8% (111.9%) more high-confidence peptides than GraphNovo, with 43.3% (35.9%) higher amino acid coverage and 15.2% (10.2%) higher protein coverage. Compared to the traditional single-protease strategy, such as Try (GraphNovo), the advantages of DiNovo using two pairs of mirror proteases were more pronounced, as evidenced by sequencing 458.9% (509.2%) more high-confidence peptides for the E. coli (yeast) datasets, corresponding to 153.8% (194.5%) higher amino acid coverage and 28.7% (34.3%) higher protein coverage. Notably, DiNovo with one pair of mirror proteases (Try+Lys) was superior to or at least comparable to the combined results of GraphNovo with four proteases (All), revealing the immense contribution of mirror spectra to peptide sequencing. Furthermore, DiNovo identified 68.4% (68.4%) of E. coli (yeast) high-confidence proteins in the database, demonstrating the capability of the multiple mirror-protease strategy in protein identification. The advantage of DiNovo over GraphNovo was greater in terms of the number of peptides than amino acid and protein coverages. This was because DiNovo sequenced many mirror peptides with large sequence redundancy.

a De novo sequencing result on the E. coli datasets. b De novo sequencing results on the yeast datasets. Single-protease spectra were analyzed by GraphNovo, and mirror spectra were analyzed by DiNovo. All(GraphNovo) represents the union of LysN(GraphNovo), LysC(GraphNovo), Lys(GraphNovo) and Try(GraphNovo), and All(DiNovo) for the union of LysC + LysN(DiNovo) and Try+Lys(DiNovo). Green hollow bars indicate all results, while orange solid bars indicate high-confidence (full fragment ion coverage) results. c Venn diagrams of high-confidence amino acids and proteins sequenced by DiNovo and GraphNovo. Source data are provided as a Source Data file.
Figure 4c further shows the intersection of high-confidence amino acids and proteins sequenced by DiNovo and GraphNovo. Across all datasets with four proteases, DiNovo covered 87.2% to 98.7% of the high-confidence amino acids and 96% to 99% of the high-confidence proteins sequenced by GraphNovo, while sequencing 49,622 to 125,526 additional high-confidence amino acids and 392 to 512 additional high-confidence proteins. More results are given in Supplementary Fig. 3, Supplementary Fig. 4.
Comparison with different de novo sequencing algorithms
To further demonstrate the advantages of the mirror-protease strategy in de novo peptide sequencing, we compared the performance of DiNovo with more single-protease de novo sequencing algorithms. The description and configuration of these various software tools are detailed in “Methods”. The comparison was conducted on the combined results of four proteases for the E. coli and yeast datasets. As in the previous section, sequenced peptides were mapped to the target-decoy database, and the estimated FDR for each algorithm was controlled at 1% using different mass thresholds (Supplementary Table 1). We first report the high-confidence results below.
The results in Fig. 5a, b show that DiNovo outperformed all competitors at the peptide, amino acid, and protein levels for both E. coli and yeast datasets. Notably, GCNovo (the updated version of Denovo-GCN) is the built-in single-protease sequencing model in DiNovo, and GCNovo* is its retrained model. Compared to GCNovo and GCNovo*, DiNovo on average sequenced 138.6% (116.7%) more peptides for the E. coli (yeast) datasets, achieving 46.3% (38.9%) higher amino acid coverage and 14.4% (9.5%) higher protein coverage. The performance advantage of DiNovo over GCNovo is due to the contribution of the mirror-protease sequencing model (MirrorNovo), as detailed in Supplementary Fig. 5b. Compared to other commonly used algorithms, DiNovo sequenced 124.7% to 184.8% more E. coli peptides and 110.2% to 195.3% more yeast peptides. In terms of amino acid coverage, DiNovo exceeded other algorithms by 40.7% to 68.6% (35.9% to 74.2%) for the E. coli (yeast) datasets. At the protein level, DiNovo possessed 15.2% to 19.9% (10.3% to 17.4%) higher protein coverage than other algorithms for the E. coli (yeast) datasets. This superior performance holds even when compared to CasanovoV2, which was trained on a large-scale dataset15. Figure 5c shows the significant overlap of high-confidence amino acids sequenced by DiNovo and other algorithms. Specifically, DiNovo covered most (96.4%–98.6%) of the amino acids sequenced by other algorithms except GCNovo, and uniquely sequenced an additional proportion (39.4%–76.1%) of amino acids than other algorithms.

a De novo sequencing result on the E. coli datasets. b De novo sequencing results on the yeast datasets. Green hollow bars indicate all results, while orange solid bars indicate high-confidence (full fragment ion coverage) results. c Venn diagrams of high-confidence amino acids sequenced by DiNovo and other algorithms on the combined results of four proteases. Algorithms for comparison include pNovo3, PointNovo, GraphNovo, Casanovo, GCNovo (the updated version of Denovo-GCN), and DiNovo. The asterisks represent models trained on the 8-species dataset (excluding yeast), such as PointNovo*, Casanovo*, and GCNovo*. CasanovoV2 was trained on a large-scale dataset derived from the MassIVE Knowledge Base (MassIVE-KB). More detailed configurations of these algorithms are given in “Methods”. Source data are provided as a Source Data file.
All results (green hollow bar) from DiNovo also surpassed those of other algorithms across the three levels for both E. coli and yeast datasets. More results are given in Supplementary Figs. 6–8. All these results demonstrate that DiNovo is not only a high-coverage but also a high-confidence de novo peptide sequencing algorithm.
In addition to complex proteomes, DiNovo also demonstrated high proficiency in sequencing purified proteins, highlighting the generality of the software (Supplementary Note 2). To be specific, we used different algorithms to sequence two monoclonal antibodies (PXL1 and PXL2), which were expressed in Chinese hamster ovary (CHO) cells23. It has been shown that DiNovo achieved the highest (83.5%) average amino acid coverage among all compared sequencing algorithms.
