
Introduction
Synthetic xeno-nucleic acids (XNAs) consist of unnatural bases (UBs) beyond the canonical A, T, C, G, and U found in naturally occurring DNA and RNA. Such non-canonical bases (NCBs) expand the information space of the genetic code, offer novel biochemical properties, and have opened up new fields in xenobiology, synthetic biology, and biotechnology. Exemplary XNAs include the bases isoG and isoC developed at the Swiss Federal Institute of Technology1, P and Z developed at the Foundation for Applied Molecular Evolution2,3, d5SICS and dNaM developed at the Scripps Research Institute4,5, Ds and Pa/Px developed at RIKEN6,7, and 7‑Deaza-2′-deoxyisoguanosine developed at the Center for Nanotechnology8. There are also naturally occurring NCBs which have been found in bacterial tRNAs9 or predicted to be part of the genomes of diverse viruses as a defense against host restriction systems9,10,11,12, though direct detection and characterization of their function has been hampered by the lack of appropriate sequencing technologies. The use of NCBs to expand the genetic alphabet has several applications in synthetic biology, including the development of sensors, aptamers, nanostructures, and semi-synthetic organisms13,14,15. With the advent of DNA storage as a paradigm to develop low-energy, ultra-high-density systems16,17, reading and writing NCBs has another potential application, however current approaches are limited in speed, nucleotide-resolution, and throughput.
A common early approach to detect NCBs (focusing on UBs) was to use Sanger sequencing and decipher UB positions through signal gaps stemming from the inability to sequence through these UBs6,7,18,19. An alternative approach has involved using PCR to replace UBs followed by sequencing and mutation analysis to detect them, but with the drawback of introducing unintended errors during amplification2,20. Both approaches suffer from the corresponding limitations of sample processing, which introduce sources of error and operational cost. Next-generation sequencing, while much less limited in throughput, largely faces similar challenges due to its inability to natively detect UBs21,22. In particular, the detection of UBs in specific sequence contexts or in the presence of multiple UBs can be challenging20,22. A potential alternative is direct single-molecule interrogation of nucleic acids with third-generation technology using nanopores, which have shown promise with canonical nucleic acid base modifications (e.g. methylated DNA23,24,25,26), though the ability to directly sequence XNAs, which contain a different nucleic acid backbone, has not been demonstrated27. Although PacBio is also a third-generation sequencing platform, its sequencing-by-synthesis process may need major adaptations with fluorescently labeled non-canonical bases to facilitate direct sequencing of XNAs.
A major obstacle for direct single-molecule sequencing of non-canonical bases is the ability to synthesize and utilize a sufficiently complex dataset for training basecalling models. In this study, we have addressed these challenges and developed technology that enables direct sequencing of XNAs using a widely available nanopore sequencing platform (Oxford Nanopore Technologies—ONT). Our results suggest that NCB-containing XNA templates can be robustly sequenced on an ONT MinION system, with yield similar to DNA controls ( > 2 million reads per flowcell), without notable truncation of reads, and with raw signals that are significantly different from canonical bases (median fold-change > 6×). To enable direct decoding of XNAs from these raw nanopore signals, we developed an XNA design and synthesis framework that generates a complex library of XNA oligonucleotides (n = 1024) containing all possible single-UB, 6-mer sequences, which provides the critical materials to train a basecaller model for XNAs. We have expanded the conventional basecaller architecture decoding states for it to be able to learn how to also decode the non-canonical bases X and Y, in addition to the canonical bases (A, T, C and G). Via our bootstrapped training approach, which retrieves additional accurate training data, and our read-splicing based data-augmentation technique, responsible for generating reads with high sequence context diversity, we show that a generalizable deep learning model based on convolutional neural networks (CNNs) can be trained to call non-canonical bases with high accuracy ( > 80%) and specificity (99%). Our work provides the first example of direct high-throughput deconvolution of non-canonical bases using nanopore sequencing, enabling the determination of nucleotide-level identities in XNAs needed for synthetic biology applications, and opening up the possibility that other classes of synthetic and natural nucleic acids may also be amenable to a bootstrapped and data-augmented learning approach.
Results
Analysis of XNAs on a high-throughput nanopore sequencer retains high fidelity
As a proof-of-concept to study the sequencing of NCB-containing templates, we leveraged the Px-Ds (X-Y) architecture, one of the three known classes of unnatural basepairs7 (Fig. 1A, Supplementary Fig. 1). The Px-Ds basepair system was designed taking into consideration hydrophobicity, shape, and electrostatic complementarity properties that enable the basepair to be efficiently amplified with PCR7, facilitating synthesis of XNAs with NCBs in diverse sequence contexts. For the initial experiment, we designed 20 individual templates with either a single NCB (XNA01-XNA12), multiple NCBs at varying distances (XNA13-XNA16), or four equidistant NCBs (XNA17-XNA20), flanked by stretches of canonical bases in different combinations and tagged via unique 24-nucleotide barcodes (Supplementary Fig. 2 and Supplementary Data 1).

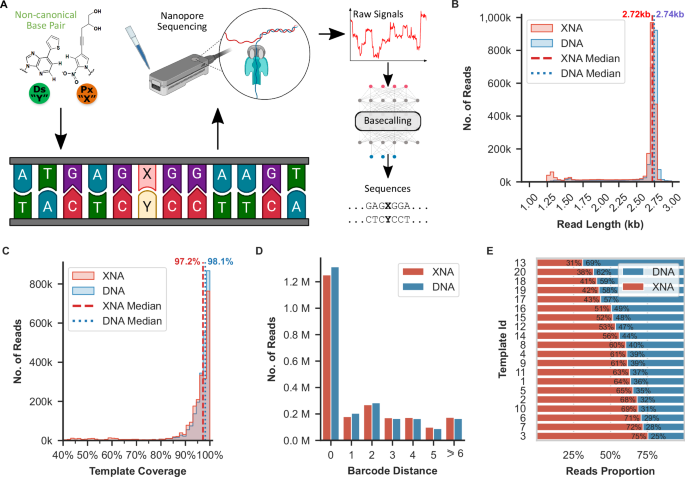
A Overview of workflow, showing the synthesis of XNA templates containing non-canonical basepairs (details in Supplementary Fig. 2), nanopore sequencing, signal-level analysis and direct deconvolution of canonical and non-canonical bases. B Histogram of read lengths for XNA and control DNA libraries (dotted lines indicate median values). C Histogram of template coverage of template-specific region for XNA and control DNA libraries (dotted lines indicate median values). D Bar chart with read counts as a function of barcode distance (computed as edit distance of sequenced barcode to the closest reference barcode). E Read proportion between XNA and DNA sequences for each template, sorted by XNA proportion. Source data are provided as a Source Data file.
We then used a commercially available high-throughput nanopore sequencer (ONT MinION) to analyze the XNA molecules and control DNA (NCB replaced by canonical base), initially using standard sequencing protocols and basecalling pipelines (Fig. 1A, “Methods”). The XNA sequencing process generated data continuously with no unexpected interruptions, with each flow cell yielding a total of >2 million raw reads ( > 5 Gbp of data), similar to yields from other sequencing runs with DNA templates28 (Supplementary Fig. 3). However, XNA containing libraries had lower throughput as a function of time relative to the DNA libraries (mean = 81%, 95% CI = 69–99%), potentially due to a higher number of pore blockage and saturation events (39% vs 27% for control DNA). Despite this, in libraries containing equal concentrations of DNA and XNA templates, no strong bias was observed against XNA templates, with similar numbers of XNA and DNA reads being sequenced and successfully aligned to reference templates (2.3 vs 2.4 million reads). Read lengths for XNAs and DNAs were also very similar (median length 2.7kbp), with a substantial fraction of XNA reads ( > 75%) providing near full-length ( ± 5%) template sequences (Fig. 1B). A slightly higher fraction of XNA reads were shorter (1.2kbp) than the DNA reads recovered, due to incomplete fusion PCR19 in the XNA synthesis process (Fig. 1B, “Methods”).
