Main
Functional nucleic acids (FNAs) are DNA and RNA molecules designed to perform specific functions beyond storing genetic information, such as aptamers for molecular recognition1,2, regulatory elements for gene regulation3,4,5 and DNAzymes or ribozymes for catalysis6,7,8. Despite their potential across multiple fields, including chemistry, biology, medicine and material science, FNA design remains a formidable challenge due to the vast nucleotide sequence space. Unlike protein design, which has benefited from well-established sequence–structure–function rules and abundant three-dimensional (3D) structural data, FNA design is hindered by high structural flexibility, which complicates nucleotide sequence–structure–function relationships9.
Traditional experimental screening methods, such as the systematic evolution of ligands by exponential enrichment (SELEX)10,11, are commonly used but they are often plagued by high cost, low success rate, incomplete sequence space in the initial library, and PCR bias that favors amplification efficiency over functional affinity12,13,14,15. Computational approaches have advanced biomolecular design, but they heavily rely on the accuracy of 3D structure prediction9,16,17,18. While computational design of proteins has been greatly facilitated by tools such as ESM19, AlphaFold20, RoseTTAFold21 and ProteinMPNN22, an analogous development in the nucleic acid field is impeded by the scarcity of experimental 3D structures of nucleic acids. Deep learning-based models such as RaptGen23 and AptaDiff24 enable a rapid exploration of the vast design space in silico without the need for 3D structures, but they are trained solely on small, target-specific SELEX data, preventing them from learning the rich semantics required to capture comprehensive sequence–function relationships.
Large language models (LLMs) trained on a vast number of biological sequences have revolutionized the paradigm of protein design19,25,26,27, yet their application in nucleic acid design remains largely uncharted. Recently, a handful of nucleic acid LLMs (NA-LLMs) have been reported18,28,29,30,31,32,33,34. These inspired us to develop a method that can instruct an NA-LLM to generate FNA sequences with better functionality. Here, we present InstructNA, a framework for de novo generative design of FNAs by leveraging advanced NA-LLMs and high-throughput SELEX (HT-SELEX) data. InstructNA introduces two notable capabilities. First, we continue to pretrain an existing NA-LLM with HT-SELEX data, resulting in a virtual library for generating better FNA sequences that are often overlooked in physical screening. Second, to iteratively refine FNA design, we develop the HC-HEBO (hill climbing–heteroscedastic and evolutionary Bayesian optimization) algorithm, which enables directed evolution of FNAs in a continuous latent space. By establishing a generation–evaluation closed-loop system, in silico and in vitro data iteratively refine the surrogate function for FNA functionality, guiding the generation of progressively optimized sequences.
We demonstrate that InstructNA can learn semantically rich, functionally relevant and robust representations for FNAs, generating DNA sequences with higher binding specificity to transcription factors (TFs) than the existing state-of-the-art model, and aptamers with higher binding affinity for protein targets than the original HT-SELEX candidates. The high efficacy, robustness and broad utility of InstructNA demonstrate a notable capability of NA-LLMs, particularly when integrated with HT-SELEX, in advancing the field of FNA design. This synergy opens up avenues for developing FNA-based molecular tools towards a broad range of applications.
Results
Architecture of InstructNA
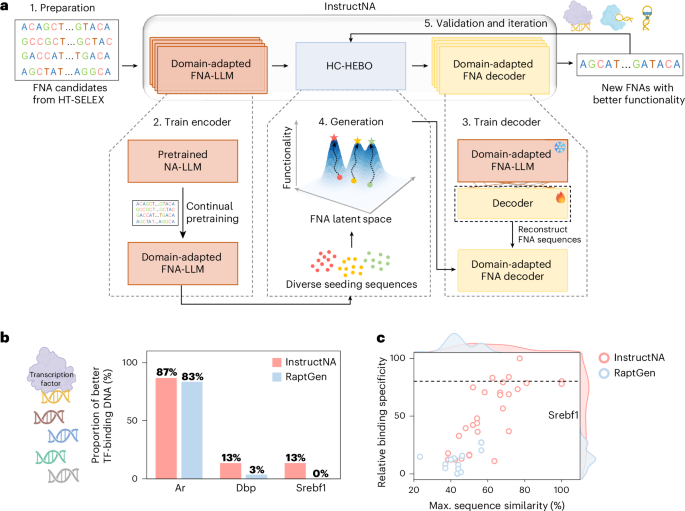
InstructNA aims to achieve de novo design of FNAs (Fig. 1a). It is a five-step framework: (1) collect FNA sequences from HT-SELEX experiments for model training (preparation), (2) continually pretrain an existing NA-LLM to be a domain-adapted FNA-LLM using the collected FNA sequences (train LLM), (3) train a lightweight decoder on top of the domain-adapted FNA-LLM using the collected FNA sequences (train decoder), (4) employ the HC-HEBO algorithm to continuously optimize the generated FNA sequences in the latent space (generation) and (5) validate the generated FNAs by experiments and perform iterative function-guided optimization (validation and iteration). Following this framework, InstructNA is applicable for generating any type of FNA, provided that its HT-SELEX data are available and its functionality can be measured. The pipeline of InstructNA is detailed in Supplementary Note 1. We thoroughly discuss the design choice of encoder, decoder and Bayesian optimization (BO) algorithm, respectively, in the following sections.

a, Architecture of the InstructNA framework: by employing a two-stage training approach, InstructNA transfers the domain knowledge of HT-SELEX FNA sequences to an existing pretrained NA-LLM to produce a domain-adapted FNA-LLM, and then generates new FNA sequences through the domain-adapted FNA decoder. Use of the HC-HEBO algorithm in the latent space enables function-guided evolution of FNAs to achieve better functionality. b, The fraction of 30 DNA sequences generated by InstructNA and RaptGen that have higher binding specificity scores than the ten top-frequency sequences in the original HT-SELEX datasets of Ar, Dbp and Srebf1. c, Scatter plot of the relative binding specificity versus maximum sequence similarity of the DNA sequences generated by InstructNA and RaptGen on the Srebf1 dataset. n = 30. The relative binding specificity is normalized to a range from 0 to 100 using min–max normalization of binding specificity scores. The maximum sequence similarity refers to the highest sequence similarity between the generated DNA sequence and ten top-frequency sequences in the original HT-SELEX dataset. The dashed line indicates the highest relative binding specificity among the ten top-frequency sequences in the original HT-SELEX dataset. Icons in a,b created in BioRender; Guo, P. https://biorender.com/xqmjipu (2026).
