Introduction
Tuberculosis (TB) is a major infectious disease caused by Mtb. Over the past few decades, millions of people have lost their lives to TB-related illnesses, far exceeding the total number of deaths caused by malaria and HIV combined1. Despite Bacillus Calmette-Guérin (BCG) being the current TB vaccine and widely administered, TB remains a serious and deadly infectious disease2. In many regions worldwide, multidrug-resistant strains of Mtb are emerging with increasing frequency, emphasizing the urgent need for the development of an effective vaccine to halt the spread and transmission of TB3.
Although BCG vaccination in infancy confers significant initial protection, its efficacy during adolescence and adulthood is not consistently reliable4,5. As a live vaccine, BCG’s efficacy relies on its replication within the host to produce a sustained immune response. In addition to factors such as climatic conditions, geographic latitude, the host’s genetic background, and the specific strain of BCG6,7, its effectiveness may be compromised by cross-reactive immune responses induced by environmental nontuberculous Mycobacteria (NTM), since shared Mycobacterial antigens elicit pre-existing immunity that inhibits BCG replication, thereby reducing its protective efficacy8. In contrast, candidate vaccines based on non-Mycobacterial vectors, such as recombinant proteins or virus-like particles, lack pre-sensitizing activity and are thus free from these constraints, offering clear advantages and providing effective host protection9,10.
Recombinant subunit vaccines, recognized for their effectiveness and safety, have been extensively investigated in developing novel TB vaccines. Four recombinant subunit vaccines, M72/AS01E, H56:IC31, ID93 + GLA-SE and GamTBvac, currently in clinical trials, incorporate a total of 11 Mycobacterial antigens in different combinations and formulations5,11. Notably, in a Phase IIb trial, GlaxoSmithKline’s (GSK’s) M72/AS01E vaccine—comprising Mtb32A and Mtb39A antigens combined with the AS01E adjuvant—demonstrated a 49.7% efficacy, thereby meeting the World Health Organization’s criteria for TB vaccines12. This outcome not only shows promise in preventing latent TB in adults but also revitalizes the prospects for TB subunit vaccine development12,13. While antibodies are essential for protection against many pathogens, there is no definitive evidence supporting their critical role in protection against Mtb14. Consequently, TB vaccine development has focused on enhancing T-cell-mediated immune responses, particularly through the activation of CD4⁺ and CD8⁺ T cells, along with cytokines like IFN-γ, IL-2 and TNF-α, which are key contributors to immune protection15. The antigen-specific T-cell immune responses elicited by advanced TB subunit vaccines typically require activation of the non-specific innate immune system through adjuvant components, which initiate host immunity by recognizing pathogen-associated and damage-associated molecular patterns via receptors such as Toll-like receptors (TLRs), NOD-like receptors (NLRs) and RIG-I-like receptors (RLRs). Based on these specific vaccine development characteristics, the subunit vaccines M72/AS01E, H56:IC31, GamTBvac and ID93/GLA-SE, which utilize adjuvants targeting TLR-4, TLR-9, TLR-9, and TLR-4, respectively16,17,18,19, primarily induce Th1, Th17, NK cell and CTL responses, demonstrating significant protective efficacy in current clinical trials16,20,21,22.
Nanoparticles (NPs), broadly defined as synthetic or naturally derived particulate structures typically 1–100 nm in diameter, encompass diverse platforms such as virus-like particles (VLPs), viral vectors, lipid nanoparticles (LNPs), liposomes, cationic polymers, inorganic materials (e.g., gold or silica), and self-assembling protein nanoparticles (e.g., ferritin, AP205, i53-50, and mi3)23,24,25. These systems offer distinct advantages in antigen delivery, structural stability and immunogenicity enhancement for vaccine development. Nanoparticle (NP) vaccines constructed based on the self-assembling structures of virus-like particles (VLPs) are composed of viral structural proteins, such as capsid proteins, and are capable of recapitulating the size and morphology of native virions while lacking pathogenic genetic material, thereby achieving potent immunogenicity and excellent safety profiles. The intrinsic ability of VLPs to form highly ordered, repetitive polyhedral structures enables efficient stimulation of both innate and adaptive immune responses24,26. Nanoparticles (NPs) based on virus-like particles (VLPs) have been shown to elicit stronger humoral and cellular immune responses compared to recombinant protein vaccines, as demonstrated in multiple clinical studies27,28,29. This enhanced immunogenicity has been exemplified by VLP-based vaccines for hepatitis B virus (HBV) and human papillomavirus (HPV), which have achieved over 90% protection in clinical trials, representing a significant breakthrough in vaccinology30,31,32,33. Similarly, the R21 malaria vaccine, a chimeric nanoparticle comprising hepatitis B surface antigen (HBsAg) and malaria circumsporozoite protein (CSP), demonstrated 77% efficacy in infants, offering substantial promise for malaria control in endemic regions34,35,36. These outcomes highlight the favorable biological properties of VLP-based platforms, including high immunogenicity, structural stability, and the absence of infectious components, which collectively position them as promising candidates for next-generation vaccine development37,38. Nanoparticle (NP) scaffolds such as ferritin, I53-50, and mi3 have been employed to effectively display viral antigens, including the SARS-CoV-2 spike protein and its receptor-binding domain (RBD), and have demonstrated robust protective efficacy in multiple preclinical studies39,40. Achieving effective antigen display on the surface of NPs is critical for eliciting optimal immune responses and often relies on either chemical conjugation or bioconjugation strategies. Among these, bioconjugation has garnered increasing attention owing to its advantages, such as eliminating the need for toxic chemical reagents and simplifying the manufacturing process. A prominent example is the SpyTag/SpyCatcher covalent bioconjugation system, derived from cleavage fragments of the fibronectin-binding protein FbaB of Streptococcus pyogenes41, which has been widely applied in the development of NP-based vaccines and has exhibited exceptional efficacy in clinical evaluations42,43.