Alignment of reads to reference sequences provided similarly high coverage for XNA and DNA templates (median > 97%; Fig. 1C, “Methods”), though a higher fraction of XNA reads (33% vs 21%) exhibited incomplete coverage ( < 95%), potentially due to higher fragmentation rate or inability of conventional analytics to decode canonical bases correctly (particularly around NCBs). Demultiplexing successfully identified templates for a high proportion of both XNA and DNA reads (92.5% vs 93.1%, edit distance < 5 bp; Fig. 1D), while the distribution of XNA and DNA reads also did not show a strong bias across the barcodes/templates where they were pooled together for sequencing (Fig. 1E). In terms of basecalling accuracy, we noted that error rates were similar but slightly higher for canonical bases in XNA libraries versus control libraries (5% vs 3%; Supplementary Fig. 4), consistent with the expectation that the presence of NCBs could induce basecalling errors in related signals. Taken together, these results highlight the ability to generate signal data for XNAs with high fidelity on a high-throughput nanopore sequencer, despite the system not being designed for it, and correspondingly, the native basecaller’s inability to appropriately analyze NCB-containing signals.
Nanopore sequencing generates distinct electrical signals and reproducible errors near NCBs
After aligning reads to templates, we analyzed the corresponding raw electrical signal data measured by the nanopore sequencing device as a function of various sequence contexts (“Methods”). XNA signals showed NCB-specific divergence in contexts containing a single NCB (XNA01), as well as multiple NCBs that were close together (XNA13) or more spaced out (XNA16), and this pattern was seen for Ds as well as Px bases (upper and lower panels; Fig. 2A). However, the largest difference was not necessarily observed when the NCB was in the middle of the kmer (Fig. 2A), and specific patterns of divergence varied in different sequence contexts (Supplementary Figs. 5–7). Overall, the average signal-level difference between XNA and DNA control sequences was observed to be greater near NCB positions (±3 bp) than away from it (mean signal difference of 5.3 vs 0.7, median fold-change > 6×), and the impact of sequence context was also seen in the form of larger variance for the distribution (43 in NCB context vs 0.4 in canonical context, Fig. 2B). These results verify that the raw electrical signals generated from nanopore sequencing in the presence of NCB-containing XNAs can be notably distinct from those generated from an analogous DNA template.

A Average signal level comparison between XNA and control DNA reads for representative templates (other templates in Supplementary Figs. 5–7). Data are presented as mean values ± standard deviation. Vertical dashed lines indicate the location of the NCB, shaded areas indicate ±3 bp windows around the NCB, and asterisks (*) indicate Bonferroni adjusted two-sided Wilcoxon p-value < 0.05 and signal fold-change > 2%. B Boxplots showing signal differences between XNA and DNA libraries for a template in the context of NCBs (±3 bp window) and in canonical sequence contexts. C Signal level distributions based on read data for kmers containing NCBs and corresponding Nanopolish model distributions for kmers where the NCB is replaced by one of the canonical bases. D Boxplots showing average signal fold-change per template between read data for NCB kmers and corresponding Nanopolish model distributions for kmers where the NCB is replaced by one of the canonical bases. E Error rates per base position for selected templates with one or two NCBs. The NCB position is indicated by a vertical dashed line and shaded areas indicate 5 bp windows around the NCB. F Boxplots showing error rates as a function of distance from the non-canonical base across all templates (****: One-sided Wilcoxon p-value < 0.0001, *”:p-value < 0.05, ns: p-value > 0.05). P-values from distances 1 to ≥10 respectively: 9e–42, 6e–33, 1e–29, 3e–19, 3e–10, 0.02, 0.1, 0.2, 0.5 and 1. Sample sizes per distance are equally divided between XNA and DNA. Summary statistics were computed based on a sub-sampling of 5k reads per template and strand. Boxplots show first and third quartiles (box), median (line), and ±1.5× the interquartile range (whiskers). Source data are provided as a Source Data file.
Subsequently, we investigated if signal-level distributions for NCB-containing kmers were distinguishable from reference distributions for kmers containing each of the canonical bases, based on a nanopore signal model25. In general, we noted a pattern where XNA-containing kmers exhibited signal distributions with distinct modes compared to canonical bases in several (but not all) kmers surrounding a NCB (similar signals when NCB is at end of the kmer and generally larger differences when NCB is at the center, Fig. 2C). The fold-change per template between the non-canonical base and the mode for each canonical base had median value > 5% for both Px and Ds bases (Fig. 2D). These observations indicate that these NCBs are associated with electrical signal traces potentially distinguishable from all four canonical bases.
We next assessed the impact of signal-level differences on basecalling with the standard basecaller model and whether they introduced reproducible errors. In agreement with the signal-level comparisons (Fig. 2A), XNA basecalling error rates also diverged from control DNA basecalling error rates in contexts containing a NCB (Fig. 2E). Several templates exhibited very high error rates ( > 60%; Fig. 2F) in the neighborhood of a single NCB (XNA01) as well as when multiple NCBs were present at varying distances to each other (XNA13 and XNA16). Notably, in contexts where canonical bases are between NCBs that are moderately spaced apart ( > 5 bp), we observed only a marginal effect for basecalling of those bases (XNA16, <10% error rate; Fig. 2E), highlighting that the basecaller model can perform reasonably well even in regions flanked by NCBs.
Overall, in comparison to control sequences, the bases surrounding a NCB ( ± 5 bp) showed substantially higher basecalling error rates (mean of 18% vs 3%; Fig. 2F). The observed error rates are particularly high for the closest neighbors but quickly decrease with increasing distance away from the NCB (from 1 to 3 bp distance, median of 30%, 18% and 12%, respectively). In contrast, basecalling for positions far from the NCB was not impacted by its presence, obtaining error rates not significantly different from those for the control DNA templates ( > 6 bp; Fig. 2F). Finally, for most templates ( > 90%) the native basecaller repeatedly miscalled the same canonical base more than 40% of the time (Supplementary Fig. 8), emphasizing the consistency of the underlying signal data. These observations demonstrate that the performance of the standard basecaller is strongly and reproducibly impacted when deconvoluting NCB signals (i.e., bases affected by a NCB), while it is not impacted at positions sufficiently distant from NCBs, a feature that is ideal for bootstrapping the preparation of training data for learning a basecaller that can natively decode NCBs.
Generation of a complex template library to enable training of XNA basecaller models
To obtain a basecaller for XNAs (i.e. capable of natively basecalling canonical as well as non-canonical bases) we needed training data with enough reads and diversity of sequence contexts containing NCBs such that a neural network model with sufficient generalization capability could be developed. Since nanopore signal levels are known to be affected by the identity of at least the 6 adjacent bases in the pore during sequencing (Fig. 2A), we designed a complex library where a NCB is flanked by 5-mers of the same sequence identity on both sides (45 = 1024 templates containing N1N2N3N4N5-M-N1N2N3N4N5 sequence, where: Ni = A, T, G, or C; M = Ds or Px; Supplementary Fig. 2, Supplementary Data 2, 3), such that a rolling 6-nt window places the NCB in every position within the 6-mer resulting in all possible 6144 single-NCB 6-mer sequence contexts being represented. Synthesizing such a large library would have been infeasible by employing the chemical synthesis approach that is typically used for obtaining high-fidelity XNAs, which was used for the initial proof-of-concept library here (20 templates). We therefore designed and developed a cost-effective enzymatic synthesis scheme to generate this much larger complex library ( > 50× in size; Fig. 3A; “Methods”).