Comparison with database search
As previously mentioned, the TD mapping method enables us to directly compare the results of de novo sequencing with those of database search at the same FDR level. To do this, we used three database search engines including pFind27,28,29 MSFragger30, and MS-GF+31 to search the mass spectra against the protein sequence databases. The FDR of the database search results was controlled at 1%, and other search parameters are detailed in “Methods”. The results reported below were not filtered by high-confidence peptides.
As shown in Fig. 6a, b, DiNovo sequenced more peptides than the three search engines for all four proteases on the E. coli and yeast datasets. This advantage of DiNovo stems primarily from its use of the complementarity of mirror spectra, which allows for the simultaneous accurate sequencing of mirror peptides that may be difficult to sequence alone. Of course, there is sequence redundancy in mirror peptides, and overall, DiNovo sequenced comparable numbers of amino acids and proteins to all search engines. It should be noted that MS-GF+ showed a deficiency in identifying N-terminal digested peptides; therefore, we retrained its scoring model on peptides digested by four different proteases (denoted as MS-GF + *), and much better results were obtained. Importantly, there are significant overlaps between the results of DiNovo and those of each search engine, demonstrating the reliability of the results (Fig. 6c, Supplementary Figs. 9–12). Specifically, DiNovo covered 85.4% (73.7%) of the amino acids and 92.4% (91%) of the proteins identified by pFind for the E. coli (yeast) datasets. For MSFragger, the proportions are 85.4% (71.3%) and 94.8% (93.1%). For MS-GF + *, the proportions are 86.2% (75.4%) and 92.9% (95.4%). Figure 6d further illustrates that the peptide sequences of spectra identified by both DiNovo and pFind are highly consistent. These results show that thanks to the mirror-protease strategy and the powerful DiNovo software, de novo sequencing is expected to become a practical and robust complement—or even an alternative—to traditional database search approach for peptide and protein identification. In this advancement, the TD mapping-based FDR control also plays a crucial role.

a De novo sequencing and database search results on the E. coli datasets. b De novo sequencing and database search results on the yeast datasets. c Venn diagrams of amino acids sequenced by DiNovo and database search engines on the combined results of four proteases. Database search engines for comparison include pFind, MSFragger, and MS-GF + . d The consistency of peptide sequences of spectra identified by both DiNovo and pFind. Gray indicates that the DiNovo sequence is consistent with the pFind sequence; otherwise, it is shown in yellow. To ensure a fair comparison, only pFind was included here, as it used the same set of spectra (mgf format) as DiNovo (exported by the pParse algorithm with the co-eluted precursors detected), while other engines directly used the raw spectra. Source data are provided as a Source Data file.
Interestingly, it can be observed that database search engines showed proficiency in the identification of C-terminal digested peptides but were not as good as DiNovo at sequencing N-terminal digested peptides. This phenomenon suggests that current search engines may have been optimized for C-terminal proteases, mainly trypsin, and can be improved for N-terminal proteases.
Comparison of mirror-spectra sequencing algorithms
There are two optional de novo peptide sequencing algorithms in DiNovo, i.e., MirrorNovo and pNovoM2. MirrorNovo is based on deep learning and runs on a GPU, while pNovoM2 is based on graph theory and runs on a CPU. pNovoM2 is an updated version of pNovoM, which we developed previously23. In this section, we compared the sequencing accuracies and running speeds of these three algorithms. Different mass thresholds were applied to control the FDR of each algorithm at 1%, as detailed in Supplementary Table 2. As illustrated in Fig. 7a, b, MirrorNovo sequenced more amino acids than pNovoM2 and pNovoM, but pNovoM2 had a much faster speed. Therefore, we recommend using MirrorNovo if one or more GPUs are available; otherwise, pNovoM2 is the better option for efficiency. Certainly, the two algorithms in DiNovo can be selected simultaneously, and their results can be combined by DiNovo to achieve optimal sequencing sensitivity.

a The number of sequenced amino acids. b The running speeds of mirror-protease de novo sequencing algorithms. c Venn diagrams of amino acids sequenced by MirrorNovo, pNovoM2 and pNovoM. MirrorNovo was run on an NVIDIA GeForce RTX 4090 GPU, while pNovoM2 and pNovoM were executed on an Intel(R) Xeon(R) Silver 4410Y CPU. The de novo sequencing results are limited to mirror-protease sequencing due to the inability of pNovoM2 and pNovoM to perform single-protease sequencing. Source data are provided as a Source Data file.
Furthermore, we analyzed the intersection of the sequencing results of MirrorNovo, pNovoM2, and pNovoM. As shown in Fig. 7c, there were significant overlaps between the three algorithms, with each identifying a number of unique amino acids. Finally, we analyzed the consistency of spectral pairs whose peptides were sequenced by both MirrorNovo and pNovoM2 (Supplementary Fig. 13). Under the 1% FDR, over 99% of these spectral pairs exhibited identical sequences, with a maximum of only 0.54% exhibiting differences, regardless of species and proteases. This outcome further demonstrated the reliability of the sequencing results and our quality control methodology.
Discussion
In recent years, although deep learning has significantly advanced de novo peptide sequencing, the coverage and reliability of sequencing results remain unsatisfactory. This is largely due to the inherent limitations of current single protease-based LC-MS/MS experiments, i.e., incomplete fragment ions and undigested peptides. To address these challenges, we developed the DiNovo software that leverages multiple mirror proteases to enable higher-coverage and confidence de novo peptide sequencing. DiNovo is based on several advanced algorithms and, to our knowledge, is the first software suite supporting the full workflow of mirror-protease MS data analysis for de novo sequencing.
Experimental results demonstrate that the mirror-protease strategy employed in DiNovo can effectively improve fragment ion coverage of mass spectra. The average ion coverage of mirror spectra reached as high as 98%, much higher than separate spectra. Consequently, DiNovo achieved significantly higher sequencing accuracy than commonly used single-protease de novo sequencing algorithms such as pNovo3, PointNovo, GraphNovo, and Casanovo, which were applied to the separate spectra of four proteases. Specifically, DiNovo sequenced 110% to 195% more high-confidence peptides, which correspond to 36% to 74% higher amino acid coverage and 10% to 20% higher protein coverage. Remarkably, compared with the trypsin protease used alone, using two pairs of mirror proteases led to 154% to 195% more high-confidence amino acids sequenced and 29% to 34% more high-confidence proteins identified.