Although tuberculosis (TB) nanoparticle vaccines have garnered increasing attention44, research in this field remains comparatively limited relative to nanoparticle vaccines targeting viral pathogens such as varicella-zoster virus (VZV), SARS-CoV-2 and respiratory syncytial virus (RSV). Two self-assembling TB nanoparticle vaccines, based on the Hepatitis B virus core (HBc) protein44,45 and displaying either Early Secreted Antigenic Target (ESAT-6) or Culture Filtrate Protein 10 (CFP-10), were tested in mouse immunization studies, demonstrating significant IFN-γ production, which is crucial for TB protection. Additionally, HPV16 L1, a well-known VLP assembly unit, was fused with the acyltransferase antigen Ag85B from Mtb, and chimeric HPV16 L1/Ag85B VLPs were successfully produced using the Pichia pastoris expression system. Mouse studies indicated that HPV16 L1/Ag85B could induce a strong Ag85B-specific T-cell immune response46. Moreover, LV20 VLPs, based on influenza virus-like particles containing Heparin-binding hemagglutinin (HBHA) and Mtb pili (MTP) antigens, generated in Sf9 insect cells, induced stronger CD4+/CD8+ T-cell responses than PBS and BCG vaccination in mice47. However, these tuberculosis (TB) nanoparticle vaccines have not yet been evaluated using the internationally recognized aerosol challenge method with the standard Mtb H37Rv strain in standard animal models, a more widely accepted approach for efficacy assessment. Therefore, continued investigation of nanoparticle (NP) vaccine delivery platforms is critical for advancing the development of more effective TB vaccines.
To present TB antigens, the mi3 nanoparticle, a self-assembling scaffold engineered from 2-Keto-3-deoxy-6-phosphogluconate (KDPG) aldolase, was selected for the display of TB antigens. Among nanoparticle (NP) platforms used in vaccine development, mi3 offers distinct advantages over conventional scaffolds such as ferritin, AP205 virus-like particles (VLPs) and the computationally designed i53-50 system48,49. Mi3 assembles into a symmetric 60-mer dodecahedron and enables high-density, site-specific antigen presentation via modular systems such as SpyTag/SpyCatcher50. Compared to the smaller 24-mer Ferritin51, mi3 allows for increased antigen-loading capacity and provides a larger surface area for multivalent antigen display. Unlike the dual-component i53-50 system, which requires antigens to be genetically fused specifically to the i53-50A subunit and necessitates careful structural design to maintain proper nanoparticle assembly48,52,53, mi3 functions as a single-component scaffold that self-assembles independently and allows conjugation of diverse antigens through modular systems such as SpyTag/SpyCatcher. This architecture offers greater design flexibility, facilitates early-stage vaccine development, and enables a more streamlined manufacturing process. Additionally, while AP205 VLPs often require dialysis-induced self-assembly48,54 and may exhibit batch-to-batch variability due to their capsid-derived architecture, their production is also associated with process complexity, which may present additional challenges for large-scale manufacturing. mi3 exhibits high structural homogeneity and manufacturing consistency owing to its engineered aldolase origin. Collectively, these features make mi3 a suitable and efficient platform for the development of nanoparticle-based TB vaccines. For simplicity and consistency purposes, we will uniformly refer to the mi3 platform as NP in the rest of this article.
Given the diverse array of structural antigens in Mtb, including secreted proteins, cell wall components, intracellular proteins, virulence factors, and metabolism-associated molecules55, the incorporation of multistage antigens is considered essential for the development of an optimal tuberculosis (TB) vaccine. Five antigens were selected, including Ag85A (Rv3804c), ESAT-6 (Rv3875), CFP10 (Rv3874), and TB10.4 (Rv0288) as early secreted antigens associated with active infection, together with Rv2660c as a latent-phase antigen. These early antigens are predominantly expressed during the initial or active phases of Mtb infection, driving robust Th1-type immune responses characterized by IFN-γ, TNF-α and IL-2 secretion that promote bacterial clearance56,57. The Ag85 complex, comprising the abundantly secreted antigens Ag85A, Ag85B, and Ag85C, plays a critical role in Mycobacterial cell wall biosynthesis as a mycolyltransferase58 and is considered a promising target for TB vaccine development59,60. In TB infection, the Ag85 complex induces IFN-γ production, promotes Th1 and cytotoxic lymphocyte responses, and confers protective immunogenicity61,62, with its highly immunodominant Ag85A subunit abundantly secreted during bacillary proliferation, leading to increased numbers of antigen-specific CD4⁺ and CD8⁺ T cells63,64. Ag85A is specifically recognized by both cell-mediated and humoral responses in infected patients and induces strong T-cell proliferation and IFN-γ production in most healthy individuals with latent Mtb infection, as well as in BCG-vaccinated mice and humans65,66. The 6-kDa early secretory antigenic target (ESAT-6) and its partner protein, culture filtrate protein 10 (CFP-10), form a heterodimer and are secreted together via the ESX-1 type VII secretion system encoded within the region of difference 1 (RD1) of Mtb67, playing a critical role in modulating host immune responses68. Acting synergistically, ESAT-6 and CFP-10 enhance T-cell recruitment, elicited the strongest IFN-γ secretion from peripheral blood cells of pulmonary TB patients69 induced both antigen-specific CD4⁺ and CD8⁺ T cells70, thereby enhancing protection against Mtb infection, serving not only as specific antigens for interferon-gamma release assays (IGRAs)71 but also as promising targets for tuberculosis vaccine development72. TB10.4 (EsxH), secreted by the ESX-3 type VII secretion system involved in iron and zinc acquisition, is essential for Mtb survival within macrophages and virulence in vivo, although its role in bacterial growth varies with culture conditions73,74. It also serves as a major target of CD4⁺ and CD8⁺ T-cell responses following infection in humans and mice75,76,77,78,79. Rv2660c, markedly upregulated during latent Mtb infection under nutrient-limited conditions80, induces memory T-cell responses targeting dormant bacilli and preferentially stimulates IFN-γ production by CD4⁺ T cells from individuals with latent infection compared to those with active disease, suggesting a role in controlling latent infection and preventing disease progression81.
In this study, a novel nanoparticle vaccine was developed using nanoparticles (NPs) and a SpyTag/SpyCatcher isopeptide linkage system to present multistage Mtb antigens82,83, which collectively engage the immune system across different stages of infection to provide broad and durable protection, and whose immunoprotective efficacy was preliminarily evaluated in a murine aerosol challenge model to assess their potential for pre-exposure protection.