Learning semantically rich and functionally relevant FNA representations with InstructNA
The encoder of InstructNA, which is the domain-adapted FNA-LLM (Fig. 1a), maps FNA sequences to continuous vector representations and plays a critical role in the de novo design of FNAs. We evaluate the quality of FNA representations learned by InstructNA in terms of sequence semantics and target functionality. For this purpose, we use HT-SELEX datasets of ten TFs35 as our benchmark datasets (Supplementary Note 2). The number of cleaned unique DNA sequences in each TF dataset is shown in Supplementary Fig. 1.
Regarding sequence semantics, we examine the relationship between the sequence similarity within the real sequence space and that within the latent space of InstructNA, compared with two baseline models: DNABERT28 and RaptGen23 (Supplementary Note 3). The results show that InstructNA encodes DNA sequences with much higher Pearson correlation coefficient values than does RaptGen across all ten TF datasets, and outperforms DNABERT in eight TF datasets, all except for Klf12 and Sox10 (Extended Data Fig. 1a). Notably, although the numbers of cleaned unique DNA sequences in the ten TF datasets vary from 1,695 (Atf4) to 205,472 (Srebf1), InstructNA constantly shows a good performance. Overall, after a continual pretraining of DNABERT with FNA sequences from HT-SELEX, InstructNA achieves a substantial enhancement in capturing FNA sequence semantics.
We then explore whether the FNA representations learned by InstructNA can effectively capture sequence–function relationships, by performing a binary classification of binding specificity (Methods). Data independence analysis is detailed in Supplementary Note 4, Supplementary Figs. 2–4 and Supplementary Table 1. For sequences in each TF dataset, we use a k-mer-based approach36 to calculate their binding specificity scores (Supplementary Fig. 5). On the basis of the median binding specificity score in the training and validation sets, we label each sequence as 0 (low binding specificity) or 1 (high binding specificity) for model training. We conduct linear probing to interpret the representations encoded by InstructNA. Specifically, we train a separate linear classifier on top of InstructNA’s representations to classify the binding specificity. InstructNA surpasses RaptGen across all ten TF datasets in terms of the area under the receiver operating characteristic curve (AUROC), F1, accuracy, precision and recall except for the precision in the Mlx dataset, and outperforms DNABERT in at least seven TF datasets regarding the five metrics (Extended Data Fig. 1b,c and Supplementary Fig. 6). These results indicate that continual pretraining enables InstructNA to gain a deeper understanding of FNA sequence–function relationships, and demonstrate the generalization ability of InstructNA on unseen sequences.
Robustness of InstructNA towards representation perturbations
As InstructNA applies BO, which introduces additional randomness to the learned latent space, we further assess the robustness of InstructNA when decoding FNA sequences from perturbed representations. For this aim, we mimic the perturbation by kernel density estimation (KDE)37. By sampling from the fitted kernel distribution, we evaluate the quality of decoded FNA sequences in terms of k-mer frequency and guanine/cytosine (G/C) content3,24 (Supplementary Note 5). A longer k-mer contains more complex sequence features that are more difficult to capture. InstructNA outperforms RaptGen in generating k-mer (k = 3–6) motifs in almost all cases, except for 5-mer and 6-mer motifs in the Mlx dataset (Extended Data Table 1). In addition, InstructNA generates sequences that are more similar to the real sequences in terms of G/C content (Supplementary Fig. 7). These results demonstrate the efficacy and robustness of InstructNA in learning and generating FNAs.
Generating TF-binding DNA with higher binding specificity with HC-HEBO
InstructNA incorporates a BO-based approach to guide optimization of seeding sequences (priors) in the latent space. The choice of BO-based approach is discussed in Supplementary Note 6. We first observe that DNA sequences optimized by a recently developed BO method named HEBO38 exhibit high sequence diversity but poor binding specificity, relative to the seeding sequences from the original HT-SELEX dataset (Extended Data Fig. 2b and Supplementary Fig. 8). We suspect that this may be due to BO’s tendency to perform global optimization. To address this, we develop HC-HEBO, which introduces hill climbing (HC)39 into HEBO to restrict its search space within a local region around the seeding sequence’s representation (Extended Data Fig. 2a). Compared with HEBO, our developed HC-HEBO is able to design DNA sequences with higher binding specificity while exhibiting lower sequence diversity (Extended Data Fig. 2b and Supplementary Fig. 8). HC-HEBO also performs better than HEBO when designing a larger number of DNA sequences (Supplementary Fig. 9). We further test different seeding strategies for HC-HEBO (Supplementary Note 7). Using seeding sequences from three different sources (S1 + S2 + S3) consistently outperforms a single source (Extended Data Fig. 2c). In addition, two rounds of HC-HEBO typically achieve satisfactory performance (Extended Data Fig. 2d). These results demonstrate that HC-HEBO can rapidly evolve FNAs towards better functionality.
We next assess the overall performance of InstructNA in generating FNAs. We randomly select Ar, Dbp and Srebf1 as benchmark datasets and generate 30 new DNA sequences for each TF. The results show that 87%, 13% and 13% of the DNA sequences generated by InstructNA exhibit higher binding specificity scores than the ten top-frequency sequences from the original HT-SELEX, compared with 83%, 3% and 0% of those generated by RaptGen on the Ar, Dbp and Srebf1 datasets, respectively (Fig. 1b,c and Supplementary Fig. 10). Both InstructNA and RaptGen perform well for the Ar dataset, and this can be attributed to the low binding specificity of the ten top-frequency sequences from the original HT-SELEX. For the Srebf1 dataset, InstructNA generates four better DNA sequences while RaptGen fails to generate any better sequences. Moreover, among the four better DNA sequences generated by InstructNA, three show maximum sequence similarities below 80% to the ten top-frequency sequences from the original HT-SELEX (Fig. 1c). These results demonstrate the ability of InstructNA to generate functionally improved FNAs with high sequence diversity.