A Schematic overview of XNA synthesis process, where DNA fragments phosphorylated at the 5’-end and containing the template and barcode sequences undergo enzymatic single-nucleotide insertion of Ds at 3’-terminus. The resulting XNA fragments are then subjected to circular self-ligation followed by PCR using canonical and non-canonical base substrates to obtain the double-stranded XNA. B Figure depicting the two alternative strategies that were explored for enzymatic single-nucleotide insertion, using a 31-mer (Pa) and a 20-mer (mini-hairpin) template. C Gel electrophoresis results for single-nucleotide insertion products using the two alternative strategies. The experiment was performed once. The uncropped gel is provided as a Source Data file. D Boxplots depicting Ds insertion efficiency per pool of templates (N = 16) for the two alternative strategies. Insertion efficiencies were estimated via replacement PCR and Ion PGM sequencing. Boxplots show first and third quartiles (box), median (line), and ± 1.5× the interquartile range (whiskers). Source data are provided as a Source Data file.
The proposed scheme consists of enzymatically inserting the Ds base at the 3’-end of a fixed DNA sequence. For this insertion, we explored two methods using different templates (31-mer and 20-mer; Fig. 3B). In the first method, we used 31-mer templates containing the Pa nucleotide (more stable alternative to Px). However, only a moderate yield for the Ds-inserted product could be obtained from the reaction mixture (even after purification by denaturing gel electrophoresis; T-Temp31Pa; Fig. 3C). Subsequently, we designed a second method, using a 20-mer template with a mini-hairpin DNA sequence to stabilize the structure at the insertion target site (20-mer; Fig. 3B). This method resulted in higher Ds insertion efficiency (T-MH-Temp20; Fig. 3C).
To assess the insertion efficiency of the proposed enzymatic synthesis scheme, we estimated the purity of Ds insertion per pool of templates (Fig. 3D, “Methods”), where each pool was comprised of subsets of templates with the same first and fifth bases in the target sequence (N1N2N3N4N5, 16 = 42 pools with 64 = 43 fragments each, Supplementary Fig. 9). Considering the proportion of pools with high NCB retention rate ( > 85%), the 20-mer (mini-hairpin) method provided a substantial improvement over the 31-mer (Pa) method (median of 94% vs 67%; Fig. 3D). Overall, a great majority of XNA templates (91%) synthesized via the mini-hairpin method were estimated to have high-purity ( > 85%). These results emphasize how an enzymatic synthesis scheme with the right design can still enable generation of a substantially more complex template library reliably, and with purity high enough that it could enable effective training of a XNA basecaller model.
Bootstrapping and data augmentation enables training of an accurate generalized XNA basecaller
The complex template library was sequenced on a MinION sequencer, generating >6 million reads, with similar level of success as the proof-of-concept library in terms of long read lengths, near full-length template coverage and successful barcode identification (Supplementary Fig. 10). The distribution of reads across templates was found to be relatively similar, with hundreds of reads being available per template for most templates, after preprocessing and filtering of short reads (Supplementary Fig. 10, “Methods”). XNA training data was prepared by pre-processing reads selected for training by splitting them into signal chunks and attributing ground-truth sequence based on the library reference template sequence, discarding chunks that matched poorly to the reference (Supplementary Fig. 12, “Methods”). A significant fraction of reads was discarded in this way in an initial round of training (29%), as the baseline DNA model is prone to high error rates within NCB regions (Fig. 2E, F, Supplementary Table 1). Nevertheless, the resulting model was able to substantially improve basecalling in bases adjacent to the NCB (93.8%, Round 1) versus the baseline model (75.6%, Round 0; Fig. 4A). The updated basecaller model was used to repeat the pre-processing of reads, resulting in much fewer reads being discarded (11%), and retraining of a basecaller model with improved accuracy for calling NCBs and neighboring bases ( > 94% in Round 2; Fig. 4A). Further rounds of train data preparation and basecaller fine-tuning had limited impact on the number of reads added ( < 1%) and improvement of model performance (Round 3, Fig. 4A), indicating that one round of bootstrapping was sufficient to obtain an accurate model for the complex library.

A Line plot depicting base-level accuracy per round of training for non-canonical bases, canonical bases within 5 bp of a non-canonical base and more distant canonical bases. The baseline round of training depicts results using the Bonito Super Accuracy DNA model. B Line plots depicting performance on the proof-of-concept (POC) library using basecaller models trained with different sets of real and artificial reads, with increasing proportion of NCBs per read. The following figures are based on models trained with spliced data and 9% NCB proportion. C Line plots depicting the performance of models trained jointly for both NCBs or separately, with an increasing number of layers frozen. Subsequent analyses are based on models with 6 frozen layers. D Boxplots depicting performance of the final model, based on the complex and proof-of-concept libraries for testing with diverse templates and held-out reads. Boxplots show first and third quartiles (box), median (line), and ± 1.5× the interquartile range (whiskers). E Basecalling confusion matrix for NCBs and canonical bases. The last column represents deletion errors. F Barplots depicting performance analysis for reads with multiple NCBs. Data are presented as mean values ± standard deviation. NCB proximity, number and order did not seem to have a strong impact on NCB accuracy, indicating that the final trained basecaller is robust in handling reads with multiple NCBs. Source data are provided as a Source Data file.
Model training based directly on XNA reads from the complex library is expected to have low generalizability as the sequence context surrounding the NCB containing kmers is fixed. Correspondingly, testing on reads from the proof-of-concept 20-member library demonstrated low accuracy for both NCBs and canonical bases (5% NCB accuracy, 71% DNA accuracy; Complex Library, Fig. 4B). To overcome this limitation, we supplemented model training with different data augmentation approaches, allowing us to generate artificial reads with raw signals corresponding to NCB containing kmers placed in diverse sequence contexts. Initially, fully synthetic signals were simulated based on a signal-level model trained on real signal data from the complex library (Fully Synthetic, Supplementary Fig. 13, “Methods”). This approach notably improved NCB and DNA accuracy (24% and 89% respectively) relative to the model trained directly on the complex library, where the proportion of NCBs was kept constant (0.3%; Fully Synthetic, Fig. 4B). However, as DNA accuracy was lower than the baseline DNA model (94%), we investigated a hybrid approach containing a mixture of real DNA signals with simulated XNA signals that were generated by replacing specific positions in real DNA chunks with synthetic signals representing NCB kmers (Hybrid, Supplementary Fig. 13, “Methods”). This approach provided slightly lower NCB accuracy relative to the fully synthetic version, but higher DNA accuracy that matched the baseline model (14% and 94% respectively; Hybrid, Fig. 4B). The final strategy that was tested involved splicing real DNA and XNA signals together to generate artificial XNA reads with high context diversity while retaining the fidelity of real sequencing data (Spliced, Supplementary Figs. 13, 14, “Methods”). Training based on these spliced chunks resulted in substantial improvement in NCB accuracy without compromising DNA accuracy (34% and 94% respectively), indicating that it provides the most robust and generalizable approach for data augmentation and model training (Spliced, Fig. 4B).