Unlike current evaluation methods for de novo sequencing that rely on prior peptide identification by database search, DiNovo adopts TD database mapping to estimate the FDR of de novo peptides. TD mapping makes de novo sequencing a parallel method to database search for peptide identification with quality control. In our experiments, DiNovo identified comparable numbers of protein sequences to database search at the same FDR, and showed a high degree of consistency. For the first time, we demonstrated the great potential of de novo sequencing as a practical alternative to database search. In this work, we ranked the de novo peptides by their precursor masses; however, an alternative and potentially better approach would be to rank them by some scoring function, which could increase the number of peptides retained at the given FDR. Additionally, the pattern of mapping can be extended, e.g., from precise mapping to error-tolerant mapping to discover sequence variations or protein modifications. These are our ongoing works and will be reported elsewhere. Of course, general database-free quality control for de novo sequencing is still an open problem in the field, with few attempts32.
Recognition of mirror spectral pairs is a key step in mirror-protease de novo sequencing. In this paper, we have proposed a new algorithm, named MirrorFinder, for this purpose. MirrorFinder directly compares two spectra and does not require pre-sequencing from separate spectra, and thus is less sensitive to spectral quality and is more efficient than a pre-sequencing-based approach23. However, when the scale of spectra is very large, considering all spectral pairs can be a significant computational burden. Possible solutions to this problem are, for example, improving the precision of peptide precursor masses, using a tighter bound on retention time difference, or reducing data size by spectral clustering and combination.
MirrorNovo, which we developed, is the first deep learning-based de novo sequencing algorithm for mirror spectra, and showed higher accuracy than the traditional graph-based approach. As we know, the performance of DNNs greatly relies on the scale of training data, as illustrated by Casanovo14,15 and most recent π-PrimeNovo19, which were trained on ~30 million spectra. Due to the limited availability of mirror spectra data, MirrorNovo has been trained on a relatively small dataset in this paper. We can expect that as more training data becomes available in the future, there will be a lot of room for improvement in the performance of MirrorNovo.
Overall, it is reasonable to believe that the use of multiple mirror proteases can provide a promising solution for high-coverage and high-confidence de novo protein sequencing, and the DiNovo software we developed can serve as a powerful tool to facilitate mirror protease-based proteomics.
Methods
Protein sample preparation and LC-MS/MS analysis
Proteins of E. coli or yeast strains were extracted using a lysis buffer containing 8 M urea, 150 mM NaCl, 50 mM Tris-HCl (pH 7.5), 1 mM phenylmethysulfonyl fluoride (PMSF), 1 mM 2-chloroacetamide, and 1× cocktail (Roche, 11697498001). The proteins were reduced with 5 mM DTT at 45 °C for 30 min, and then fully alkylated with 15 mM IAA at room temperature in the dark for 30 min. Samples were processed by in-solution digestion or the FASP method as previously described, with slight modifications33,34 After reducing the urea concentration in the lysate to <1 M by dilution or displacement, the proteins were aliquoted into four portions and digested with different proteases at an enzyme-to-protein ratio of 1:50. For trypsin/Lys-C or Lys-C alone digestion, the sample was solubilized in a buffer of 50 mM ABC (pH 8.3), and trypsin and/or Lys-C were added for overnight digestion at 37 °C. For LysargiNase/Lys-N digestion, the sample was solubilized in a buffer of 20 mM HEPES, 10 mM CaCl2, and 1 mM Zn(Ac)2 (pH 8.3); after digestion with LysargiNase at 37 °C for 4 h, Lys-N was added for overnight digestion. For Lys-N alone digestion, the sample was solubilized in a buffer of 50 mM ABC and 1 mM Zn(Ac)2 (pH 7.5–8.5), and Lys-N was added for overnight digestion at 37 °C. All of trypsin, LysargiNase, Lys-C and Lys-N were provided by Enzyme & Spectrum (Beijing, China)23,35,36,37. After digestion, 1% formic acid (FA) was added to stop the reaction. The digested peptides were desalted using an optimized StageTip38.
LC-MS/MS analysis was conducted using an Orbitrap Q-Exactive HF mass spectrometer (Thermo Fisher Scientific, USA) coupled with an EASY-nLC 1000 system (Thermo Fisher Scientific, USA). The resulting peptides were resolved in buffer containing 1% acetonitrile and 1% FA, then loaded onto a 75 μm I.D. ×20 cm LC column packed with 1.9 μm C18 reverse phase packing particles (Dr. Maisch GmbH, Germany). LC separation was performed with a 120 min gradient from 6% to 45% buffer B (80% acetonitrile with 0.1% FA) at a flow rate of 300 nL/min. MS measurements were performed in data-dependent acquisition mode. For trypsin/Lys-C digested peptides, MS1 spectra were acquired with a survey scan (300–1400 m/z) at a resolution of 60,000 at 200 m/z, with an AGC target of 3e6 and a maximum injection time (MIT) of 30 ms. Precursors with an intensity >3.3e4 were selected for MS2 analysis, collected at an MIT of 60 ms or an AGC target of 5e4, then fragmented at a normalized collision energy (NCE) of 27. The resulting fragment ions were analyzed in the Orbitrap at a resolution of 15,000 at 400 m/z. For LysargiNase/Lys-N digested peptides, MS1 spectra were acquired with the same survey scan (300–1400 m/z) at a resolution of 60,000 at 200 m/z, an AGC target of 3e6 and an MIT of 50 ms. Precursors with an intensity >1.3e4 were selected for MS2 analysis, collected with an MIT of 150 ms or an AGC target of 1e5, then fragmented with an NCE of 27. The resulting fragment ions were analyzed in the Orbitrap at a resolution of 15,000 at 400 m/z.