Incorporating other NA-LLMs into InstructNA
While we initially build InstructNA upon DNABERT28 as a demonstration, it is capable of integrating any NA-LLM as its foundation model. We further test incorporation of Evo131 and Nucleotide Transformer32 (NT), which have larger parameters than does DNABERT. InstructNA-DNABERT, InstructNA-NT and InstructNA-Evo1 show comparable performance on the mean binding specificity; InstructNA-DNABERT and InstructNA-Evo1 achieve the highest binding specificity on Dbp/Srebf1 and Ar datasets, respectively (Extended Data Fig. 2e). Possible reasons for the negligible improvement when incorporating larger DNA-LLMs are discussed in Supplementary Note 8. As an autoregressive NA-LLM, Evo1 is capable of generating NA sequences. We compare the sequences generated by InstructNA-Evo1 with those directly generated by Evo1. We first continue to pretrain Evo1 using the DNA sequences collected from HT-SELEX and then sample sequences from the continually pretrained Evo1 without running BO. InstructNA-Evo1 consistently outperforms Evo1 in generating high-quality sequences (Extended Data Fig. 2e), underscoring the necessity of BO in InstructNA.
Generating aptamers with high binding affinity and sequence diversity with InstructNA
To further demonstrate the universal applicability of InstructNA, we next turn to aptamer design. Protein-binding aptamers have shown broad applications in clinical diagnosis and therapeutics40,41,42. To this end, we conduct HT-SELEX experiments for two protein targets, LOX1 and CXCL5, and use sequencing data from the last round of HT-SELEX to train InstructNA (Fig. 2a). The HT-SELEX experiments are detailed in Supplementary Note 9 and Supplementary Table 2. We evaluate the binding affinity of screened or designed DNA aptamers for the protein target using a surface plasmon resonance (SPR) experiment (Supplementary Note 10). Among the ten top-frequency aptamer candidates from HT-SELEX, namely T1L to T10L for LOX1 and T1C to T10C for CXCL5 (Supplementary Tables 3 and 4), only two and one strong aptamer binders (dissociation constant KD ≤ 100 nM) are identified for LOX1 and CXCL5, respectively, and the remaining ones are weak aptamer binders (KD > 100 nM) or have no measurable binding to the protein target (Fig. 2b,c and Supplementary Figs. 11 and 12).

a, Schematic showing HT-SELEX experiments for screening DNA aptamers that bind to a specific protein target (LOX1 and CXCL5 in this study): incubation of single-stranded DNA library and protein target, removal of unbound DNA, amplification of bound DNA by PCR, and HT sequencing. b,c, SPR binding assays of ten top-frequency aptamer candidates from HT-SELEX and ten aptamer candidates generated by InstructNA for LOX1 (b) and CXCL5 (c). KD values are plotted for strong aptamer binders (KD ≤ 100 nM). #, weaker binders (KD > 100 nM); *, no measurable binding. Data in b,c are presented as mean ± s.d. (n = 3 independent experiments), with the error bar indicating s.d. d,e, SPR sensorgrams for binding of T1L and G1L to LOX1 (d) and binding of T1C and G1C to CXCL5 (e). Data in d,e are presented as mean ± s.d. (n = 3 independent experiments). Icons in a created in BioRender; Guo, P. https://biorender.com/xqmjipu (2026).
InstructNA generates ten aptamer sequences for each protein, namely G1L to G10L for LOX1, and G1C to G10C for CXCL5 (Supplementary Tables 5 and 6). For LOX1, InstructNA generates four strong aptamer binders (G1L, G2L, G3L, G4L) with KD values of 12.9, 19.3, 57.4 and 75.0 nM, respectively, among which G1L and G2L exhibit higher binding affinity than the original T1L (KD = 23.1 nM) and T2L (KD = 37.6 nM) (Fig. 2b,d and Supplementary Fig. 13). For CXCL5, InstructNA generates three strong aptamer binders (G1C, G2C, G3C) with KD values of 6.6, 18.6 and 40.3 nM, respectively, among which G1C exhibits higher binding affinity than the original T1C (KD = 18.3 nM) (Fig. 2c,e and Supplementary Fig. 14).
We further analyze sequences and structures of InstructNA-generated aptamers. Using LOX1 as an illustration, the strong aptamers generated by InstructNA and screened from HT-SELEX are distributed within different clusters (Extended Data Fig. 3a), with G1L exhibiting a sequence similarity to T1L as low as 38% (Extended Data Fig. 3b). From a structural perspective, T1L forms a plain stem–loop structure whereas G1L exhibits a more intricate fold at the apical loop, and they interact with different domains of LOX1 (Extended Data Fig. 3c–e). The LOX1–G1L complex exhibits extensive hydrogen-bonding interactions at the binding interface (Extended Data Fig. 3f) and a more favorable Gibbs free-energy change than the LOX1–T1L complex (Supplementary Fig. 15). The sequence and structural analyses are detailed in Supplementary Note 11. Among the strong aptamer binders for CXCL5, the InstructNA-generated G1C, G2C and G3C show sequence similarities of 97%, 49% and 97% to the original T1C, respectively (Supplementary Fig. 16). In addition to these two protein targets, we also show that InstructNA generates strong aptamer binders for protein tyrosine kinase 7 (Supplementary Fig. 17), a widely used protein model for aptamer development42,43. These results demonstrate that InstructNA can efficiently generate aptamers with high binding affinity for protein targets.
Discussion
In this work, we introduce the InstructNA framework for de novo design of FNAs by leveraging NA-LLMs and HT-SELEX. Several potential extensions of InstructNA can be envisioned. First, here we employ the pretrained DNABERT28, Evo131 and Nucleotide Transformer32 as NA-LLMs to illustrate the approach of customizing domain-adapted FNA-LLMs. As the field advances rapidly, more powerful and sophisticated NA-LLMs are being developed, and they can be adapted to the InstructNA framework. Second, integrating 3D structure prediction tools such as AlphaFold320 and RoseTTAFoldNA44, complemented with molecular dynamics (MD) simulations as additional evaluation criteria, is expected to improve the efficiency of FNA design. As acquiring HT-SELEX data is time consuming, relevant works demonstrate their FNA design approaches on a few HT-SELEX datasets23,45,46,47. Although here we demonstrate InstructNA on relatively large datasets, de novo design of other types of FNA such as DNAzymes and functional RNA molecules needs to be explored in future work. These will expand InstructNA’s ability to design FNAs with tailored and enhanced functionality.