Results
Generation and characterization of nanoparticle 85A-NP, EC-NP, and RT-NP
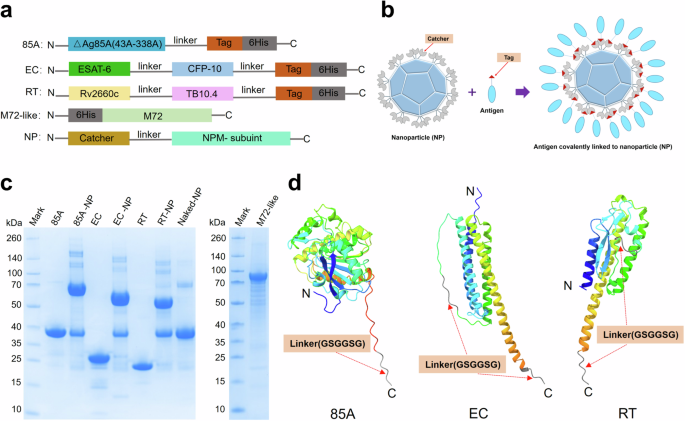
In this study, recombinant Mtb antigens 85A, EC and RT were conjugated to naked nanoparticles (NPs) using the Tag/Catcher system, successfully preparing three nanoparticle-based vaccines: 85A-NP, EC-NP and RT-NP, which display multistage Mtb antigens. The purified 85 A, EC and RT fusion proteins, along with naked NP (Catcher-NP) and the antigen-conjugated 85A-NP, EC-NP and RT-NP, exhibited high purity and uniformity, as evidenced by distinct single bands in reducing SDS-PAGE and single major peaks in size exclusion chromatography (SEC) (Figs. 1c and 2a). The M72-like antigen, serving as a control vaccine antigen, was also successfully prepared. Theoretical molecular weights of 85A, EC, RT, M72-like and naked NP (Catcher-NP) were 34.4, 23.5, 20.9, 72.4, and 33.0 kDa, respectively. SDS-PAGE and Western blot analyses confirmed protein expression near expected sizes, although 85A, EC, RT and naked NP bands exhibited a slight upward shift, likely due to altered electrophoretic mobility (Fig. 1c and Supplementary Fig. 1a–c). To verify the identities and intactness of these proteins, intact mass analysis by liquid chromatography–mass spectrometry (LC-MS) was performed, confirming that the experimentally measured molecular weights matched the theoretical values (Supplementary Fig. 1d). In addition, we observed variability in the binding efficiency of 85 A, EC and RT fusion proteins to naked NP, presumably due to differences in structural design and steric hindrance on the particle surface. Nevertheless, densitometric analysis revealed that the overall conjugation efficiencies of 85A-NP, EC-NP and RT-NP exceeded 75% (Fig. 2c). Dynamic light scattering (DLS) was further used to measure the particle properties of naked nanoparticles (NPs), 85A-NP, EC-NP and RT-NP, revealing a uniform particle size distribution across all samples, note that the hydrodynamic diameters of 85A-NP, EC-NP and RT-NP increased due to antigen conjugation or display (Fig. 2b). The negative-staining electron microscopy (EM) examination demonstrated that all nanoparticles were clearly visible on the surface of monodispersed particles, as shown in Fig. 2d.

a Schematic representation of the 85A, EC, RT, M72-like antigens and NP carriers. b Schematic representation of the covalent linkage between nanoparticle (NP) and antigen. c SDS-PAGE results of 85A, EC, RT, 85A-NP, EC-NP, RT-NP, naked NP and M72-like under reduced conditions. d 3D structure prediction of 85A, EC and RT antigens using the AlphaFold 3 Server.

a Size exclusion chromatography results (SEC) of 85A, EC and RT antigens, 85A-NP, EC-NP, RT-NP and naked NP on a Superdex200 increase 10/300GL column. b Dynamic light scattering (DLS) of 85A-NP, EC-NP and RT-NP and naked NP. c Characterization of 85A-NP, EC-NP, RT-NP and naked NP. d Negative-staining EM of 85A-NP, EC-NP, RT-NP and naked NP. The average measurement of diameter is indicated by the arrows (n = 3 individual measurements), and the negative-staining electron microscope image of all nanoparticles is presented with a scale bar of 200 nm.
Nanoparticle 85A-NP, EC-NP, and RT-NP induce higher IgG and IgG avidity than their corresponding recombinant protein
Humoral immune evaluation is often emphasized as a critical aspect in vaccine development. Under low-dose immunization conditions, we compared the immunogenicity of 85A, EC and RT with their corresponding nanoparticle formulations, 85A-NP, EC-NP and RT-NP. Groups receiving 85A, EC and RT as individual antigens or their respective nanoparticle formulations (85A-NP, EC-NP and RT-NP) were administered 0.6 μg per dose, while the mixed recombinant protein and nanoparticle groups received 0.6 μg (0.2 μg per antigen) per dose, all formulated with the AS01E-bio adjuvant, and control group (M72-like) received 0.6 μg per dose. Serum samples were collected and spleens were harvested 6 weeks post-initial immunization, followed by endpoint ELISA to assess antigen-specific IgG, IgG1, and IgG2c titers against 85A, EC, RT and M72-like molecules. The results showed the IgG antibody levels induced by the 85A-NP, EC-NP and RT-NP nanoparticle groups were significantly higher than those induced by the corresponding 85A, EC and RT recombinant protein groups, with statistical significance (P < 0.001 or P < 0.0001, Fig. 3b). Similarly, the control vaccine M72-like also induced high levels of IgG antibodies in immunized mice (Fig. 3b). In mice, IgG isotype switching is regulated by cytokines secreted by T-helper (Th) cell subsets: IFN-γ produced by Th1 cells promotes switching to IgG2a or IgG2c, whereas IL-4 from Th2 cells induces IgG1 production84. In C57BL/6 mice, where the IgG2a gene is disrupted due to the Igh1-b allele, IgG2c serves as its functional equivalent and is widely used as a surrogate marker for assessing Th1-type immune responses85,86. To evaluate the Th1/Th2 bias elicited by the vaccine candidates, antigen-specific IgG1 and IgG2c levels were quantified using endpoint ELISA87,88. The results showed that immunization with 85 A, EC and RT antigens, as well as their respective nanoparticle formulations (85A-NP, EC-NP and RT-NP), induced higher IgG2c than IgG1 antibody titers, with IgG2c/IgG1 ratios greater than 1.0 (Fig. 3c), suggesting that these candidate vaccines formulated with AS01E-bio predominantly elicit Th1-type immune responses, consistent with the profile observed for the M72-like control group.