Datasets and preprocessing
The LC-MS/MS analysis of the E. coli and yeast proteins digested by four proteases resulted in eight datasets of MS2 spectra. The number of spectra in each dataset is shown in Supplementary Table 3. In addition, we conducted a database search using pFind v3.2.0 and calculated the pairing rate of the dataset at both spectrum and peptide levels (Supplementary Table 3). For simplicity, we refer to trypsin/Lys-C mixed digestion as trypsin digestion, and LysargiNase/Lys-N mixed digestion as LysargiNase digestion.
Centroided spectra were extracted in Mascot generic format from RAW files using pParse39. In order to better recognize the mirror spectra, all mass spectra were preprocessed with the following steps. First, we retained the most abundant N peaks ((N=200) by default) for each spectrum. Second, we detected isotopic clusters and transformed each cluster into a singly charged monoisotopic peak with intensities combined. To enhance the signals of backbone fragment ions, we also accumulated the intensities from their isotopic and neutral-loss (−H2O and −NH3) peaks. Third, we normalized the peak intensities and then took their square roots to reduce variance effects. Fourth, we performed local denoising by retaining the most abundant K peaks ((K=7) by default) per mass bin (with a default bin width of 100 Da). Additionally, we removed peaks corresponding to precursor ions and immonium ions. At last, to alleviate the problem of imbalanced ion types, we generated a complementary peak for each peak after denoising, while maintaining the same ion intensity.
It should be noted that these preprocessing steps were applied only to the mirror-spectra recognition process in DiNovo and not to the other algorithms in order to avoid any unexpected influence on their performance.
Mirror-spectra recognition algorithm
In DiNovo, we developed a novel mirror-spectra recognition algorithm, named MirrorFinder, which is free of prior peptide sequencing from separate spectra as done in pNovoM23. The algorithm directly utilizes the information of precursor and fragment ions to recognize spectra generated from mirror peptides. To be specific, it first selects candidate spectral pairs whose precursor masses comply with the rule of mirror peptides (as outlined in Supplementary Table 4) and whose retention time difference falls in a reasonable range. The rule states that each type of mirror peptides has a theoretical precursor mass difference and two theoretical fragment ion mass differences. Each candidate spectral pair is then assigned a matching score, reflecting the confidence level that they are indeed a pair of mirror spectra. Notably, the parameters for preprocessing and mirror-spectra recognition were optimized through extensive testing to ensure better performance, as detailed in Supplementary Note 3. Furthermore, the results in Supplementary Note 3 show that MirrorFinder was more accurate than pMerge, the undisclosed mirror-spectra recognition algorithm used in pNovoM.
To begin with, we select candidate spectral pairs with precursor mass differences matching the theoretical values (as listed in Supplementary Table 4) within a specified tolerance. We set the tolerance to 20 ppm, and calculate the maximum and minimum values of precursor mass difference, which are
$$begin{array}{c}{D}_{1}=left[{m}_{1}left(1+20cdot {10}^{-6}right)right]-left[{m}_{2}left(1-20cdot {10}^{-6}right)right]\ {D}_{2}=left[{m}_{1}left(1-20cdot {10}^{-6}right)right]-left[{m}_{2}left(1+20cdot {10}^{-6}right)right]end{array}$$
(1)
where ({m}_{1}) and ({m}_{2}) are the precursor masses of the two spectra, respectively. If one of the theoretical precursor mass differences in Supplementary Table 4 falls within the interval (left[{D}_{2},{D}_{1}right]), then the spectral pair is a candidate of a certain category (e.g., A-G).
After that, each candidate spectral pair is assigned a matching score, which is calculated through the following steps (as illustrated in Fig. 1b). First, a fixed mass range (−200 to +200 Da in this work) is divided into equal small intervals (e.g., 0.02 Da), with the total number of intervals denoted as (N). Next, for the (i)-th interval, the number of fragment ion pairs whose mass differences fall in this interval is counted, and their intensities are summed, denoted as ({{mbox{count}}}_{i}) and ({{mbox{sum_inten}}}_{i}) ((i=1,cdots,N)), respectively. Then, a statistic ({s}_{i}) is calculated for the (i)-th interval by multiplying the number of pairs by the summed intensity, which is
$${s}_{i}={{mbox{sum_inten}}}_{i}*{{mbox{count}}}_{i},i=1,cdots,N$$
(2)
Finally, two Bonferroni-corrected (p)-values are estimated for the intervals corresponding to two theoretical fragment ion mass differences for a certain category (as shown in Supplementary Table 4) from the distribution of the observed statistics40. The smaller of the two (p)-values is denoted as ({p}^{*}), and the matching score (S) is obtained by
$$S=-log left({p}^{*}right)$$
(3)
Obviously, a higher matching score indicates stronger evidence that the spectral pair is a pair of mirror spectra.
Subsequently, we use a target-decoy strategy25,26 to filter spectral pairs according to their matching scores. In MirrorFinder, the matching score is closely related to the theoretical fragment ion mass differences, allowing us to directly use the score derived from a random mass difference as the decoy score to compete with the corresponding original (target) score. For each spectral pair, if the original score is higher than the decoy score, the spectral pair is labeled as a target spectral pair, and its final score is set as the original score. Otherwise, it is labeled as a decoy spectral pair, and its final score is set as the decoy score. Then the final scores are sorted in descending order, and the FDR of target spectral pairs with final scores above a given threshold t can be estimated as
$${{mbox{FDR}}}left(tright)=frac{{{{rm{D}}}}left(tright)+1}{{{{rm{T}}}}left(tright)}$$
(4)
where T(t) and D(t) are the numbers of target and decoy spectral pairs, respectively. Through this strategy, we can filter target spectral pairs and control the FDR under a specified level, e.g., 2%. This approach eliminates the need to construct additional decoy spectra and offers a reasonable quality control standard for the recognized mirror spectra. We also verified that our quality control method satisfies the assumption of the target-decoy strategy in Supplementary Note 4.
DNN-based de novo sequencing algorithm for mirror spectra
We proposed a DNN-based de novo sequencing algorithm called MirrorNovo, which fully leverages complementary fragment ions in mirror spectra without the need to explicitly merge the spectra. In MirrorNovo, we preprocess each spectrum in a mirror spectral pair individually, padding the peak list with zeros if there are fewer than the top N peaks. To improve computational efficiency and reduce intensity variance, we convert the m/z values of all peaks to neutral masses and normalize their square-root-transformed intensities to the base peak.