Methods
Preparation of HT-SELEX data of TF-binding DNA for model training
The HT-SELEX datasets of ten TFs (Ar, Atf4, Dbp, Egr3, Foxg1, Klf12, Mlx, Nr2e1, Sox10 and Srebf1) used in this study were downloaded from the European Nucleotide Archive database (accession number: ERP001824)35. The FASTQ files were further processed by removing the variable-length and redundant sequences, to obtain the cleaned sequences used for further continual pretraining of NA-LLM. The lengths and numbers of cleaned unique DNA sequences in each TF dataset are summarized in Supplementary Fig. 1.
Evaluating the binding specificity of TF-binding DNA sequences
The binding specificity score of TF to a DNA sequence is calculated using the 8mer_sum algorithm as described previously36:
$${S}_{mathrm{total}}=mathop{sum }limits_{i=1}^{n}{S}_{8mathrm{mer}}^{i}$$
(1)
where Stotal is the binding specificity score of the full-length DNA sequence, ({S}_{8mathrm{mer}}^{i}) is the protein-binding microarray fluorescence intensity of the ith 8-mer motif, which is extracted using a sliding window of length 8 and stride 1, and n represents the total number of 8-mer motifs in the full-length DNA sequence.
The relative binding specificity is normalized to a range from 0 to 100 using min–max normalization of binding specificity score:
$${S}_{{rm{norm}}}=frac{S-{S}_{min }}{{S}_{max }-{S}_{min }}times 100$$
(2)
where ({S}_{min }) and ({S}_{max }) represent the minimum and maximum binding specificity scores in the dataset, respectively. This scaling transforms the raw scores to the [0, 100] range.
InstructNA training and inference using DNABERT (3-mers)
Tokenization in DNABERT (3-mers)
In the domain of language modeling, tokens represent the fundamental semantic units that a model employs to interpret linguistic data. They can represent discrete units such as words or even more granular semantic elements such as individual characters. The act of tokenization involves transforming these linguistic units into unique integer identifiers, each corresponding to an entry within a reference lookup table. Subsequently, these integers are translated by embedding layers into vectorial representations, which the model then processes in a comprehensive, end-to-end manner. For the specific application within the InstructNA framework using DNABERT (3-mers), DNA sequences were tokenized using a trinucleotide (3-mers) resolution method. This approach results in a combinatorial diversity of 64 (43) possible distinct 3-mers. Additionally, special utility tokens such as [CLS], [UNK], [SEP], [MASK] and [PAD] were integrated into the model for specific functional roles within the modeling process.
Training
The training process of InstructNA using DNABERT (3-mers) consists of two stages. In the first stage, we conducted continual pretraining on a pretrained DNABERT DNA-LLM using HT-SELEX data, with the training objective aligned with that of the original DNABERT pretraining (Supplementary Fig. 18). Specifically, 15% of the tokens were masked, and the model was trained to predict the masked tokens. To facilitate BO, a dimensionality reduction layer was incorporated during training, reducing the dimensionality of each token embedding to 8 (Supplementary Fig. 19). This process results in a domain-adapted FNA-LLM (encoder).
In the second stage, we froze the encoder and introduced a randomly initialized decoder composed of six-layer transformer blocks. We then performed a sequence reconstruction task, where HT-SELEX sequence data are fed into the model again. The encoder transformed these sequences into embeddings, which were subsequently passed to the decoder, enabling it to reconstruct the original sequence tokens. The objective of this training stage is to reconstruct the original input sequence from the embeddings. Specifically, the decoder outputs a vector of logits for the ith position token based on the corresponding embedding ei, and a softmax layer is applied to obtain the probability distribution over the vocabulary. The loss function for this stage is
$${L}_{mathrm{recon}}=-sum log ,P({x}_{i}|{mathbf{e}}_{{{i}}})$$
(3)
where P(xi|ei) represents the probability assigned to the ground-truth token xi given the embedding ei after the softmax transformation.
Inference
Using embeddings derived from clustering centers or BO, the decoder processes these embeddings to produce an initial generated sequence. Next, the 15% of tokens with the lowest probabilities in this sequence were replaced with a special token, [MASK], and the modified sequence was fed back into the encoder to predict and refine the masked-out portions. This iterative refinement process is repeated several times, progressively enhancing the accuracy of the generated sequence.
HC-HEBO in InstructNA
The HC-HEBO algorithm combines HC with HEBO to efficiently optimize FNAs.
- (1)
Initial solution construction. Sequences from diverse sources are evaluated and their embeddings are denoted as E0 = {e0, e1, e2,…,en}. E0 is divided into n groups and each ei ∈ E0 (0 ≤ i ≤ n) represents the initial solution for the group respectively.
- (2)
Search space definition. For the n groups, define the initial search space as a neighborhood centered around E0, with a search radius R0 (default ΔR0 = 5). The sensitivity experiment of the search radius and minimum threshold is shown in Supplementary Fig. 20. The n initial search space S0 = {s0, s1, s2, …, sn} is defined as
$${E}_{0},({rm{initial}},{rm{solution}})$$
(4)
$${S}_{0}={mathbf{e}|parallel mathbf{e}-{E}_{0}parallel le Delta {R}_{0}},({rm{initial}},{rm{search}},{rm{space}}).$$
(5)
- (3)
Searching and generation. Use HEBO to search next embeddings in the search space with the hyperparameters in HEBO kept as default. InstructNA generates and evaluates a total of n new sequences, with one new sequence generated per group.
- (4)
Greedy refinement. At each iteration, the solution (embedding) with the best evaluated performance within its group is chosen as the candidate solution for the next optimization. The ith group solution obtained at the tth iteration is Ei,t:
$${mathbf{E}}_{i,t}=text{arg}mathop{max }limits_{textbf{e}in {G}_{t,i}}f(textbf{e})$$
(6)
where Gt,i represents the ith group of evaluated solutions at iteration t and f(e) represents the evaluated performance.
- (5)
Shrinking search radius. After each iteration, the search radius is reduced by half, but once it reaches a minimum threshold ΔRmin (default ΔRmin = 1.25), the search radius will no longer shrink and remains constant:
$${S}_{t+1}=left{textbf{e}|Vert textbf{e}-{E}_{t}Vert le frac{Delta {R}_{t}}{2}right},,mathrm{if},Delta {R}_{t} > Delta {R}_{min }$$
(7)
$${S}_{t+1}={mathbf{e}|Vert mathbf{e}-{E}_{t}Vert le Delta {R}_{min }},,mathrm{if},Delta {R}_{t}le Delta {R}_{min }.$$
(8)
- (6)
Obtain the tth iteration best solution Et, search space St. Repeat steps 3–5 for iteration.