a Schematic diagram of the vaccination timeline. C57BL/6 mice were immunized twice with a 3-week interval, and serum samples were collected in the sixth week. b, c Antigen-specific IgG, IgG1 and IgG2c antibody titers were measured using ELISA. d The avidity index (AI) of antigen-specific IgG was determined by ELISA, reflecting the percentage of antibody titers retained after NaSCN treatment compared to untreated controls. e The avidity of Mtb whole cell (inactivated H37Rv lysate protein)-specific IgG, as measured by AI, reflects the percentage of antibody titers retained after NaSCN treatment versus untreated controls. Data for antibody titers and avidity index (AI) are shown as geometric means (GM) with 95% CI, with n = 6 mice per group. Statistical significance was assessed by one-way ANOVA with Tukey’s multiple comparisons test, where **P < 0.01, ***P < 0.001 and ****P < 0.0001.
Previous studies have suggested a correlation between the avidity of IgG antibodies and the protective efficacy of TB vaccines89. In this study, the IgG avidity of different antigens in the TB nanoparticle vaccine was explored using the ELISA method. The results showed that the IgG avidity in the 85A-NP, EC-NP and RT-NP groups was significantly higher than that in the corresponding recombinant protein groups (85A, EC and RT) when these proteins were coated individually (P < 0.01, P < 0.001 or P < 0.0001, Fig. 3d). Furthermore, when 85A, EC and RT proteins were co-coated in a 1:1:1 ratio, the IgG avidity in the nanoparticle mixture group (85A-NP:EC-NP:RT-NP) was significantly higher than that in the recombinant antigen mixture group (85A:EC:RT) (P < 0.0001, Fig. 3d). Additionally, the IgG avidity observed for the M72-like-coated protein was significantly lower than that in the nanoparticle mixture group (85A-NP:EC-NP:RT-NP) (P < 0.001, Fig. 3d). These results suggest that higher-affinity antibodies are induced by the 85A, EC and RT antigens displayed on nanoparticles, and under identical immunization doses and adjuvant conditions, superior IgG avidity is exhibited by the nanoparticle mixture (85A-NP:EC-NP:RT-NP) compared to M72-like vaccine. Additionally, coating with inactivated H37Rv lysate protein for analysis revealed that the IgG avidity of the nanoparticle mixture (85A-NP:EC-NP:RT-NP) was significantly higher than that of M72-like and BCG (P < 0.01 and P < 0.001, Fig. 3e). The above indicates that 85A-NP, EC-NP, RT-NP and 85A-NP:EC-NP:RT-NP not only induce high levels of humoral immunity but also demonstrate high IgG avidity, particularly with 85A-NP:EC-NP:RT-NP.
Nanoparticle 85A-NP, EC-NP, and RT-NP induce higher IFN-γ, IL-2, and TNF-α than their corresponding recombinant protein
The host defense against intracellular Mtb infection depends critically on the essential roles of IFN-γ, IL-2 and TNF-α as key cytokines. To quantify the IFN-γ, IL-2, TNF-α and IL-4-secreting cells induced by the TB nanoparticle vaccines developed in this study, ELISPOT assays were performed on splenocytes (Fig. 4). The results showed that, under the same specific antigen stimulation, all immunized groups had significantly higher numbers of IFN-γ⁺, IL-2⁺, TNF-α⁺ and IL-4⁺ splenocytes compared to the saline group, with IFN-γ⁺, IL-2⁺ and TNF-α⁺ splenocytes significantly outnumbering IL-4⁺ splenocytes (Fig. 4a–c). This indicates that recombinant protein and nanoparticle vaccines prepared using 85A, EC and RT with the AS01E-bio adjuvant induce strong Th1-type immune responses. Upon stimulation with individual 85A, EC or RT antigens, the numbers of IFN-γ⁺, IL-2⁺ and TNF-α⁺ splenocytes were significantly higher in the 85A-NP, EC-NP and RT-NP groups compared to the respective 85A, EC, RT and 85 A:EC:RT groups (P < 0.0001 or P < 0.01; Fig. 4a–c). These findings demonstrate that nanoparticle vaccines (85A-NP, EC-NP and RT-NP), prepared using the Tag/Catcher system, elicit stronger cellular immune responses compared to recombinant protein vaccines. Specifically, splenocytes stimulated with a mixture of 85A, EC and RT antigens at equal concentrations showed that the nanoparticle mixture group (85A-NP: EC-NP: RT-NP) had a significantly higher total number of IFN-γ⁺, IL-2⁺ and TNF-α⁺ splenocytes than the recombinant antigen mixture group (85A: EC: RT) and any individual nanoparticle group (85A-NP, EC-NP or RT-NP) (P < 0.01, P < 0.001 or P < 0.0001; Fig. 4d). Notably, although no significant difference was observed between the nanoparticle mixture group (85A-NP:EC-NP:RT-NP) and the M72-like group (P > 0.05), the nanoparticle mixture still elicited the highest frequencies of IFN-γ⁺, IL-2⁺ and TNF-α⁺ splenocytes among all candidate vaccine groups.

C57BL/6 mice were immunized twice with a 3-week interval, and spleen cells were collected in the sixth week. a IFN-γ, IL-2, TNF-α and IL-4 cytokine were measured following 85A antigen stimulation in the 85A-NP:EC-NP:RT-NP, 85A:EC:RT, 85A-NP, 85A and saline groups. b IFN-γ, IL-2, TNF-α and IL-4 cytokine were assessed after EC antigen stimulation in the 85A-NP:EC-NP:RT-NP, 85A:EC:RT, EC-NP, EC and saline groups. c IFN-γ, IL-2, TNF-α and IL-4 cytokine were evaluated following RT antigen stimulation in the 85A-NP:EC-NP:RT-NP, 85A:EC:RT, RT-NP, RT and saline groups. d IFN-γ, IL-2, TNF-α and IL-4 cytokine levels were measured following stimulation with a mixture of 85A, EC and RT antigens at equal concentrations in the nanoparticle mixture group (85A-NP: EC-NP: RT-NP) and recombinant antigen mixture group (85A: EC: RT), and then compared with the levels in the corresponding individual groups (85A-NP, EC-NP, RT-NP) and the M72-like group. Data are presented as the mean ± SEM (n = 6 mice per group). Statistical analysis was performed using one-way ANOVA followed by Tukey’s multiple comparison test. “Ns” indicates no statistical significance (P > 0.05), while *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 indicate significant differences.