As illustrated in Fig. 1c, the input to the MirrorNovo model consists of two peak-ion matching matrices, each corresponding to one spectrum in a mirror spectral pair, along with the category of the spectral pair. We compute the two matrices by comparing the observed peak masses in each spectrum to 18 types of theoretical fragment ion masses (i.e., singly and doubly charged b, y, and a ions, their singly charged neutral-loss ions, and 6 types of internal ions), and then concatenate them along the peak dimension. Theoretical fragment ions are generated by enumerating all possible subsequent amino acids based on the already predicted prefix sequence. To ensure meaningful matrix concatenation, we align the two matrices according to the corresponding fragmentation sites in the overlapping regions of the mirror peptides. For example, when iteratively predicting a trypsin-digested peptide of length L with either lysine or arginine (identified by MirrorFinder) at its C-terminal end, we align its bi (yL–i) fragments with the bi + 1 (yL–i-1) fragments derived from the LysargiNase-digested peptide, which has either lysine or arginine at its N-terminal end.
MirrorNovo employs a T-Net architecture13 with three one-dimensional convolutional layers to extract peak features from the concatenated matching matrices. A subsequent GRU layer captures both short- and long-term dependencies across peaks while maintaining the dimensionality of the input and output matrices. Next, three fully connected (FC) layers are utilized to learn higher-level feature representations, with batch normalization applied between them to enhance training stability and accelerate convergence. The Rectified Linear Unit (ReLU) activation function introduces non-linearity to prevent the collapse of adjacent linear layers. Finally, a softmax layer outputs probabilities for 20 amino acids, where isoleucine and leucine are not distinguished.
MirrorNovo predicts complete peptide sequences from mirror spectral pairs through iterative amino acid inference. At each iteration, it employs a beam search strategy to avoid local optima and utilizes a knapsack algorithm to reduce the number of candidate amino acids. A bidirectional prediction strategy is integrated to mitigate the loss of correct sequences. The iteration terminates when the difference between the prefix mass and precursor mass falls within 20 ppm. The top (M) ((M=10) by default) candidate peptides are retained based on peptide probability scores, calculated as the average of individual amino acid probabilities. To enhance data utilization, DiNovo also includes a single-protease de novo sequencing algorithm, GCNovo (the updated version of Denovo-GCN18), which allows peptide sequencing from spectra that cannot form mirror spectral pairs. As an independent sequencing algorithm, the running speed of GCNovo is similar to that of PointNovo and Casanovo, while GraphNovo and pNovo3 have significantly faster speeds (Supplementary Fig. 5a). To process the sequencing results, we first resolved conflicts where a single spectrum was paired with different spectra to produce different sequences. After assigning a unique mirror sequence to each spectrum, we further filtered out results where the mirror sequence was inconsistent with the sequence derived from the single spectrum.
In this study, MirrorNovo was trained on three high-resolution tandem mass spectrometry datasets from protein samples of Vero, MC2155, and human testis. Annotated mirror spectral pairs were constructed based on pFind (v3.2.0) search results at 1% FDR. Since a pair of mirror peptides may correspond to multiple mirror spectral pairs, and to ensure high-quality PSMs and sequence diversity, we ranked the mirror spectral pairs by pFind’s final scores and selected up to the top (K) highest-scoring pairs. Only spectral pairs of types A, B, and C with charge states of 2+ or 3+ were retained for the final annotated dataset. The dataset was split into training and validation sets at a 9:1 ratio based on peptide sequences (as detailed in Supplementary Table 5), ensuring no sequence overlap between the sets. The single-protease sequencing model, GCNovo, was trained on all trypsin- and LysargiNase-digested PSMs from the same dataset.
Graph-based de novo sequencing algorithm for mirror spectra
In addition to the DNN-based sequencing algorithm MirrorNovo, we also developed a graph-based sequencing algorithm, pNovoM2, which generates candidate peptides through spectrum graphs. Building on pNovoM23, we have enhanced the performance of peptide sequencing and expanded the types of mirror peptides that can be sequenced, enabling pNovoM2 to deliver more results than pNovoM. Furthermore, we have optimized the speed and memory management of pNovoM2, facilitating rapid de novo peptide sequencing on large-scale datasets without the need for GPU support.
Supplementary Fig. 1 illustrates the schematic diagram of pNovoM2. Unlike pNovoM, pNovoM2 interprets not only the merged mirror spectra but also the separate spectra. This strategy allows pNovoM2 to sequence peptides even when MirrorFinder fails to find the mirror spectral pairs. In the workflow of pNovoM2, all spectra are first preprocessed, with each generating a spectrum graph similar to pNovo8. If MirrorFinder recognizes the corresponding mirror spectrum for the input spectrum, the spectrum graphs of the two spectra will be merged and used for subsequent analysis. Otherwise, the spectrum graph of the input spectrum will be used directly. For the merged spectrum graph, an edge is created between two nodes if their mass difference matches the total mass of any two or three amino acids. However, for the spectrum graph from a separate spectrum, the maximum number of amino acids is increased to five, due to the relatively lower abundance of signal peaks. Our scoring function aligns with that of pNovo, but we’ve made slight adjustments to better accommodate the merged spectrum graph. If the mass of a node in one spectrum graph differs from the mass of a node in another spectrum graph by a specific value, such as the mass of the amino acid K or R in the case of trypsin and LysargiNase, the peaks corresponding to these nodes are likely signal peaks of fragment ions. These nodes are then assigned a higher score. The pDag41 algorithm is subsequently used to identify the top (N) highest-scoring paths from the source node to the target node. For the merged spectrum graph, the value of (N) is set to 20; for the spectrum graph from a separate spectrum, it is set to 40. All peptides corresponding to these paths from the spectrum graphs are rescored together, and a final list of candidate peptides is generated.