Leveraging the flexibility of this approach for data augmentation, we next explored various ways to improve model performance. By increasing the proportion of NCBs in each generated read, in contrast to using a single NCB per read, substantial gains in NCB accuracy were noted ( ~ 2× to 64% at 4% NCB) with only a slight reduction in DNA accuracy ( ~ 1%; Fig. 4B). While NCB accuracy can be further improved, this comes with diminishing returns at higher NCB proportions (66% NCB accuracy and 90% DNA accuracy; trained with 11% NCB proportion, Fig. 4B). As baseline models are well-adapted to call DNA bases, we next explored a domain adaptation approach by preserving these pre-trained weights for some layers during fine-tuning (“Methods”). Successively freezing layers of the basecaller architecture up to layer 6 (out of 9; Supplementary Fig. 15) helped boost model accuracy without substantially altering DNA accuracy (70% NCB accuracy for XY model with 6 frozen layers; Fig. 4C). Not surprisingly, models trained separately for each of the NCBs achieved even higher accuracy (77% for X and 81% for Y), which could be leveraged for highly accurate joint basecalling when both strands of the XNA are sequenced in duplex mode (Fig. 4C).
Using the complex library to evaluate model performance across diverse sequence contexts further confirmed that high NCB accuracies are typically obtained by our model across templates (median accuracy = 86% and 82% for X and Y, and 73% for XY model; Fig. 4D). In addition, this comes with very high specificity (99%), and thus canonical bases are rarely miscalled as one of the NCBs (Fig. 4E). Instead, a significant proportion of the errors involve NCBs being missed (i.e. a base deletion), similar to what is seen in nanopore basecalling models for canonical bases as well (4–7%; Fig. 4E). In addition, NCBs are more likely to be miscalled to a canonical base, relative to the canonical-base-to-canonical-base miscall rate (2-6% vs 0–2%; Fig. 4E), suggesting that our models are conservative. Nevertheless, our models achieve good recall rates (77% and 81% for X and Y models, and 75% for XY model). Finally, we did not observe a strong influence from the order of NCBs in a template, or the number of canonical bases between consecutive NCBs, in terms of X and Y basecalling accuracy (Fig. 4F). Moreover, model performance was also comparable for templates containing multiple NCBs (including a 6-mer with two NCBs), despite these configurations not being present in the training data (Fig. 4F), highlighting the generalization capability of the model. Overall, these results highlight the ability to learn a high-accuracy generalized direct basecalling model for XNA sequences using bootstrapping and data augmentation to increase data complexity.
Discussion
In this work, we provide the first demonstration of direct high-throughput sequencing and deconvolution of non-canonical bases that can be readily performed on a commercial sequencer. This effectively expands the set of bases that can be natively read by nanopore sequencers paving the way for in-depth exploration of other classes of non-canonical bases as well29,30. The workflow used here can serve as a general template for this, including (i) systematic assessment of the ability of a nanopore sequencing system to process NCB containing templates with high fidelity (section 1), (ii) evaluation of the resulting signals for distinctness and reproducibility (section 2), (iii) synthesis of a complex training library using a mini-hairpin sequence to enable high efficiency (section 3), and (iv) training of a high-accuracy model using bootstrapping and signal splicing to boost data complexity (section 4). More specifically, our results highlight that ONT’s nanopore sequencing device can be applied out-of-the-box to XNAs containing multiple Ds-Px unnatural base pairs, processing them to generate raw electrical signals without significant disruption, and with only a slight reduction in throughput to produce >2 million reads and >5Gbp of data in a single MinION run (Supplementary Fig. 3). The resulting read sets were frequently near full-length ( > 75% within 5% of expected length) and showed no notable biases in data generation efficiency compared to their DNA controls (Fig. 1B–E). These results bode well for the use of nanopore architectures as versatile systems for natively reading and decoding diverse non-canonical bases.
The raw signal data, however, does not directly provide nucleotide base identities. To assess if it could be used for training a basecaller that could directly deconvolve signals into bases, we first systematically assessed the distinguishability, fidelity and reproducibility of the signal data. We found that the native basecaller was robust enough to produce DNA reads that were largely accurate, except in regions surrounding the NCB (Fig. 2F). This enables accurate alignment of signal data to reference bases, and consequently preparation of training data for machine learning with NCB containing XNA signals. Notably, signal data around a NCB ( ± 3 bp) is substantially different relatively to control DNA reads (median fold-change > 6×; Fig. 2A, B), and these differences are not apparent outside the NCB region. We further noted that the native basecaller makes reproducible errors in XNA containing reads indicating that the underlying signal patterns could be reproducibly re-interpreted with an appropriately trained basecaller (Supplementary Fig. 8).
Training a basecaller that directly deconvolves NCBs along with canonical bases requires training data that contains signals from XNA reads with known sequences in a sufficiently complex library. While chemical synthesis approaches have high efficiency, they can be prohibitively expensive for generating large-scale libraries. Enzymatic synthesis approaches are generally known to provide modest insertion efficiencies (7%-73%)30 at a lower cost. Here we demonstrated that a complex XNA library (n = 1024 templates) can be generated with high insertion efficiency ( > 85%) by designing a custom insertion template and a mini-hairpin to stabilize the NCB at the target site (Fig. 3C, D). A similar strategy could be useful for designing complex libraries for other NCBs as well. We chose to design our library to capture signals generated by all 6144 single-NCB 6-mers sequence contexts (Fig. 3A, Supplementary Fig. 2). This improves on previous work based on 4-mers30, and is expected to benefit model training by capturing greater sequence context diversity in critical bases impacting signal data31. In principle, generating data with 6-mers containing multiple and consecutive NCBs would also benefit model training. However, this is currently infeasible with even modest levels of efficiency7 and therefore, a limitation of our work is that we are currently unable to assess the capabilities of our model with such XNAs. Since directly sequencing XNAs containing consecutive NCBs would expand the utility of this model for synthetic biology applications, such as while creating XNA enzymes or genomes, we aspire to address such limitations when advances in the synthesis process allows the generation of data with such characteristics. Noteworthily, prior work using different classes of NCBs and a non-commercial nanopore demonstrated that having homopolymers with NCBs increases premature nanopore dissociation, resulting in many incomplete reads32. Thus, in order to successfully sequence NCB homopolymers, future adaptations to nanopore chemistry or the NCB structure itself might be required.
Despite the sequence diversity in the immediate neighborhood of the NCB in our complex library, the XNAs that we sequence still have a predictable sequence backbone (Supplementary Fig. 2). Not surprisingly, therefore, using this read data directly provides a high-accuracy model for reads from the library (94% for UBs; Fig. 4A), but does not generalize to other sequence contexts. Nevertheless, this initial model based on bootstrapped learning allows us to align signal data from the complex library to its corresponding reference bases accurately and thus enables further robust training. As generalizability is a common problem for training XNA models with limited training data complexity, we explored several strategies for data augmentation, including generating fully synthetic signals, as well as signals that splice combinations of real and synthetic data. Our results suggest that retaining signals from real XNA reads while embedding them in diverse DNA read contexts provides higher accuracy relative to synthetic data ( > 80%) and robust generalizability for the model (Fig. 4B–F). The models reported here also exhibit very high specificity (99%) relative to prior work30 (80–93%), while demonstrating the capability for the first time, to the best of our knowledge, to directly deconvolve signals and read NCBs and canonical bases in an XNA. Our strategy for data augmentation is versatile and can be adjusted to different requirements. For example, lower NCB proportions can be used when prioritizing DNA accuracy over NCB accuracy, while NCBs that were not synthesized together can be incorporated into the same artificial read to improve model accuracy for more densely packed NCBs and different classes of NCBs. Notably, data augmentation might be even more important as we seek to expand the alphabet of bases that can be directly read by nanopores 5,30, and the corresponding demands for library complexity go up rapidly.