Nanoparticles 85A-NP, EC-NP and RT-NP induced strong functional antigen-specific polyfunctional CD4⁺ T-cell responses
Although IFN-γ, IL-2 and TNF-α can be secreted by both CD4⁺ and CD8⁺ T cells, with some cells simultaneously producing multiple cytokines, the precise cellular sources induced by the TB nanoparticle vaccine remain unresolved, despite the elevated cytokine levels observed in ELISPOT assays. To further characterize the immune profile induced by the vaccine, intracellular cytokine staining (ICS) was performed to evaluate cellular immune responses. Upon stimulation with 85A, EC or RT antigens individually (Supplementary Fig. 3a–c), markedly higher frequencies of IFN-γ⁺, IL-2⁺ and TNF-α⁺ CD4⁺ T cells (0.20–3.00%) were observed compared to CD8⁺ T cells (0.02–0.09%). CD8⁺ T-cell responses were consistently more than 10-fold lower than CD4⁺ T-cell responses across all vaccine groups, indicating that the cytokine response induced by these antigens and their AS01E-bio–adjuvanted nanoparticle formulations was predominantly CD4⁺ T-cell-mediated. Similarly, ICS analysis of M72-like–stimulated splenocytes showed significantly higher frequencies of IFN-γ⁺, IL-2⁺ and TNF-α⁺ CD4⁺ T cells compared to CD8⁺ T cells (Supplementary Fig. 3d), with CD4⁺ T-cell responses being approximately 20-fold greater overall. Consistent with the ELISPOT results, stimulation with a mixture of 85A, EC and RT antigens showed that the nanoparticle mixture group (85A-NP: EC-NP: RT-NP) had significantly higher proportions of IFN-γ⁺, IL-2⁺ and TNF-α⁺ CD4⁺ T cells compared to the recombinant antigen mixture group (85A: EC: RT) and individual nanoparticle groups (P < 0.0001, P < 0.001, P < 0.01 or P < 0.05; Fig. 5a), while the corresponding proportions of IFN-γ⁺, IL-2⁺ and TNF-α⁺ CD8⁺ T cells remained notably lower (Supplementary Fig. 4). While no significant differences were observed between the nanoparticle mixture group and the M72-like group for IL-2⁺ and TNF-α⁺ CD4⁺ T cells (P > 0.05), the nanoparticle mixture group showed significantly higher proportions of IFN-γ⁺ CD4⁺ T cells (P < 0.01). Additionally, we analyzed the secretion of IFN-γ, IL-2 and TNF-α by single and polyfunctional CD4⁺ T cells across different groups. The highest proportion of CD4⁺ T cells expressed IFN-γ⁺IL-2–TNF-α⁺, followed by IFN-γ⁺IL-2+TNF-α⁺ CD4⁺ T and IFN-γ–IL-2+TNF-α⁺ CD4⁺ T cells (Fig. 5c, d). In contrast, IFN-γ–IL-2+TNF-α– CD4⁺ T cells were almost undetectable, while IFN-γ–IL-2–TNF-α+ CD4⁺ T cells were relatively abundant (Fig. 5c, d). Additionally, despite each component in the nanoparticle mixture group (85A-NP: EC-NP: RT-NP) being administered at a dose of 0.2 μg, lower than the 0.6 μg used in individual nanoparticle groups, the total polyfunctional CD4⁺ T-cell response remained higher in the nanoparticle mixture group, even exceeding that of the M72-like group (Fig. 5b, c). These findings highlight that the robust production of key cytokines (IFN-γ, IL-2 and TNF-α), crucial for combating Mtb infection, is predominantly driven by polyfunctional CD4⁺ T cells, rather than single-cytokine CD4⁺ T cells, and primarily hinges on CD4⁺ T-cell activation induced by the candidate nanoparticle vaccines.

C57BL/6 mice were immunized twice at 3-week intervals, and splenocytes were collected 6 weeks after the initial immunization for ICS analysis. a Under stimulation with a mixture of 85A, EC and RT antigens at equal concentrations, the proportions of IFN-γ⁺, IL-2⁺and TNF-α⁺ CD4⁺T cells were evaluated using an ICS assay in the nanoparticle mixture group (85A-NP: EC-NP: RT-NP) and the recombinant antigen mixture group (85A: EC: RT), with comprehensive comparisons made against individual nanoparticle groups (85A-NP, EC-NP and RT-NP), M72-like and saline groups. b Activation marker combinations overall. c Polyfunctional CD4⁺T-cell frequencies overall. d Polyfunctionality proportions by different groups are presented as mean percentages. Bars are presented as the mean ± SEM, with n = 6 mice per group. Statistical analysis was performed using two-way ANOVA with Tukey’s multiple comparison test. Ns indicates no statistical significance (P > 0.05), while *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 denote significant differences.
Mixed-form 85A-NP:EC-NP:RT-NP nanoparticles generated significantly better protection compared to their corresponding single nanoparticles, M72-like and BCG
Building on the evaluation methods established for other TB recombinant vaccines90, we conducted an initial study to assess the protective efficacy of the TB nanoparticle vaccine in a high-dose (1500 CFU) aerosol challenge mouse model to screen and confirm the potential vaccine candidate. The results demonstrated that the bacterial load in the lungs of mice from the nanoparticle mixture group (85A-NP:EC-NP:RT-NP), the recombinant antigen mixture group (85A:EC:RT), and the individual nanoparticle groups (85A-NP, EC-NP or RT-NP), as well as the M72-like group, was significantly reduced compared to the saline group (P < 0.0001 or P < 0.05; Fig. 6b). Moreover, the bacterial load in the 85A-NP:EC-NP:RT-NP group was lower than that in the recombinant antigen mixture group (P < 0.01) and the individual nanoparticle groups (85A-NP, EC-NP or RT-NP). Notably, a significant difference was observed when comparing the 85A-NP:EC-NP:RT-NP group to the RT-NP group (P < 0.0001). Additionally, the challenge results showed that the bacterial load in the lungs of the 85A-NP:EC-NP:RT-NP group was reduced by 1.4 log compared to the saline group, whereas the M72-like and BCG groups showed reductions of only 0.8 log and 1.0 log, respectively. The bacterial load in the 85A-NP:EC-NP:RT-NP group was reduced by 0.6 log compared to the M72-like group (P < 0.01), although the reduction compared to the BCG group was only 0.4 log, with no significant difference observed (ns, P > 0.05).