De novo sequencing results evaluation
In this paper, we invented a simple yet effective method for evaluating de novo sequencing results, called TD mapping (as illustrated in Fig. 2a). This method requires the existence of a sequence database of proteins to be sequenced, but eliminates the need for peptide identification by traditional database search. Through the introduction of a decoy database, it addresses the lack of quality control in existing mapping-based methods16.
Building on the TD method25,26 we first generate a decoy protein database by simply reversing or shuffling the sequences in the original database. Then, we map all the de novo sequences separately to the protein sequences in the target and decoy databases. Those successfully mapped peptides are retained and subjected to subsequent analysis. Unmatched peptides are excluded, as well as those mapped to both target and decoy sequences. Next, mapped peptides are ordered according to their masses, and the FDR of target peptides with masses above a threshold m is estimated as
$${{mbox{FDR}}}left(mright)=frac{{{{rm{D}}}}left(mright)+1}{{{{rm{T}}}}left(mright)}$$
(5)
where T(m) and D(m) are the numbers of target and decoy peptides, respectively. Finally, an appropriate mass threshold is selected so that the FDR is controlled under the desired level.
Configuration of de novo sequencing and database search
To fairly compare the performance of de novo sequencing software tools, we configured different tools with the same parameters. In addition, to perform a horizontal comparison between de novo sequencing and database search, all raw files were analyzed using pFind, MSFragger, and MS-GF+ for peptide identification. Both precursor and fragment ion mass tolerances were set to 20 ppm. Carbamidomethyl of cysteine (C) was specified as a fixed modification, and oxidation of methionine (M) was considered as a variable modification. The detailed configurations for each software tool are as follows:
-
DiNovo: We offer two optional de novo sequencing algorithms in DiNovo, i.e., MirrorNovo and pNovoM2. By default, MirrorNovo is used for optimal performance, unless otherwise specified. The MirrorNovo model was trained on an NVIDIA GeForce RTX 3090 GPU with the following parameters: optimizer = Adam, loss function = Focal, maximum epochs = 5, batch size = 20, learning rate = 1 × 10−3, decay factor = 0.5. For the mirror-spectra recognition experiment, we first retained the 200 most abundant peaks and then selected 7 peaks per 100 Da. For the sequencing experiment, we only retained spectra with a precursor mass in the range of [300, 3500] Da. When comparing these two algorithms (as discussed in “Results”), we set the number of processes of pNovoM2 as 15, and the batch size of MirrorNovo as 350. Additionally, MirrorNovo was run on an NVIDIA GeForce RTX 4090 GPU, while pNovoM2 was executed on an Intel(R) Xeon(R) Silver 4410Y CPU.
-
pNovo3: pNovo312 uses the pDag41 algorithm to find the maximum scoring path within the spectrum graph, after which it rescores candidate peptides using an SVM model. The peptide with the highest score is then reported as the output. For our analysis in “Results”, we used the latest version of pNovo3, pNovo v3.1.5, which was released on December 6, 2023.
-
PointNovo*: PointNovo13 is a neural network model that employs a T-Net structure, specifically designed for order-invariant data. This design allows it to be applicable to mass spectra of varying resolutions without increasing computational complexity. Since the authors of PointNovo did not provide a trained model file, and the original 9-species training dataset used in13 included the yeast species, we excluded the yeast species and trained a PointNovo model from scratch on the remaining 8-species dataset for our analysis.
-
GraphNovo: We acquired the commercial software Peaks-Online 12 and used its built-in de novo peptide sequencing algorithm to analyze E. coli and yeast datasets in “Results”. Peaks-Online 12 utilizes an integrated deep learning-based de novo peptide sequencing algorithm (GraphNovo17), which addresses the missing fragments problem in de novo sequencing by using graph deep learning to accurately reconstruct full peptide sequences from incomplete mass spectrometry data. However, the specifics of the training set used to train the model remain undisclosed to us.
-
Casanovo* and CasanovoV2: Casanovo14,15 is a deep learning algorithm that employs a transformer neural network architecture to translate the sequence of peaks in a tandem mass spectrum into the corresponding amino acid sequence of the generating peptide. Notably, Casanovo does not constrain the peptides it generates by precursor mass, so the precursor masses of its generated peptides may not be equal to those provided by the spectra. In contrast, all the other algorithms mentioned above use precursor mass as a constraint for de novo peptides. Therefore, it would be unfair to Casanovo to directly use the highest-scoring results without considering the precursor mass error. However, if we filter out peptides generated by Casanovo with precursor mass errors exceeding 20 ppm, some low-quality spectra will not yield any sequencing results. As a result, these spectra will be discarded in the results of Casanovo, and the TD mapping method will be applied only to the remaining spectra with higher quality. This could unfairly underestimate the FDR of Casanovo compared to other software tools. For a fairer comparison, we adopted a compromise strategy: we filtered out peptides generated by Casanovo with precursor mass errors exceeding 20 ppm and applied the same peptide mass thresholds to the Casanovo results as we did to DiNovo. For our analysis in “Results”, we used two models of Casanovo 4.2.0, which was released on May 14, 2024. The first model, referred to as Casanovo*, was trained by us from scratch on the 8-species dataset, similar to the approach used with PointNovo. The second model, referred to as CasanovoV2, is the latest version provided by its authors, trained on a large-scale dataset derived from the MassIVE Knowledge Base (MassIVE-KB), which consists of 30 million PSMs. It is important to note that CasanovoV2 is a trained model that does not allow for the specification of modifications. For a fair comparison, we selected the highest-ranking peptide without unexpected modifications as the result for each spectrum.
-
pFind: In our analysis, we used pFind v3.2.027,28,29 for database search. The E. coli database includes 4789 protein entries along with contaminant protein sequences from UniProt. The yeast database contains 7021 protein entries from UniProt, also with contaminant protein sequences. For the closed search parameters, the maximum number of modification sites per peptide sequence was limited to 3. The length of identified peptides was set to 6 to 100. The digestion mode was set as full specificity with a maximum of 3 missed cleavage sites allowed. The search results were filtered to achieve a 1% FDR at both the PSM and protein levels.