Assessing correlation between embeddings and sequences
We randomly sampled 1,000 unique sequences from the HT-SELEX dataset. From these sequences, we generated all possible pairs (({C}_{1,000}^{2})), resulting in a total of 499,500 sequence pairs. We explored the relationship between sequences and embeddings by analyzing the cosine similarity and pairwise alignment similarity of these sequence pairs.
Cosine similarity calculation
For InstructNA and DNABERT, we calculated the cosine similarity of token embeddings at corresponding positions for each pair of sequences, and then averaged the cosine similarities across all tokens to obtain the final cosine similarity score representing the full embedding similarity of the sequence pair.
$${{rm{Similarity}}}_{{rm{sequence}}{rm{pair}}}({S}_{1},{S}_{2})=frac{1}{N}displaystyle mathop{sum }limits_{i=1}^{N}frac{{mathbf{e}}_{i}^{1}cdot {mathbf{e}}_{i}^{2}}{| {mathit{e}}_{mathit{i}}^{1}| | {mathit{e}}_{mathit{i}}^{2}| }$$
(9)
where S1 and S2 represent the sequences in the sequence pair, ({mathbf{e}}_{i}^{1}) and ({mathbf{e}}_{i}^{2}) represent the embedding of the ith token in S1 and S2, and · denotes the vector inner product.
For RaptGen, we compute the cosine similarity between the 2D embeddings of sequence pairs directly.
Sequence similarity
Sequence similarity is calculated using the EMBOSS Needle tool48 that creates an optimal global alignment of two sequences using the Needleman–Wunsch algorithm with default parameters: matrix = DNA full, gap open penalty = 10, gap extended penalty = 0.5, end gap open penalty = 10 and end gap extend penalty = 0.5. For the DNA sequences binding to TFs, the sequence similarity is calculated on the 14-nucleotide or 20-nucleotide random DNA (Supplementary Fig. 1). For the DNA aptamers binding to LOX1 or CXCL5, the sequence similarity is calculated on the 36-nucleotide random DNA (Supplementary Table 2).
Binary classification of binding specificity
For the cleaned unique sequences in each TF dataset as shown in Supplementary Fig. 1, we cluster them using cd-hit49 at an 80% sequence similarity cutoff18,50, collect representative sequences from each cluster and randomly split the collected sequences into training (40%), validation (10%) and test (50%) sets. The numbers of sequences in training, validation and test sets are summarized in Supplementary Table 1. On the basis of the median binding specificity score in the training and validation sets, we label each sequence as 0 (low binding specificity) or 1 (high binding specificity) and train the model.
In the classification pipeline, encoders pretrained by InstructNA and RaptGen remain frozen, with a trainable linear layer appended for binary classification. Model performance is assessed on the test set using the following metrics:
$$mathrm{Precision}=frac{mathrm{TP}}{mathrm{TP}+mathrm{FP}}$$
(10)
$$mathrm{Accuracy}=frac{mathrm{TP}+mathrm{TN}}{mathrm{TP}+mathrm{TN}+mathrm{FP}+mathrm{FN}}$$
(11)
$$mathrm{Recall}=mathrm{TPR}=frac{mathrm{TP}}{mathrm{TP}+mathrm{FN}}$$
(12)
$${F}_{1}=2times frac{{rm{Precision}}times {rm{Recall}}}{{rm{Precision}}+{rm{Recall}}}$$
(13)
$$mathrm{FPR}=frac{mathrm{FP}}{mathrm{FP}+mathrm{TN}}$$
(14)
$$mathrm{AUROC}={int }_{0}^{1}mathrm{TPR}(mathrm{FPR}),{rm{d}}(mathrm{FPR})$$
(15)
where TP, TN, FP and FN represent true positive, true negative, false positive and false negative respectively in the confusion matrix.
KDE perturbation generation experiment
A perturbation generation experiment was performed separately on the ten HT-SELEX datasets of TF-binding DNA sequences. The embeddings of all DNA sequences from each HT-SELEX dataset were obtained using the InstructNA and RaptGen encoders. These embeddings were then used to train a KDE37 model to learn the probability density distribution of the embeddings. Two thousand new embeddings were sampled from the trained KDE model based on the learned probability density function. These new embeddings were input into the decoder for inference. Finally, InstructNA and RaptGen generated 2,000 sequences for each HT-SELEX dataset, respectively. For the real sequences, we performed stratified sampling to select 2,000 sequences on the basis of sequence frequency from each HT-SELEX dataset. Then, k-mer frequency and G/C content of these sequences were calculated. The k-mer counts were obtained using sliding windows of lengths 3–6 with a stride of 1 for each sequence, and frequency was calculated on the basis of the k-mer counts. The G/C content was determined by calculating the proportion of G and C nucleotides in each sequence.
Pipeline of InstructNA to generate new DNA sequences for TF binding
To generate new DNA sequences that have high binding specificity to a TF, we trained InstructNA as stated above. During the generation and optimization by HC-HEBO, 30 starting seeds were collected from three sources, including ten top-frequency sequences in the original HT-SELEX data (S1), ten sequences decoded from each of the ten cluster centers of the entire HT-SELEX embeddings clustered by the Gaussian mixture model (GMM)51 (S2) and ten sequences with similar embeddings (calculated by the minimum Euclidean distance) to the DNAs with the highest binding specificity from S1 and S2 (S3). Notably, the number of seeding sequences can vary. The 30 candidates were then clustered into ten clusters using GMM, and the sequence of the highest binding specificity score in each group was chosen to constitute the initial ten seeding sequences for HC-HEBO.