With ongoing advances in nanopore sequencing technology, there are several opportunities to further enhance capabilities to sequence NCB-containing templates by directly optimizing the design of motor enzymes, pores and signal modalities (e.g. variable-voltage33). Additional measurements (e.g., duplex-basecalling, translocation time and backwards step) could also substantially aid in closing the gap between NCB and canonical basecalling accuracy. Since the signal trace corresponding to a particular nucleobase sequence greatly influences basecalling performance, a deeper understanding of the underlying physicochemistry could help to significantly improve basecalling of XNAs. This knowledge would enable the development of NCBs that produce electrical signals more distinct from those produced by canonical bases, thereby reducing or eliminating ambiguities that often cause sequencing errors. Advances in the deep learning architecture of basecallers can also be beneficial for modeling NCB signals, especially if future models become more generalizable without needing extensive amounts of training data.
High-throughput NCB sequencing can, in turn, pave the way for advances in various fields. For example, DNA storage applications can greatly benefit from the much higher information density that an expanded alphabet enables17,29. In synthetic biology, the development of new UBs, novel XNA synthesis methods, and the creation of synthetic genomes that exploit UBs can be transformed by the ability to directly sequence XNAs. Finally, the development of NCB-containing aptamers21,34 and other DNA therapeutics would benefit from easier access to direct XNA sequencing. More broadly, we hope that the proof-of-concept shown in this work further accelerates the use of nanopore sequencing techniques to interrogate the role of non-canonical basepairs across various domains of life, particularly for viral genomics11 as well as for sequencing tRNAs9.
Methods
Preparation of XNA fragments containing canonical and non-canonical bases
XNA fragments containing Ds or Diol-Pa were chemically synthesized with an automated DNA synthesizer (H-8 SE DNA/RNA synthesizer, K&A Laborgeraete) using Ds and Diol-Pa phosphoramidite prepared in-house6,35 and commercially available canonical base phosphoramidites (Glen Research), or purchased from Gene Design. In-house synthesized oligonucleotides were purified by denaturing gel electrophoresis after deprotection with concentrated ammonia solution. The non-canonical base substrates, dDsTP, dPxTP, and dPa’TP, were prepared in-house as described previously6,36. DNA fragments consisting of canonical base sequences were purchased from Integrated DNA Technologies (IDT) and used to prepare short and long XNAs (Supplementary Data 4).
Preparation of proof-of-concept 20-member XNA library
Using chemically synthesized DNA fragment sets, we first prepared short XNAs with barcode sequences, through primer extension in the presence of dPxTP and then PCR amplification in the presence of dDsTP and dPxTP as additional non-canonical base substrates, as described previously19. Combinations of Ds-containing templates and the barcode fragments are summarized in Supplementary Data 5. The presence of non-canonical bases at specific positions in the PCR-amplified short XNAs ( ~ 97% purity) was confirmed by an in-house Sanger gap sequencing method19. For nanopore sequencing analysis, we prepared long XNAs by fusion PCR as described previously19, in the presence of dDsTP and dPxTP, using three DNA blocks: short XNAs prepared as described above, left-arm DNA fragments, and right-arm DNA fragments. The presence of non-canonical bases at specific positions was confirmed by an in-house Sanger gap sequencing method19. The control long DNAs, where the non-canonical bases were replaced by canonical bases, were prepared by fusion PCR in the absence of non-canonical substrates, using the short DNA blocks with chemically-synthesized templates comprising of canonical base only (for XNA01–XNA16) or prepared by replacement PCR in the presence of dPa’TP22 (for XNA17–XNA20).
Preparation of complex XNA library
Ds insertion
Synthetic 90-mer oligonucleotide pools (oPoolsTM Oligo Pool, Supplementary Data 2,3 and Supplementary Fig. 9) were purchased from IDT, as mixtures of 50pmol fragments. DNA fragments with phosphorylation at the 5’-end (ordered with phosphorylation or phosphorylated by T4 DNA polynucleotide kinase followed by denaturing gel purification) were directly used for single-Ds nucleotide insertion by exo-nuclease-deficient Klenow Fragment DNA polymerase (KFexo-, New England Biolabs).
In the initial trial, each 90-mer DNA pool was annealed with the corresponding 31-mer template (5’-TG(Diol-Pa)NNNNNTATGGCAGCTGTTTCATGTGTGA-3’, N = A, G, C, or T), positioning Pa opposite the site intended for Ds insertion. The Ds insertion was performed through the incubation of the 90-mer DNA pool (2 μM) with 31-mer template (20 μM) in the presence of 100 μM dDsTP and 0.2U/μl KFexo- in the reaction buffer (25 mM Tris-HCl pH 7.5, 10 mM MgCl2, 1 mM DTT, 50 mM NaCl) for 60 min at room temperature. The inserted products were purified by denaturing PAGE. However, we found that Ds insertion rate was relatively low, so we designed a template for Ds insertion using a 20-mer containing a mini-hairpin DNA sequence at 5’ terminus, X-MH-temp20 (5’- GCGAAGCGTNNNNNTATGG(ddC)-3’, N = A, G, C, or T, underlined: mini-hairpin sequence). The design was based on three points: (1) the mini-hairpin sequence at the 5’-terminus allowed specific and stabilized hybridization to the 3’-region of 90-mer pool, (2) the use of 2’,3’-dideoxy C at the 3’-terminus prevents additional Ds insertion, and (3) Ds insertion opposite T is allowed since we add only dDsTP (Fig. 3A, B). As expected, the Ds insertion in the presence of the 20-mer template was much improved compared with that in the presence of the 31-mer (Fig. 3C, D). Finally, the Ds insertion to 90-mer pool was performed through the incubation of the 90-mer DNA pool with 20-mer template (75 μM) in the presence of 100 μM dDsTP and 0.5U/μl KFexo- in the reaction buffer for 30 min at 37 °C.
Circligation
The prepared Ds-inserted DNA pools (around 1.5 to 2.5 pmol) were subjected to self-ligation (10 μl) by 50 Units of Circligase II (Lucigen) in the circligation buffer supplemented with 2.5 mM MnCl2 and 1 M Betain for 16 h at 60 °C, followed by heat inactivation of the ligase at 80 °C for 10 min. The unligated fragments were removed by treatment with Exonuclease I (NEB) and Exonuclease III (NEB) at 37 °C for 45 min, followed by heat inactivation of the nucleases at 80 °C for 20 min.
Preparation of short and long XNA pools
The reaction aliquots were subjected to 12-cycle PCR amplification in the presence of dDsTP and dPxTP, using two primers, pUC19rev-47 and pUC19fwd-42. The PCR-amplified products (short XNA pools) corresponding to 135-bp (without Ds-Px) /136-bp (with Ds-Px) were purified by denaturing PAGE, followed by fusion PCR to prepare long XNAs, as described above. The presence of non-canonical bases at specific positions in the PCR-amplified short/long XNA pools were confirmed by an in-house Sanger sequencing method supplemented with ddPa’TP19 or Ion PGM deep sequencing after replacing the non-canonical bases with canonical bases22.