-
MSFragger: In our analysis, we used FragPipe 23.0, in which the version of MSFragger30 is v4.2. The searched E. coli and yeast databases were the same as those used in pFind, and the basic search parameters were consistent with those of pFind.
-
MS-GF+ and MS-GF + *: In our analysis, we used two models of MS-GF+ v2024032631. The first model, referred to as MS-GF + , was the original model provided by its authors. The second model, referred to as MS-GF + *, was retrained on peptides digested by four different proteases. Specifically, we used the program provided in the original text to train scoring parameters for each protease. The searched E. coli and yeast databases were the same as those used in pFind, and the basic search parameters were consistent with those of pFind.
Design of DiNovo software
Previously, the only software available for mirror-protease de novo sequencing was pNovoM, which required users to manually carry out step-by-step operations for pre-sequencing, mirror-spectra recognition, and de novo sequencing (note that the programs for the first two steps were not publicly released). This makes its use of it cumbersome and not user-friendly. As a result, there remains a need for software that can recognize and sequence mirror spectra with a simple, one-click operation.
To meet this need, we integrate the various algorithms developed in this paper into a complete software solution named DiNovo, making it easily accessible to participants in the field. The DiNovo software includes the following modules: configuration settings, MS data I/O, spectra preprocessing, mirror-spectra recognition, de novo sequencing, and real-time log output during the process. As illustrated in Supplementary Fig. 14, parallelization is fully supported in each analytical component of DiNovo, significantly improving computational efficiency and reducing overall processing time.
In DiNovo, error-tolerant precursor mass indexes are built for all spectra to enable high-speed mirror-spectra recognition. As a result, the time complexity of DiNovo for selecting candidate spectral pairs involving (n) trypsin-digested spectra and (m) LysargiNase-digested spectra is (O(m+n)), in contrast to (O({mn})) by brute force. For example, DiNovo takes approximately 20 s to select candidate spectral pairs from a pool of 10 million spectral pairs, 1000 times faster than brute force. On the 16 million spectral pairs of E. coli dataset, DiNovo performed mirror-spectra recognition 2.1 and 46.5 times faster than pMerge (the mirror-spectra recognition module of pNovoM, unreleased) with 1 and 30 processes on a workstation, respectively. In combination of mirror-spectra recognition and sequencing, DiNovo completed the whole task four to five times faster than pMerge and pNovoM.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The proteomic mass spectrometry data have been deposited to the ProteomeXchange Consortium via the iProX partner repository42 with the dataset identifier PXD059331. The antibody data used in this study are available in the ProteomeXchange Consortium via the PRIDE partner repository with the identifier PXD008690. Source data are provided with this paper.
Code availability
The standalone software package and source code for DiNovo (developed in Python3 and JAVA) are publicly available at [https://github.com/YanFuGroup/DiNovo] and permanently archived under https://doi.org/10.5281/zenodo.1841428343.
References
-
Tran, N. H. et al. Personalized deep learning of individual immunopeptidomes to identify neoantigens for cancer vaccines. Nat. Mach. Intell. 2, 764–771 (2020).
-
Beslic, D., Tscheuschner, G., Renard, B. Y., Weller, M. G. & Muth, T. Comprehensive evaluation of peptide de novo sequencing tools for monoclonal antibody assembly. Brief. Bioinform. 24, bbac542 (2023).
-
Ng, C. C. A., Zhou, Y. & Yao, Z. P. Algorithms for de-novo sequencing of peptides by tandem mass spectrometry: a review. Anal. Chim. Acta 1268, 341330 (2023).
-
Bittremieux, W. et al. Deep learning methods for de novo peptide sequencing. Mass Spectrom. Rev. https://doi.org/10.1002/mas.21919 (2025).
-
Van Den Bossche, T. et al. Metaproteomics beyond databases: addressing the challenges and potentials of de novo sequencing. Proteomics 25, 51–61 (2024).
-
Ma, B. et al. PEAKS: powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Commun. Mass Spectrom. 17, 2337–2342 (2003).
-
Frank, A. & Pevzner, P. PepNovo: de novo peptide sequencing via probabilistic network modeling. Anal. Chem. 77, 964–973 (2005).
-
Chi, H. et al. pNovo: de novo peptide sequencing and identification using HCD spectra. J. Proteome Res. 9, 2713–2724 (2010).
-
Ma, B. Novor: real-time peptide de novo sequencing software. J. Am. Soc. Mass Spectrom. 26, 1885–1894 (2015).
-
Tran, N. H., Zhang, X., Xin, L., Shan, B. & Li, M. De novo peptide sequencing by deep learning. Proc. Natl. Acad. Sci. USA 114, 8247–8252 (2017).
-
Tran, N. H. et al. Deep learning enables de novo peptide sequencing from data-independent-acquisition mass spectrometry. Nat. Methods 16, 63–66 (2019).
-
Yang, H., Chi, H., Zeng, W.-F., Zhou, W.-J. & He, S.-M. pNovo 3: precise de novo peptide sequencing using a learning-to-rank framework. Bioinformatics 35, 183–190 (2019).
-
Qiao, R. et al. Computationally instrument-resolution-independent de novo peptide sequencing for high-resolution devices. Nat. Mach. Intell. 3, 420–425 (2021).
-
Yilmaz, M., Fondrie, W., Bittremieux, W., Oh, S. & Noble, W. S. De novo mass spectrometry peptide sequencing with a transformer model. in Proceedings of the 39th International Conference on Machine Learning. 25514-25522 (PMLR, 2022).
-
Yilmaz, M. et al. Sequence-to-sequence translation from mass spectra to peptides with a transformer model. Nat. Commun. 15, 6427 (2024).
-
Liu, K., Ye, Y., Li, S. & Tang, H. Accurate de novo peptide sequencing using fully convolutional neural networks. Nat. Commun. 14, 7974 (2023).
-
Mao, Z., Zhang, R., Xin, L. & Li, M. Mitigating the missing-fragmentation problem in de novo peptide sequencing with a two-stage graph-based deep learning model. Nat. Mach. Intell. 5, 1250–1260 (2023).