HT-SELEX for screening protein-binding DNA aptamers
The ssDNA library and primers shown in Supplementary Table 2 were purchased from Sangon Biotech. The ssDNA library was denatured at 95 °C for 10 min, 4 °C for 6 min and 25 °C for 2 min. COOH-modified magnetic beads (400 μl; Zecen Biotech) were washed with 1 ml of 10-mM NaOH once and 1 ml of double-distilled water twice, and activated in 800 μl of 0.4-M 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide (EDC) and 0.1-M N-hydroxysuccinimide (NHS) (1:1, v/v) for 15 min. Then, ~80 μg of protein (PTM Bio) diluted in sodium acetate was added to the magnetic beads and incubated for 2 h. Supernatant was discarded and the beads were washed three times with 1 ml of Dulbecco’s phosphate-buffered saline (DPBS). Then, the beads were blocked in 1 ml of 10 mM ethanolamine (pH 8.0), incubated for 15 min, washed three times with 800 μl of 1× washing buffer and then resuspended in 400 μl of antibody diluent buffer.
Each round of counter-selection and positive selection was conducted using the conventional magnetic bead-based HT-SELEX method. The His–magnetic beads were washed with 1× DPBS, and then incubated with the ssDNA library for 1 h. The mixture was washed with 1× washing buffer, and then the His–magnetic beads were discarded, leaving the remaining DNA pool for positive selection. DNAs bound to the target protein were eluted by boiling the protein–magnetic beads in 100 μl of double-distilled water at 95 °C for 10 min, amplified with PCR using rTaq DNA polymerase (Takara) and used for the next round of selection. For PCR amplification, we used MIX configuration with the forward primer and biotin-modified reverse primer, and the ssDNA eluted from the positive selection as the template. Double-stranded DNA (1 ml) obtained from PCR was incubated with 80 μl of streptavidin-modified magnetic beads (Smart-Lifesciences Biotech) in 1-M NaCl for 30 min, washed with 1× washing buffer three times and treated with 100 μl of 40-mM NaOH. The biotin-modified strands left on streptavidin-modified magnetic beads were removed by magnetic suction, neutralized with 4 μl of 1-M HCl and combined with an equal volume of 2× DPBS buffer. For high-throughput sequencing, each round of the ssDNA library was amplified using the forward and reverse primers, followed by the use of a standard library construction kit (E7335S NEB). Polyclonal amplicons were purified using VAHTS DNA Clean Beads (Vazyme) following the manufacturer’s protocol, quantified with QUBIT (Invitrogen). The sequencing was performed using the GeneMind sequencer. The high-throughput sequencing data from the last round of SELEX were used for InstructNA.
Pipeline of InstructNA to generate DNA aptamers
The last round of aptamer HT-SELEX data was used to train InstructNA as stated above. The 30 seeding sequence candidates from S1, S2 and S3 were engineered in the same way as stated above for those of TF-binding DNAs. The 30 candidate sequences were clustered into ten clusters using GMM, and the sequence with the smallest KD measured by SPR to the target protein was chosen to constitute the initial seeding sequences for HC-HEBO optimization.
SPR experiments
The DNA oligonucleotides used for SPR experiments shown in Supplementary Tables 3–6 were purchased from Sangon Biotech. The SPR experiments were performed on a Biacore 8K instrument (GE Healthcare). The sensor chip (CM5, Cytiva) was activated by injecting a 50:50 (v/v) mixture of NHS (0.1 M) and EDC (0.4 M) for 5 min. The protein (50 µg ml−1) was diluted in sodium acetate and injected into flow cell 2 at a flow rate of 10 µl min−1 for 5 min. Next, the His protein (5 µg ml−1) diluted in sodium acetate was injected into flow cell 1 as a control channel. Finally, the sensor chip was blocked using ethanolamine-HCl (1 M, pH 8.5) for 2 min. The DNA aptamers were diluted to different concentrations using 1× PBS containing 5-mM magnesium chloride. Aptamers at various concentrations in running buffer (DPBS supplemented with 5-mM MgCl2) were injected into the analyte channel at a flow rate of 30 μl min−1, a contact time of 120 s and a dissociation time of 120 s. After each analysis cycle, the analyte channels were regenerated via a 30-s injection of 1.5-M NaCl. The KD was obtained by fitting the sensorgram with a 1:1 binding model using Biacore evaluation software (GE Healthcare).
Docking and unrestrained MD simulations
The AlphaFold3 webserver (https://alphafoldserver.com/) was utilized to predict the 3D structures of DNA aptamers T1L, G1L (nucleotides 1–76) and LOX1 (amino acids 61–237), which are consistent with the sequence lengths used in SPR experiments. Initially, we attempted to predict the complex structure directly from the input protein and DNA sequences, but the resulting structures were visibly not bound together. Therefore, on the basis of the monomer structures of the protein and DNAs, the LOX1–T1L and LOX1–G1L complexes were obtained via the molecular docking using HDOCKlite v.1.1, based on a hybrid algorithm of template-based modeling and ab initio free docking. The LOX1 protein was set as the receptor, and the DNA aptamer was treated as the ligand. Blind docking was performed, and the top docking pose based on the docking score was selected as the initial structure for MD simulations.
The complex structure was subjected to unrestrained MD simulations on the Amber22 package. The complex was modeled using the tleap module in Amber22. The Amber ff19SB force field was used for the protein and the OL15 force field was employed for DNA. After adding Na+ to neutralize the phosphate backbone charges, the system was then solvated with TIP3P water in a 12-Å cuboid box. Initial minimizations were carried out with 2,000 steps of steepest descent followed by 3,000 steps of conjugated gradient, during which the DNA positions were fixed. The particle mesh Ewald algorithm was employed to efficiently treat the long-range electrostatic interaction. Then the system was heated to 300 K under constant volume in 50 ps, followed by a 50-ps constant-pressure relaxation using the Langevin thermostat at 300 K. All covalent bonds involving hydrogen atoms were constrained using the SHAKE algorithm. The cutoff was set to 12.0 Å for the electrostatic and van der Waals interactions. The final production was performed at 300 K without restraints on DNA for 100 ns. Ten independent runs of MD simulations were performed for each complex.
Statistics and reproducibility
No statistical method was used to predetermine sample size. No data were excluded from the analyses. Training, validation and test sets for training models of binary classification were generated via random splits of the full datasets. No randomization was applied in the comparative experiments. Blinding was not relevant to this study.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The HT-SELEX datasets of Ar, Atf4, Dbp, Egr3, Foxg1, Klf12, Mlx, Nr2e1, Sox10 and Srebf1 used in this study are available from the European Nucleotide Archive under accession number ERP001824 (ref. 35). All data in this study are provided in the main text and Supplementary Information. Source data for Figs. 1 and 2 and Extended Data Figs. 1–3 are provided with this paper. Source data are provided with this paper.