a Schematic diagram of the vaccination and aerosol challenge timeline. b Lung bacterial load in the high-dose infection model, C57BL/6 mice were immunized twice (0.6 μg/dose, IM) with mixed nanoparticles (85A-NP:EC-NP:RT-NP), mixed recombinant proteins (85A:EC:RT), individual nanoparticles (85A-NP, EC-NP and RT-NP) and M72-like, a single BCG (SC) and saline (IM) as controls. Four weeks after full immunization, mice were challenged with a high-dose H37Rv aerosol (1,500 CFU), and four weeks later, they were sacrificed. Their lungs were collected, homogenized, and plated onto 7H10 agar for CFU determination after 4 weeks of incubation. c Lung bacterial load in the low-dose infection model, C57BL/6 mice were divided into groups and immunized with two intramuscular doses (0.6 μg per dose) of either the mixed nanoparticle vaccine (85A-NP:EC-NP:RT-NP) or the M72-like vaccine, a single BCG (SC) and saline (IM) as controls. Four weeks post low-dose H37Rv aerosol challenge (100 CFU), the mice were sacrificed, and lung tissues were collected. Viable Mtb colony-forming units (CFU) were determined by plating homogenized lung tissues, following the same method as used in the high-dose infection model. d–i Representative H&E-stained mouse lung tissue sections from high-dose H37Rv aerosol challenge (1,500 CFU): d H&E staining of lung tissue from non-infected mice. e H&E staining of lung tissue of Mtb infection mice from the saline group. f–i H&E staining of lung tissue from infected mice immunized with mixed nanoparticles (85A-NP:EC-NP:RT-NP), single nanoparticle EC-NP, M72-like and BCG. Results are presented as the mean ± SEM, n = 6. Asterisks indicate statistical significance between two groups, where *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001, ns indicates no statistical significance (P > 0.05), using one-way ANOVA with Tukey’s multiple comparison test. The image is presented at 150× magnification, with a scale bar of 200 μm.
Lung immunopathology was assessed 4 weeks post-challenge by analyzing inflammatory lesions in H&E-stained tissue sections from the right caudal lung lobes. The results show that, compared to the normal lung architecture observed in healthy mice (Fig. 6d), there were marked differences in the degree of inflammation and alveolar structural damage among the groups receiving saline, 85A-NP:EC-NP:RT-NP, EC-NP, M72-like and BCG vaccines (Fig. 6e–i). Similar to the BCG reference vaccine, both the 85A-NP:EC-NP:RT-NP and EC-NP formulations partially preserved the integrity of the alveolar structure. Also, histopathological analysis revealed that, compared to the saline group, which exhibited diffuse pulmonary inflammation with macrophage-dominated infiltration (Supplementary Fig. 5a), the 85A-NP:EC-NP:RT-NP group showed attenuated inflammation, characterized by increased mononuclear cells infiltration and reduced macrophage accumulation (Supplementary Fig. 5b). A similar histological profile was observed in the BCG group (Supplementary Fig. 5c). In contrast, although the M72-like vaccine elicited marked perivascular mononuclear cells infiltration, prominent macrophage infiltration persisted in the alveolar and interstitial regions (Supplementary Fig. 5d), indicating unresolved pulmonary inflammation. These findings indicate that the 85A-NP:EC-NP:RT-NP vaccine conferred histopathological protection comparable to BCG, characterized by infiltration of mononuclear cells, which mainly consist of monocytes and lymphocytes involved in anti-tuberculosis immunity, and relatively preserved alveolar architecture, whereas the M72-like vaccine provided only partial protection, with persistent macrophage infiltration despite lymphocyte recruitment.
To further evaluate the protective efficacy of the nanoparticle mixture group (85A-NP:EC-NP:RT-NP), a low-dose (100 CFU) aerosol infection experiment in mice was conducted using the same immunization dosage. The results showed that the bacterial loads in lung of the 85A-NP:EC-NP:RT-NP group, M72-like group and BCG group were significantly reduced compared to the saline group (P < 0.0001, P < 0.01 and P < 0.0001, respectively; Fig. 6c). Specifically, the bacterial load in the 85A-NP:EC-NP:RT-NP group was reduced by 1.8 log, whereas the reductions observed in the M72-like and BCG groups were 0.8 log and 1.2 log, respectively. Moreover, the bacterial load in the 85A-NP:EC-NP:RT-NP group was 1.0 log lower than in the M72-like group and 0.6 log lower than in the BCG group, with statistical significance observed for both comparisons (P < 0.0001 and P < 0.05, respectively; Fig. 6c).
The results demonstrate that the nanoparticle vaccines 85A-NP, EC-NP and RT-NP, prepared using the Tag/Catcher system with recombinant Mtb antigens, induce robust immune responses and provide significant protection against H37Rv infection in mice. Furthermore, the mixed nanoparticle formulation (85A-NP:EC-NP:RT-NP) exhibited superior efficacy in reducing lung bacterial burden and alleviating inflammatory tissue damage, outperforming both the M72-like and BCG vaccines. These findings highlight its potential and support its further development as a promising TB vaccine candidate.