-
Wu, R., Zhang, X., Wang, R. & Wang, H. Denovo-GCN: de novo peptide sequencing by graph convolutional neural networks. Appl. Sci. 13, 4604 (2023).
-
Zhang, X. et al. -PrimeNovo: an accurate and efficient non-autoregressive deep learning model for de novo peptide sequencing. Nat. Commun. 16, 267 (2025).
-
Muth, T. & Renard, B. Y. Evaluating de novo sequencing in proteomics: already an accurate alternative to database-driven peptide identification? Brief. Bioinform 19, 954–970 (2017).
-
Huesgen, P. F. et al. LysargiNase mirrors trypsin for protein C-terminal and methylation-site identification. Nat. Methods 12, 55–58 (2015).
-
Jiang, S. et al. Mirror proteases of Ac-Trypsin and Ac-LysargiNase precisely improve novel event identifications in Mycolicibacterium smegmatis MC2 155 by proteogenomic analysis. Front. Microbiol. 13, 1015140 (2022).
-
Yang, H. et al. Precision de novo peptide sequencing using mirror proteases of Ac-LysargiNase and Trypsin for large-scale proteomics. Mol. Cell. Proteomics 18, 773–785 (2019).
-
Mao, Y., Daly, T. J. & Li, N. Lys-Sequencer: an algorithm for de novo sequencing of peptides by paired single residue transposed Lys-C and Lys-N digestion coupled with high-resolution mass spectrometry. Rapid Commun. Mass Spectrom. 34, e8574 (2020).
-
Elias, J. E. & Gygi, S. P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 (2007).
-
He, K. et al. A theoretical foundation of the target-decoy search strategy for false discovery rate control in proteomics. Preprint at arXiv https://doi.org/10.48550/arXiv.1501.00537 (2015).
-
Fu, Y. et al. Exploiting the kernel trick to correlate fragment ions for peptide identification via tandem mass spectrometry. Bioinformatics 20, 1948–1954 (2004).
-
Li, D. et al. pFind: a novel database-searching software system for automated peptide and protein identification via tandem mass spectrometry. Bioinformatics 21, 2949–3050 (2005).
-
Chi, H. et al. Comprehensive identification of peptides in tandem mass spectra using an efficient open search engine. Nat. Biotechnol. 36, 1059–1061 (2018).
-
Kong, A. T., Leprevost, F. V., Avtonomov, D. M., Mellacheruvu, D. & Nesvizhskii, A. I. MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry–based proteomics. Nat. Methods 14, 513–520 (2017).
-
Kim, S. & Pevzner, P. A. MS-GF+ makes progress towards a universal database search tool for proteomics. Nat. Commun. 5, 5277 (2014).
-
Tran, N. H. et al. NovoBoard: a comprehensive framework for evaluating the false discovery rate and accuracy of de novo peptide sequencing. Mol. Cell. Proteomics 23, 100849 (2024).
-
Wiśniewski, J. R., Zougman, A., Nagaraj, N. & Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 (2009).
-
Ding, C. et al. A fast workflow for identification and quantification of proteomes. Mol. Cell. Proteomics 12, 2370–2380 (2013).
-
Zhao, M., Wu, F. & Xu, P. Development of a rapid, high-efficiency, scalable process for acetylated Sus scrofa cationic trypsin production from Escherichia coli inclusion bodies. Protein Expr. Purif. 116, 120–126 (2015).
-
Zhao, M. et al. Recombinant expression, refolding, purification and characterization of Pseudomonas aeruginosa protease IV in Escherichia coli. Protein Expr. Purif. 126, 69–76 (2016).
-
Zhang, J. et al. Recombinant expression, purification and characterization of acetylated LysargiNase from Escherichia coli with high activity and stability. Rapid Commun. Mass Spectrom. 33, 1067–1075 (2019).
-
Zhai, L. et al. Systematic research on the pretreatment of peptides for quantitative proteomics using a C18 microcolumn. Proteomics 13, 2229–2237 (2013).
-
Yuan, Z. F. E. et al. pParse: a method for accurate determination of monoisotopic peaks in high-resolution mass spectra. Proteomics 12, 226–235 (2012).
-
Fenyö, D. & Beavis, R. C. A method for assessing the statistical significance of mass spectrometry-based protein identifications using general scoring schemes. Anal. Chem. 75, 768–774 (2003).
-
Chi, H. et al. pNovo+: de novo peptide sequencing using complementary HCD and ETD tandem mass spectra. J. Proteome Res. 12, 615–625 (2013).
-
Ma, J. et al. iProX: an integrated proteome resource. Nucleic Acids Res 47, D1211–D1217 (2019).
-
Cao, Z. X. et al. DiNovo enables high-coverage and high-confidence de novo peptide sequencing via mirror proteases and deep learning (repository name: DiNovo). Zenodo https://doi.org/10.5281/zenodo.18414283 (2026).
Acknowledgments
This work was supported by the National Key R&D Program of China (Grant No. 2022YFA1304600 to Y.F. and P.X., and Grant No. 2022YFA1004801 to Y.F.), the National Natural Science Foundation of China (Grant No. 32070668 to Y.F. and Grant No. 32371503 to P.X.), the CAMS Innovation Fund for Medical Sciences (Grant Nos. 2019-I2M-5-017 and 2022-I2M-CandT-B-082 to P.X.), Beijing Science Foundation (Grant No. L246037 to P.X.), the Foundation of State Key Lab of Proteomics (Grant Nos. SKLP-O202201, SKLP-K202201 and SKLP-C202002 to P.X.), and the Young Innovation Team Development Program for Higher Education Institutions in Shandong Province (Grant No. 2019KJN048 to H.W.).
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cao, Z., Peng, X., Zhang, D. et al. DiNovo enables high-coverage and high-confidence de novo peptide sequencing via mirror proteases and deep learning. Nat Commun 17, 2203 (2026). https://doi.org/10.1038/s41467-026-70224-6
-
Received:
-
Accepted:
-
Published:
-
Version of record:
-
DOI: https://doi.org/10.1038/s41467-026-70224-6