Code availability
The code of InstructNA is deposited to GitHub (https://github.com/zhimingzhang275/InstructNA) and Code Ocean52.
References
-
He, A. et al. Structure-based investigation of a DNA aptamer targeting PTK7 reveals an intricate 3D fold guiding functional optimization. Proc. Natl Acad. Sci. USA 121, e2404060121 (2024).
-
Cheng, E. L. et al. Discovery of a transferrin receptor 1-binding aptamer and its application in cancer cell depletion for adoptive T-cell therapy manufacturing. J. Am. Chem. Soc. 144, 13851–13864 (2022).
-
Zhang, P. et al. Deep flanking sequence engineering for efficient promoter design using DeepSEED. Nat. Commun. 14, 6309 (2023).
-
Seo, E., Choi, Y. N., Shin, Y. R., Kim, D. & Lee, J. W. Design of synthetic promoters for cyanobacteria with generative deep-learning model. Nucleic Acids Res. 51, 7071–7082 (2023).
-
Gosai, S. J. et al. Machine-guided design of cell-type-targeting cis-regulatory elements. Nature 634, 1211–1220 (2024).
-
Okuda, T., Lenz, A.-K., Seitz, F., Vogel, J. & Höbartner, C. A SAM analogue-utilizing ribozyme for site-specific RNA alkylation in living cells. Nat. Chem. 15, 1523–1531 (2023).
-
Papastavrou, N., Horning, D. P. & Joyce, G. F. RNA-catalyzed evolution of catalytic RNA. Proc. Natl Acad. Sci. USA 121, e2321592121 (2024).
-
Svehlova, K., Lukšan, O., Jakubec, M. & Curtis, E. A. Supernova: a deoxyribozyme that catalyzes a chemiluminescent reaction. Angew. Chem. Int. Ed. 61, e202109347 (2022).
-
Wong, F. et al. Deep generative design of RNA aptamers using structural predictions. Nat. Comput. Sci. 4, 829–839 (2024).
-
Kohlberger, M. & Gadermaier, G. SELEX: critical factors and optimization strategies for successful aptamer selection. Biotechnol. Appl. Biochem. 69, 1771–1792 (2022).
-
Sun, D. et al. Computational tools for aptamer identification and optimization. Trends Anal. Chem. 157, 116767 (2022).
-
Hu, Q. et al. DNAzyme-based faithful probing and pulldown to identify candidate biomarkers of low abundance. Nat. Chem. 16, 122–131 (2024).
-
Yang, K. et al. A functional group-guided approach to aptamers for small molecules. Science 380, 942–948 (2023).
-
Chang, D. et al. A high-dimensional microfluidic approach for selection of aptamers with programmable binding affinities. Nat. Chem. 15, 773–780 (2023).
-
Tsuji, S. et al. Effective isolation of RNA aptamer through suppression of PCR bias. Biochem. Biophys. Res. Commun. 386, 223–226 (2009).
-
Zacco, E. et al. Probing TDP-43 condensation using an in silico designed aptamer. Nat. Commun. 13, 3306 (2022).
-
Li, T. et al. Blocker-SELEX: a structure-guided strategy for developing inhibitory aptamers disrupting undruggable transcription factor interactions. Nat. Commun. 15, 6751 (2024).
-
Shen, T. et al. Accurate RNA 3D structure prediction using a language model-based deep learning approach. Nat. Methods 21, 2287–2298 (2024).
-
Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023).
-
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
-
Baek, M. et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871–876 (2021).
-
Dauparas, J. et al. Robust deep learning-based protein sequence design using ProteinMPNN. Science 378, 49–56 (2022).
-
Iwano, N., Adachi, T., Aoki, K., Nakamura, Y. & Hamada, M. Generative aptamer discovery using RaptGen. Nat. Comput. Sci. 2, 378–386 (2022).
-
Wang, Z. et al. AptaDiff: de novo design and optimization of aptamers based on diffusion models. Brief. Bioinform. 25, bbae517 (2024).
-
Madani, A. et al. Large language models generate functional protein sequences across diverse families. Nat. Biotechnol. 41, 1099–1106 (2023).
-
Nijkamp, E., Ruffolo, J. A., Weinstein, E. N., Naik, N. & Madani, A. ProGen2: exploring the boundaries of protein language models. Cell Syst. 14, 968–978.e3 (2023).
-
Hayes, T. et al. Simulating 500 million years of evolution with a language model. Science 387, 850–858 (2025).
-
Ji, Y., Zhou, Z., Liu, H. & Davuluri, R. V. DNABERT: pre-trained Bidirectional Encoder Representations from Transformers model for DNA-language in genome. Bioinformatics 37, 2112–2120 (2021).
-
Wang, N. et al. Multi-purpose RNA language modelling with motif-aware pretraining and type-guided fine-tuning. Nat. Mach. Intell. 6, 548–557 (2024).
-
Sanabria, M., Hirsch, J., Joubert, P. M. & Poetsch, A. R. DNA language model GROVER learns sequence context in the human genome. Nat. Mach. Intell. 6, 911–923 (2024).
-
Nguyen, E. et al. Sequence modeling and design from molecular to genome scale with Evo. Science 386, eado9336 (2024).
-
Dalla-Torre, H. et al. Nucleotide Transformer: building and evaluating robust foundation models for human genomics. Nat. Methods 22, 287–297 (2025).
-
Zhou, Z. et al. DNABERT-S: pioneering species differentiation with species-aware DNA embeddings. Bioinformatics 41, i255–i264 (2025).
-
Zhou, Z. et al. DNABERT-2: efficient foundation model and benchmark for multi-species genomes. In Proc. Twelfth International Conference on Learning Representations (OpenReview, 2024); https://openreview.net/forum?id=oMLQB4EZE1
-
Jolma, A. et al. DNA-binding specificities of human transcription factors. Cell 152, 327–339 (2013).
-
DREAM5 Consortium et al. Evaluation of methods for modeling transcription factor sequence specificity. Nat. Biotechnol. 31, 126–134 (2013).
-
Węglarczyk, S. Kernel density estimation and its application. ITM Web Conf. 23, 00037 (2018).