Discussion
An ideal tuberculosis (TB) vaccine is expected to incorporate multistage antigens derived from Mycobacterium tuberculosis (Mtb)91. Nanoparticle (NP)-based vaccines offer enhanced immunogenicity and safety compared to other commonly used vaccine platforms. In this study, five Mycobacterium tuberculosis antigens, including Ag85A, ESAT-6, CFP10, Rv2660c and TB10.4, which represent early secreted and latency-associated proteins, were either fused or individually expressed and subsequently displayed on the surface of mi3 nanoparticles, a self-assembling scaffold derived from KDPG aldolase, using the isopeptide bond-forming SpyTag/SpyCatcher system29,49. Ag85A, a key component of the antigen 85 complex in Mtb, is highly expressed during active infection, playing a critical role in cell wall synthesis and virulence, and is widely recognized as a protective antigen for TB vaccine development22,92. Similarly, ESAT-6, CFP-10 and TB10.4, early secreted antigens, are prominently expressed during active infection, closely associated with bacterial virulence and host immune responses, and capable of inducing strong Th1-type and T-cell responses, including IFN-γ secretion93,94,95,96. In contrast, Rv2660c, a biphasic antigen expressed during the latent phase and the transition to active infection, elicits robust IFN-γ-driven T-cell responses and is incorporated into vaccines like H56:IC31 for latent infection protection97. The 85A-NP, EC-NP and RT-NP nanoparticles, based on the above antigens and formulated individually or as a mixture with the AS01E-biosimilar adjuvant, were first evaluated in C57BL/6 mice following aerosol Mtb challenge, demonstrating their effectiveness.
The clearance of Mtb, an intracellular pathogen, has been demonstrated in numerous studies to primarily rely on cell-mediated immunity (CMI), especially the Th1-type immune response98,99. Although the mechanisms underlying protective immunity against Mtb remain incompletely defined, conventional T cells, particularly CD4⁺ T-helper (Th) cells, are pivotal for defense100. Evidence from murine models and clinical studies underscores the indispensable role of functional Th1 CD4⁺ T cells in effective immunity against Mtb101. Leveraging MPL and QS-21, AS01 enhances innate and adaptive immunity, promoting CD4⁺ T-cell differentiation and IFN-γ/IL-2 secretion via TLR-4 activation. This Th1-driven response strengthens CMI, making AS01 suitable for vaccines against Mtb and Plasmodium102,103. A comparison of immune responses induced by six TB candidate vaccines, including M72/AS01E, revealed a common immunological feature of increased generation of IFN-γ-expressing CD4⁺ T cells, with the response induced by M72/AS01E being the most pronounced104.
CD4+ T cells secreting IFN-γ, IL-2 and TNF-α, known as Th1 cells, play a pivotal role in initiating effector functions to control intracellular Mtb105,106, with these cytokines performing distinct yet interdependent roles in the immune response and being closely associated with protective immunity107, Phase I clinical trials of several TB vaccines, including M72/AS01E, H4:IC31, and MVA85A, have placed a strong emphasis on evaluating the levels of the cytokines IFN-γ, IL-2 and TNF-α108,109,110. IFN-γ, produced by CD8⁺ T cells, and NK cells, activates macrophages to produce nitric oxide (NO) and reactive oxygen species (ROS) for Mtb elimination, promotes granuloma formation to limit infection spread, and amplifies Th1 immunity, while IL-2, secreted by activated Th1 cells, enhances the expansion of both CD4⁺ and CD8⁺ T cells, supports effector differentiation, sustains memory T-cell survival, and boosts IFN-γ and TNF-α production, with TNF-α being essential for granuloma formation, maintenance, and regulating macrophage apoptosis to clear infected cells. In this study, ICS analysis revealed that the 85A-NP, EC-NP and RT-NP vaccines, driven by the AS01E-biosimilar adjuvant, predominantly induced a high proportion of IFN-γ⁺, IL-2⁺ and TNF-α⁺CD4⁺T cells, while IFN-γ⁺, IL-2⁺ and TNF-α⁺ CD8⁺T cells were minimal (Fig. 5 and Supplementary Fig. 3, 4). The 85A-NP, EC-NP and RT-NP nanoparticle vaccines elicited significantly higher levels of IFN-γ, IL-2 and TNF-α in vivo compared to their corresponding recombinant vaccines (85A, EC and RT) (Fig. 4a–c), with these cytokines coordinating a robust immune response against Mtb111. Notably, the mixed nanoparticle formulation (85A-NP:EC-NP:RT-NP) elicited the highest levels of IFN-γ, IL-2 and TNF-α, with the predominant contribution from IFN-γ⁺IL-2–TNF-α⁺, IFN-γ⁺IL-2+TNF-α⁺ and IFN-γ–IL-2+TNF-α⁺ polyfunctional CD4+ T cells, as well as IFN-γ–IL-2–TNF-α+ single-cytokine CD4+ T cells (Fig. 5b-d). The secretion of IFN-γ, IL-2 and TNF-α by polyfunctional CD4+ T cells has long been recognized as a key correlate of protective immunity and is commonly used as a critical marker in the assessment of clinical immune responses in various TB vaccine trials108,109,110. These findings underscore the pivotal roles of both the AS01E-biosimilar adjuvant and the NP carrier (mi3) in enhancing cellular immunity, demonstrating a notable capacity to promote polyfunctional CD4⁺ T-cell responses, as evidenced in previous studies49 where the mi3-based shingles nanoparticle vaccine elicited significantly higher frequencies of IFN-γ– and IL-2–secreting splenocytes compared to vaccines based on I53 − 50, ferritin or AP205 nanoparticles, thereby highlighting the superior T-cell immunogenicity conferred by the mi3 platform.
The aerosol challenge assessment in mice revealed a significant reduction in lung bacterial load and inflammation, validating the effective protective efficacy of this novel nanoparticle vaccine against Mtb (Fig. 6b-i and Supplementary Fig. 5). Firstly, a higher dose (1,500 CFU) of virulent Mtb was used in the challenge model, compared to the typical aerosol challenge dose (50–100 CFU)112, to induce acute infection for vaccine evaluation. Four weeks post-infection, the mixed nanoparticle vaccine (85A-NP:EC-NP:RT-NP) reduced lung Mtb burden by 1.4 log, significantly outperforming a recombinant live vector vaccine, which showed only a 0.8–1.0 log reduction after a 400 CFU challenge111. The BCG group showed a 1.0 log reduction, which is lower than that reported in other studies113,114, likely due to the higher challenge dose used in this model. The average Mtb burden in the mixed nanoparticle group (85A-NP:EC-NP:RT-NP) was significantly lower than in both the mixed recombinant protein group (85 A:EC:RT) and the single nanoparticle groups (85A-NP, EC-NP and RT-NP) (Fig. 6b), highlighting the mixed 85A-NP, EC-NP and RT-NP as the optimal antigen formulation.