Nanopore library preparation and sequencing
XNA library samples were sequenced on Oxford Nanopore Technologies (ONT) MinION Mk1B or Mk1C devices using MinION R9.4.1 flow cells. These samples were prepared using the ONT Ligation Sequencing Kit (SQK-LSK109 or SQK-LSK110) following standard library preparation protocol instructions provided by the vendor. The nucleic acid concentration of each sample was measured using the QubitTM dsDNA HS kit on a QubitTM 3 Fluorometer (Invitrogen).
In total, we ran five different synthesis and sequencing batches: 1) XNA + DNA #01-16; 2) XNA #17-20; 3) DNA #17-20; 4) Complex library; 5) Complex library subset (Supplementary Fig. 2 and Supplementary Fig. 9). Run #5 was needed to replenish some of the templates (n = 214) with low read counts in run #4.
Nanopore data pre-processing
To prepare XNA sequence reads for raw nanopore signal analysis and basecaller training, we used Guppy (v4.0.14) with the dna_r9.4.1_450bps_hac profile for initial basecalling. The output basecalled sequences were then aligned to template references using Minimap237 (v2.17-r941; parameters -x map-ont -c -secondary = no -w 5). For alignment, we used only template-specific regions as references, i.e., the sequences corresponding to the left and right arms were omitted from the reference targets, as this helped to avoid misalignment to the wrong target. After alignment, the barcode region was located and used to compute the number of mismatches with the reference barcode to obtain edit distances. Reads with low template coverage ( < 85%), high barcode distance ( > 5) or unexpected read length ( < 1k or >3k) were filtered out, before counting the final number of reads per template (Supplementary Fig. 11). Nanopolish25 (v0.13.0; parameters eventalign -samples -scale-events -print-read-names) was used to obtain the mapping between read bases and raw signals.
XNA training data preparation
We randomly sampled 316k reads from the complex XNA library sequencing batches, of which 297k reads were used for training and 19k reads for validation, with respective medians of 170 and 10 reads per template and strand. We prepared the input for training the basecaller by first dividing the raw signals into chunks with a chunk size of 3600 and an overlap of 900 signal points. Each chunk was then basecalled and mapped to the library reference to retrieve the ground-truth sequence. We retained only the chunks that contained an NCB and had at least 90% coverage and accuracy relative to the aligned reference (Supplementary Table 1). Each chunk of signals ({{bf{S}}}={{S}_{1},ldots,{S}_{{chunksize}}}) was normalized via SMAD (Scaled Median Absolute Deviation) normalization38, as defined below:
$${{rm{smad}}}left({{bf{S}}}right)=k*{{rm{median}}}(left|{S}_{i}-{{rm{median}}}left({{boldsymbol{S}}}right)right|)$$
(1)
$${{{bf{S}}}}_{{{bf{norm}}}}=frac{{{{bf{S}}}}_{{{bf{raw}}}}-{{rm{median}}}left({{{bf{S}}}}_{{{bf{raw}}}}right)}{{{rm{smad}}}({{{bf{S}}}}_{{{bf{raw}}}})}$$
(2)
where, for consistency with the Gaussian distribution, we used 1.4826 as the scale factor (k). We employed SMAD normalization to comply with Bonito’s signal pre-processing step before basecalling using its pre-trained models.
XNA basecaller architecture and training
We used Bonito39 (v0.5.0) Super Accuracy (SUP) as the backbone architecture for our XNA basecaller models. We chose Bonito for the backbone because it is the state-of-the-art in terms of basecalling accuracy40, and ONT’s recommended basecaller for training new models and method development. We made certain adaptations to Bonito’s implementation and basecaller architecture (Supplementary Fig. 15) to enable training with data containing a six-letter alphabet. The baseline model, trained solely on DNA data, was used as the default set of initial weights for fine-tuning with the XNA data. After round 1, rounds 2 and 3 of the bootstrapping strategy used the weights from the previous round to extract training chunks and initialize the model for fine-tuning (Fig. 4A). Unless otherwise specified, we fine-tuned the weights from all layers. However, in some cases, we partially froze the network and only fine-tuned the weights from the top layers (Fig. 4C). We fine-tuned the network for 5 epochs with a learning rate of 5e-4, applying dropout rates of 50% to the top layer and 5% to any remaining unfrozen layers.
Performance evaluation
To comprehensively evaluate our basecaller models, we assessed their performance on both XNA libraries (proof-of-concept and complex). For performance evaluation over the proof-of-concept XNA library, we randomly sampled 10k reads, consisting of 250 reads per NCB and template (Supplementary Fig. 16). For the complex library, we randomly sampled 40k reads (median of 20 reads per NCB and template). Each model was used to basecall the two test sets, and evaluation was conducted on the reads successfully aligned to reference templates. To evaluate how well our models can basecall canonical bases (ACGT) and NCBs (XY), we calculated base-level accuracy and reported the average accuracy per template and strand for each group of bases (canonical and non-canonical). We categorized canonical bases within 5 bases of an NCB into a separate group (i.e., NCB-adjacent bases) due to their susceptibility to the influence of the NCB at the signal level and, consequently, in terms of basecalling. We computed recall and specificity metrics considering NCBs as positives and canonical bases as negatives.
Refined signal-to-sequence alignment
We used Nanopolish on the reads from the complex XNA library to obtain an initial signal-to-sequence alignment, which was subsequently refined. In order to obtain an alignment with higher quality, we used the sequences basecalled by the XNA model from round 2 of the bootstrapped training procedure, and we substituted the NCBs in the reference sequences with the canonical base that minimized the average standard deviation per kmer (A for both X and Y). We filtered out reads with alignments that have coverage <75%, a position with signal count >50, an average variance per position >20 or a signal match score >2. We also computed an alternative score by matching the signals to a reference sequence without the NCB, representing the mis-insertion case, and discarded the reads where the difference between the alternative and original scores was less than −0.25, capping discarded reads at 15% of the total. The signal match score for a given sequence of signals grouped by kmer is the average z-score based on kmer model mean and standard-deviation and is defined as:
$${{rm{score}}}=frac{{sum }_{i}^{L}left|frac{{hat{{{rm{mu }}}}}_{{ki}}-{{{rm{mu }}}}_{k}}{{sigma }_{k}}right|}{L}$$
(3)
where L is the sequence length, ({hat{upmu }}_{{ki}}) is the observed mean signal level for kmer k at position i of the sequence, ({upmu }_{{k}}) is the model mean signal level for kmer k and ({sigma }_{k}) the model standard deviation. We used the kmer signal model parameters employed by Nanopolish.
Since Nanopolish was designed solely for canonical bases, the output alignment is not very accurate around non-canonical bases, necessitating additional steps to improve it. After filtering the reads, we employed dynamic time warping (DTW)41 in two iterations to refine the signal-to-sequence alignment in the region comprising the NCB kmers and the 5 kmers before and after. Initially, we substituted both the NCBs with the canonical base A (since it minimized kmer standard deviation for both X and Y) and used the model mean signal level as the reference signal for each kmer. Subsequently, we updated the reference corresponding to the NCB kmers with the observed mean signal level, computed from the signals aggregated across all reads after the first iteration. We ran the DTW algorithm with Euclidean distance and an asymmetric step pattern, which ensures that every signal is always matched to a single kmer. Additionally, to enforce that each kmer would be matched to at least three signals, we repeated each reference signal three times.