-
Cowen-Rivers, A. I. et al. HEBO: pushing the limits of sample-efficient hyper-parameter optimisation. J. Artif. Intell. Res. 74, 1269–1349 (2022).
-
Li, L. et al. Machine learning optimization of candidate antibody yields highly diverse sub-nanomolar affinity antibody libraries. Nat. Commun. 14, 3454 (2023).
-
Chen, X. et al. Visualizing RNA dynamics in live cells with bright and stable fluorescent RNAs. Nat. Biotechnol. 37, 1287–1293 (2019).
-
Zhu, L., Yang, J., Ma, Y., Zhu, X. & Zhang, C. Aptamers entirely built from therapeutic nucleoside analogues for targeted cancer therapy. J. Am. Chem. Soc. 144, 1493–1497 (2022).
-
Tian, Y., Miao, Y., Guo, P., Wang, J. & Han, D. Insulin-like growth factor 2-tagged aptamer chimeras (ITACs) modular assembly for targeted and efficient degradation of two membrane proteins. Angew. Chem. Int. Ed. 63, e202316089 (2024).
-
Albright, S., Cacace, M., Tivon, Y. & Deiters, A. Cell surface labeling and detection of protein tyrosine kinase 7 via covalent aptamers. J. Am. Chem. Soc. 145, 16458–16463 (2023).
-
Baek, M. et al. Accurate prediction of protein–nucleic acid complexes using RoseTTAFoldNA. Nat. Methods 21, 117–121 (2024).
-
Ishida, R. et al. RaptRanker: in silico RNA aptamer selection from HT-SELEX experiment based on local sequence and structure information. Nucleic Acids Res. 48, e82 (2020).
-
Zhang, Y. et al. Single-step discovery of high-affinity RNA ligands by UltraSelex. Nat. Chem. Biol. 21, 1118–1126 (2025).
-
Chen, B. et al. Selection of allosteric DNAzymes that can sense phenylalanine by expression-SELEX. Nucleic Acids Res. 51, e66 (2023).
-
Madeira, F. et al. The EMBL-EBI Job Dispatcher sequence analysis tools framework in 2024. Nucleic Acids Res. 52, W521–W525 (2024).
-
Li, W. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006).
-
Yu, H. et al. An interpretable RNA foundation model for exploring functional RNA motifs in plants. Nat. Mach. Intell. 6, 1616–1625 (2024).
-
Xuan, G., Zhang, W. & Chai, P. EM algorithms of Gaussian mixture model and hidden Markov model. In Proc. 2001 International Conference on Image Processing 145–148 (IEEE, 2001).
-
Zhang, Z. De novo design of functional nucleic acids of aptamers. Code Ocean https://doi.org/10.24433/CO.7821298.v1 (2026).
Acknowledgements
This work is supported by the National Natural Science Foundation of China (22225402, D.H.; 32341017, D.H.; 32341018, G.C.; 22374132, P.G.; 62306290, J.Q.), Fundamental and Interdisciplinary Disciplines Breakthrough Plan of the Ministry of Education of China (JYB2025XDXM602, D.H.) and Leading Innovative and Entrepreneur Team Introduction Program of Zhejiang Province (2024R01005, D.H.). We acknowledge the support from the Scientific Experiment Center of Hangzhou Institute of Medicine (HIM), Chinese Academy of Sciences.
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Computational Science thanks Cheng Zhao and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Ananya Rastogi and Fernando Chirigati, in collaboration with the Nature Computational Science team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Capturing the complex sequence patterns in FNAs by InstructNA.
a, The Pearson correlation between the cosine distance of pairwise embeddings and the sequence similarity of embeddings derived from the latent space of InstructNA, DNABERT and RaptGen on the ten TF datasets. b–c, The performance (AUROC and F1) of InstructNA versus RaptGen (b) and DNABERT (c) for the binary classification of binding specificity on the ten TF datasets. The performance on precision, accuracy and recall is shown in Supplementary Figure 6. Statistical analysis is performed using a one-sided paired t-test.
Extended Data Fig. 2 Ablation study of BO strategies and NA-LLM models.
a, Schematics of HEBO and HC-HEBO algorithms. Arrows illustrate the direction of FNA optimization. b, Ablation experiment on the BO strategies, that is, HEBO versus HC-HEBO. Max sequence similarity refers to the highest sequence similarity between the generated sequence and the seeding sequences. n = 20. c, Ablation experiment on the sources of seeding sequences, that is, the S1, S2 or S3 single seeding source versus the S1 + S2 + S3 diverse seeding sources. n = 20. d, Ablation experiment on the HC-HEBO rounds. n = 10. e, Performance of InstructNA-DNABERT, InstructNA-NT, InstructNA-Evo1, and Evo1. n = 30. Data in c–e are shown as bar plots, with the bar height indicating the mean value and the up line indicating the maximum value. The relative binding specificity in b–e is normalized to a range from 0 to 100 using min–max normalization of binding specificity scores.
Extended Data Fig. 3 Sequence diversity of the generated aptamer binders.
a, Clustering of the embeddings of InstructNA-generated strong aptamer binders and all the HT-SELEX candidates for the LOX1 protein target. The embeddings are clustered into ten clusters using GMM followed by t-SNE to reduce the embedding’s dimension to a 2D for visualization. b, Sequence similarity (%) between the strong aptamer binders generated by InstructNA (G1L, G2L, G3L and G4L) and screened from original HT-SELEX (T1L and T2L) for the LOX1 target. The red dotted line highlights that G1L has the lowest maximum sequence similarity to T1L and T2L, with a sequence similarity of as low as 38% to T1L. c, The superimposed 3D structures of T1L and G1L as predicted by AlphaFold3. d-e, Cartoon representations of the LOX1-T1L (d), and LOX1-G1L (e) complex structures through MD simulations. f, Hydrogen bonds formed between LOX1 and G1L. The LOX1, T1L and G1L are shown in light purple, blue and red colors, respectively.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, Z., Jiang, M., He, A. et al. De novo design of functional nucleic acids of aptamers. Nat Comput Sci (2026). https://doi.org/10.1038/s43588-026-00965-3
-
Received:
-
Accepted:
-
Published:
-
Version of record:
-
DOI: https://doi.org/10.1038/s43588-026-00965-3