Synthetic signal generation
We simulated nanopore signals using the modeled signal distribution (mean and standard deviation) of each 6-mer. For DNA kmers, we used the model available with the Nanopolish tool. However, for XNA kmers, we had to generate our own model covering the NCBs explored in this work (Px-Ds). We modeled the XNA kmers by first applying our refined signal-to-sequence alignment on the sequenced reads from the complex XNA library. We then grouped the observed XNA signals per 6-mer, removed outliers ((pm 3times {{rm{stdev}}})), and extracted the mean and standard deviation from the remaining samples (Supplementary Data 6).
Using the 6-mer signal model for DNA and XNA, we simulated the signals for a specific kmer by sampling from a truncated normal distribution, with the kmer model mean and standard deviation as parameters, and then adding Gaussian noise42. The generation of a synthetic signal ({widetilde{s}}_{k}) for kmer k is defined as follows:
$${widetilde{s}}_{k}={{{rm{mu }}}}_{k}+{sigma }_{k}times {{rm{truncnorm}}}left(-omega+Delta,omega+Delta right)+{{rm{noise}}}$$
(4)
where (omega) and (Delta) are the window size and shift value, respectively, for defining the truncation lower and upper bounds. We used a window size of 1.5 and a shift value randomly chosen from -0.5, 0 and 0.5. The Gaussian noise was also sampled from a truncated normal distribution, with a standard deviation scale randomly selected within the range of 0 to 1 and lower and upper bounds of (pm 3). The random parameters (window shift and noise standard deviation) vary between reads but are chosen once and fixed for each read.
Fully synthetic XNA chunks
We used the DNA training data from Bonito as the source for the reference sequences for which we generated fully synthetic nanopore reads. Adhering to a target NCB proportion for the simulated XNA reads, we randomly selected multiple positions from the source reference sequence to be replaced by X or Y, ensuring that no 6-mer contained more than one NCB by maintaining a minimum of 5 bases between selections. We also used Bonito’s data to determine the number of signal points each kmer instance should have. To estimate this information, we segmented the chunk of raw signals corresponding to the source sequence into kmers. Finally, we normalized the resulting chunk of signals using SMAD normalization.
Hybrid XNA chunks
We created hybrid reads by replacing specific signals in DNA chunks from Bonito training data with synthetically generated XNA signals. Given a DNA chunk and its reference sequence, we selected multiple positions from the sequence to be replaced by NCBs and then substituted the signals corresponding to these positions with nanopore simulations for the NCB kmers resulting from the replacement. To determine which signals correspond to the replaced positions, we segmented the DNA signal chunks into kmer instances employing DTW, with the model mean signal level serving as the reference signal for each kmer. Similar to the final step of our refined signal-to-sequence alignment, we ran the DTW algorithm with Euclidean distance, an asymmetric step pattern, and repeating each reference signal three times.
Spliced XNA chunks
To achieve more diverse sequence contexts while maintaining high nanopore signal fidelity, we assembled spliced reads by replacing specific signals in DNA chunks from Bonito training data with XNA signal slices extracted from our nanopore XNA reads. We used the chunks from the complex XNA library, obtained with the basecaller model from round 2 of bootstrapped training, as the source for the signal slices for NCB kmers. The refined signal-to-sequence alignment approach was used to identify XNA signal slices that likely contain an NCB. To extract the XNA slices, we kmer-segmented the chunks employing DTW with our XNA kmer model. When selecting which XNA slices will be used to substitute the DNA signals, we randomly sampled five slices and then selected the one with the number of signals closest to the length of the insertion region. If the selected XNA slice was longer than the insertion region, we removed evenly spaced signal points as needed. Conversely, if the slice was shorter, we used linear interpolation to add evenly spaced signal points.
Statistics and reproducibility
No statistical method was used to predetermine sample size. The Investigators were not blinded to allocation during experiments and outcome assessment. All statistical analyses performed are described in the figure legends or “Methods” section. The exclusion criteria for aligned reads and training signal chunks are described in the Methods section. These data were excluded to eliminate noisy samples that might mislead the analyses and compromise basecaller performance. Signal and error rate analyses presented in Fig. 2 were conducted on a randomly selected sample of reads. Training and validation sets were randomly sampled from reads in the complex XNA library. Test sets were randomly sampled from reads in the proof-of-concept and complex XNA libraries. Reads sampled for the test and validation sets were randomly selected from a pool of reads with high template coverage and low barcode distance. The input order of the training data was randomized during training, which can lead to minor variations in final performance. The data augmentation techniques proposed in this work include random components (described in the Methods section), which are controlled by a hard-coded random seed in the provided code (see the ‘Code Availability’ statement).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Nanopore sequencing data is available from the European Nucleotide Archive (ENA—https://www.ebi.ac.uk/ena/browser/home) under project accession number PRJEB82716. Source data for figures are provided with this paper. Source data are provided with this paper.
Code availability
Source code for scripts used to obtain the XNA basecaller and analyze the data are available in a GitHub project at https://github.com/CSB5/XNA_Basecaller43.
References
-
Switzer, C., Moroney, S. E. & Benner, S. A. Enzymatic incorporation of a new base pair into DNA and RNA. J. Am. Chem. Soc. 111, 8322–8323 (1989).
-
Yang, Z., Chen, F., Alvarado, J. B. & Benner, S. A. Amplification, mutation, and sequencing of a six-letter synthetic genetic system. J. Am. Chem. Soc. 133, 15105–15112 (2011).
-
Hoshika, S. et al. Hachimoji DNA and RNA: a genetic system with eight building blocks. Science 363, 884–887 (2019).
-
Malyshev, D. A., Seo, Y. J., Ordoukhanian, P. & Romesberg, F. E. PCR with an expanded genetic alphabet. J. Am. Chem. Soc. 131, 14620–14621 (2009).
-
Li, L. et al. Natural-like replication of an unnatural base pair for the expansion of the genetic alphabet and biotechnology applications. J. Am. Chem. Soc. 136, 826–829 (2014).
-
Hirao, I. et al. An unnatural hydrophobic base pair system: site-specific incorporation of nucleotide analogs into DNA and RNA. Nat. Methods 3, 729–735 (2006).
-
Kimoto, M., Kawai, R., Mitsui, T., Yokoyama, S. & Hirao, I. An unnatural base pair system for efficient PCR amplification and functionalization of DNA molecules. Nucleic Acids Res 37, e14–e14 (2009).
-
Xia, Z., Kondhare, D., Budow-Busse, S., Leonard, P. & Seela, F. 7-Deaza-2′-deoxyisoguanosine, a noncanonical nucleoside for nucleic acid code expansion and new DNA constructs: nucleobase functionalization of inverse watson–crick and purine–purine base pairs. Bioconjug. Chem. 35, 1233–1250 (2024).
-
Hutinet, G. et al. 7-Deazaguanine modifications protect phage DNA from host restriction systems. Nat. Commun. 10, 5442 (2019).
-
Kirnos, M. D., Khudyakov, I. Y., Alexandrushkina, N. I. & Vanyushin, B. F. 2-Aminoadenine is an adenine substituting for a base in S-2L cyanophage DNA. Nature 270, 369–370 (1977).
-
Zhou, Y. et al. A widespread pathway for substitution of adenine by diaminopurine in phage genomes. Science 372, 512–516 (2021).
-
Pezo, V. et al. Noncanonical DNA polymerization by aminoadenine-based siphoviruses. Science 372, 520–524 (2021).
-
McKenzie, L. K., El-Khoury, R., Thorpe, J. D., Damha, M. J. & Hollenstein, M. Recent progress in non-native nucleic acid modifications. Chem. Soc. Rev. 50, 5126–5164 (2021).
-
Kimoto, M., Cox, R. S. & Hirao, I. Unnatural base pair systems for sensing and diagnostic applications. Expert Rev. Mol. Diagn. 11, 321–331 (2011).
-
Zhang, Y. et al. A semi-synthetic organism that stores and retrieves increased genetic information. Nature 551, 644–647 (2017).
-
Goldman, N. et al. Towards practical, high-capacity, low-maintenance information storage in synthesized DNA. Nature 494, 77–80 (2013).
-
Lim, C. K., Nirantar, S., Yew, W. S. & Poh, C. L. Novel modalities in DNA data storage. Trends Biotechnol. 39, 990–1003 (2021).
-
Malyshev, D. A. et al. A semi-synthetic organism with an expanded genetic alphabet. Nature 509, 385–388 (2014).
-
Kimoto, M., Soh, S. H. G. & Hirao, I. Sanger gap sequencing for genetic alphabet expansion of DNA. ChemBioChem 21, 2287–2296 (2020).
-
Kimoto, M. & Hirao, I. Genetic alphabet expansion technology by creating unnatural base pairs. Chem. Soc. Rev. 49, 7602–7626 (2020).
-
Kimoto, M., Yamashige, R., Matsunaga, K., Yokoyama, S. & Hirao, I. Generation of high-affinity DNA aptamers using an expanded genetic alphabet. Nat. Biotechnol. 31, 453–457 (2013).
-
Hamashima, K., Soong, Y. T., Matsunaga, K., Kimoto, M. & Hirao, I. DNA sequencing method including unnatural bases for DNA aptamer generation by genetic alphabet expansion. ACS Synth. Biol. 8, 1401–1410 (2019).
-
Liu, Y. et al. DNA methylation-calling tools for Oxford Nanopore sequencing: a survey and human epigenome-wide evaluation. Genome Biol. 22, 295 (2021).
-
Nguyen, T. A. et al. Direct identification of A-to-I editing sites with nanopore native RNA sequencing. Nat. Methods 19, 833–844 (2022).
-
Simpson, J. T. et al. Detecting DNA cytosine methylation using nanopore sequencing. Nat. Methods 14, 407–410 (2017).
-
Zhang, Y.-Z. et al. On the application of BERT models for nanopore methylation detection. In IEEE International Conference on Bioinformatics and Biomedicine (BIBM) 320–327 (IEEE, 2021).
-
Ledbetter, M. P. et al. Nanopore Sequencing of an Expanded Genetic Alphabet Reveals High-Fidelity Replication of a Predominantly Hydrophobic Unnatural Base Pair. J. Am. Chem. Soc. 142, 2110–2114 (2020).
-
Wang, Y., Zhao, Y., Bollas, A., Wang, Y. & Au, K. F. Nanopore sequencing technology, bioinformatics and applications. Nat. Biotechnol. 39, 1348–1365 (2021).
-
Tabatabaei, S. K. et al. Expanding the molecular alphabet of DNA-based data storage systems with neural network nanopore readout processing. Nano Lett. 22, 1905–1914 (2022).
-
Kawabe, H. et al. Enzymatic synthesis and nanopore sequencing of 12-letter supernumerary DNA. Nat. Commun. 14, 6820 (2023).
-
Rang, F. J., Kloosterman, W. P. & de Ridder, J. From squiggle to basepair: computational approaches for improving nanopore sequencing read accuracy. Genome Biol. 19, 90 (2018).
-
Thomas, C. A. et al. Assessing readability of an 8-letter expanded deoxyribonucleic acid alphabet with nanopores. J. Am. Chem. Soc. 145, 8560–8568 (2023).
-
Noakes, M. T. et al. Increasing the accuracy of nanopore DNA sequencing using a time-varying cross-membrane voltage. Nat. Biotechnol. 37, 651–656 (2019).
-
Kimoto, M., Matsunaga, K. & Hirao, I. DNA Aptamer Generation by Genetic Alphabet Expansion SELEX (ExSELEX) Using an Unnatural Base Pair System. in Methods in Molecular Biology book series (MIMB, volume 1380) 47–60 (2016).
-
Kimoto, M., Soh, S. H. G., Tan, H. P., Okamoto, I. & Hirao, I. Cognate base-pair selectivity of hydrophobic unnatural bases in DNA ligation by T4 DNA ligase. Biopolymers 112, e23407 (2021).
-
Yamashige, R. et al. Highly specific unnatural base pair systems as a third base pair for PCR amplification. Nucleic Acids Res. 40, 2793–2806 (2012).
-
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
-
Stoiber, M. H. et al. De novo Identification of DNA Modifications Enabled by Genome-Guided Nanopore Signal Processing. Preprint at bioRxiv https://doi.org/10.1101/094672 (2016).
-
Seymour, C. & Oxford Nanopore Technologies Ltd. Bonito: a PyTorch Basecaller for Oxford Nanopore reads. https://github.com/nanoporetech/bonito (2019).
-
Pagès-Gallego, M. & de Ridder, J. Comprehensive benchmark and architectural analysis of deep learning models for nanopore sequencing basecalling. Genome Biol. 24, 1–18 (2023).
-
Giorgino, T. Computing and Visualizing Dynamic Time Warping Alignments in R: The dtw Package. J. Stat. Softw. 31, 1–24 (2009).
-
Gamaarachchi, H., Ferguson, J. M., Samarakoon, H., Liyanage, K. & Deveson, I. W. Simulation of nanopore sequencing signal data with tunable parameters. Genome Res. 34, 778–783 (2024).
-
Perez, M. et al. Direct high-throughput deconvolution of non-canonical bases via nanopore sequencing and bootstrapped learning. Zenodo https://doi.org/10.5281/zenodo.15751758 (2025).
Acknowledgments
This work was funded by the Advanced Manufacturing and Engineering (AME) Programmatic grant A18A9b0060 (to I.H., W.L.C., N.N.). Additional funding supporting manpower came from Singapore Ministry of Health’s National Medical Research Council under its Open Fund—Individual Research Grants (NMRC/OFIRG/MOH-000649-00 to N.N.), Agency for Science, Technology and Research (A*STAR)’s Manufacturing, Trade And Connectivity (MTC) Individual Research Grant (M24N7c0088 to W.L.C), and the Program for Research in Epidemic Preparedness and Response (PREPARE), under its Joint Strategic Open Grant Call (Environmental Transmission & Mitigation and Diagnostics Co-operatives; PREPARE-OC-ETM-Dx-2023-006 (to N.N.) and PREPARE-OC-ETM-Dx-2023-005 (to W.L.C.)). This research was supported by a National Research Foundation Investigatorship grant NRFI09-0015 (to N.N.). Additional compute resources were provided by the A*STAR Computational Resource Center through the use of its high-performance computing facilities. The authors acknowledge program management and helpful discussions with Hong-Ting Prekop. Figure 1A was created in BioRender; Suphavilai, C. (2025) https://BioRender.com/c423rgo.
Ethics declarations
Competing interests
M.H., H.P.T., and I.H. declare competing interests in Xenolis Pte Ltd, a company commercializing XNAs. N.T.X.O. declares a competing interest in current employment with Oxford Nanopore Technologies; all contributions in this study were provided prior to this employment. The other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Perez, M., Kimoto, M., Rajakumar, P. et al. Direct high-throughput deconvolution of non-canonical bases via nanopore sequencing and bootstrapped learning. Nat Commun 16, 6980 (2025). https://doi.org/10.1038/s41467-025-62347-z
-
Received:
-
Accepted:
-
Published:
-
DOI: https://doi.org/10.1038/s41467-025-62347-z