In the subsequent low-dose challenge with 100 CFU, the mixed nanoparticle group (85A-NP:EC-NP:RT-NP) exhibited a 1.8 log reduction in lung Mtb burden, which was greater than the 1.4 log reduction observed under high-dose challenge conditions. Furthermore, the candidate vaccine group showed a significantly lower Mtb burden in the lungs compared to the BCG group (P < 0.05), whereas no significant difference was observed in the high-dose challenge model. This suggests that the candidate vaccine 85A-NP:EC-NP:RT-NP provides more robust protection at the typical 50–100 CFU challenge dose. Moreover, M72, the only protective antigen currently in phase III clinical trials, is often used as a control vaccine in research evaluations115. In this study, the M72-like vaccine, formulated with an AS01E-biosimilar adjuvant, showed no difference in protective efficacy compared to BCG under both high- and low-dose challenge conditions, consistent with a previous report90. In contrast, the candidate vaccine 85A-NP:EC-NP:RT-NP demonstrated significantly better protection than the in-house M72-like control vaccine (P < 0.01 and P < 0.0001), highlighting its potential for improving TB vaccine development.
In this study, each self-assembled 60-subunit icosahedral nanoparticle was engineered to display a single recombinant antigen via the Tag/Catcher system116, thereby constituting a modular and flexible platform for the rational design of multistage tuberculosis vaccines. However, careful attention to the compatibility of different antigens is crucial during vaccine development. Our results show that the 85A-NP, EC-NP and RT-NP nanoparticle vaccines induce higher levels of IgG antibodies, IgG avidity and key cytokines such as IFN-γ, IL-2 and TNF-α, compared to the individual recombinant antigens. Importantly, the combination of 85A-NP, EC-NP and RT-NP did not interfere with each other, demonstrating excellent immunological compatibility between the TB antigens displayed on these nanoparticles. In multi-antigen vaccine candidates, the total antigen dose and the relative proportions of individual antigens are closely associated with protective efficacy117,118. Accordingly, future studies on this TB nanoparticle vaccine should systematically investigate and optimize both antigen dosing and ratio design. In addition, Mtb contains a wide variety of protective antigens that can stimulate specific CD4+ or CD8+ T-cell responses. While less defined than Th1 CD4+ T cells, CD8+ T cells are crucial for effective immunity against TB. Studies in mice119, cattle120, guinea pigs121, rabbits122 and macaques123 have shown their essential role in defending against Mtb infection. Building on the candidate nanoparticle vaccine 85A-NP:EC-NP:RT-NP, which predominantly triggers CD4+ T-cell responses, our future research will incorporate Mtb antigens that stimulate CD8+ T-cell responses into the vaccine and conduct in-depth evaluations in animal models, including guinea pigs, rabbits and macaques. However, the prevention and control of Mycobacterium tuberculosis infections extend beyond human health and encompass zoonotic and reverse zoonotic dimensions124. Various animal species—including farmed animals such as cattle (M. bovis) and goats (M. caprae)125,126, as well as wildlife species such as birds (M. avium) and marine mammals (M. pinnipedii)127,128—can harbor Mycobacteria and serve as direct or indirect sources of human infection129. These considerations highlight the need for an integrated approach in future TB vaccine design and development that takes into account both human and animal reservoirs within a one health framework.
Approximately one-quarter of the global population has latent tuberculosis infection (LTBI), with a 5-10% lifetime risk of progression to active TB, underscoring the urgent need for Prevention of Disease (POD) vaccines, such as GSK’s M72/AS01E130. Given the superior efficacy of the nanoparticle vaccine evaluated in this study, it shows promising potential for POD vaccine development. Therefore, evaluating the protective efficacy of the candidate vaccine 85A-NP:EC-NP:RT-NP or new antigen combinations displayed by NP in latent infection models, such as guinea pigs or macaques, is of critical significance for advancing POD vaccine strategies. Many subunit vaccines have shown promising results as a BCG-booster strategy131,132, so our next step will focus on evaluating the protective efficacy of the candidate vaccine 85A-NP:EC-NP:RT-NP in this direction.
The cost of goods (COGS) associated with the vaccine under development, which is a key determinant of commercial viability133,134 and is influenced by factors such as raw material inputs, process complexity, production workflows and yield, is a particularly critical consideration for commercial TB vaccines, which are primarily intended for high-burden, low- and middle-income countries135,136. Although the TB nanoparticle vaccine described in this study requires both recombinant antigen expression and nanoparticle carrier production, process optimization and scale-up can make its antigen manufacturing cost comparable to that of conventional recombinant protein vaccines. The primary contributor to the overall cost is the AS01E-bio adjuvant, due to the high raw material costs associated with its key components137, QS-21 and MPLA. Given this vaccine’s favorable protective efficacy, future efforts will focus on reducing production costs by optimizing antigen yield, streamlining adjuvant manufacturing, and lowering raw material expenses, possibly by using synthetic analogs138,139, thereby supporting the commercial development and global accessibility of this TB vaccine.
In conclusion, the mixed nanoparticles (85A-NP:EC-NP:RT-NP), in combination with the AS01E-biosimilar adjuvant, significantly enhanced Th1 immune responses, notably increasing the levels of IFN-γ, IL-2 and TNF-α, and provided robust protection against aerosolized acute Mtb infection. Compared to recombinant antigens (85A:EC:RT) or the M72-like control vaccine, the mixed nanoparticles demonstrated superior efficacy, highlighting the enhanced immune delivery and potent Th1-type cellular immunity mediated by the nanoparticle platform. These findings highlight the promising potential of the multistage nanoparticle vaccine, which incorporates Ag85A, ESAT-6, CFP10, Rv2660c and TB10.4, as a viable candidate for TB vaccine development. Furthermore, the results underscore the advantages of nanoparticle-based antigen presentation in advancing more effective TB vaccine strategies.
Methods
Ethics statement
C57BL/6 mouse experiments were conducted in strict accordance with ethical regulations and approved by the Guangzhou Forevergen Biosciences Animal Experimentation Ethics Committee, in compliance with its Institutional Animal Care and Use Committee (IACUC) guidelines (permit number: IACUC-AEWC-F240801005). The challenge experiments followed ethical standards for animal research, adhering to IACUC guidelines, and were approved by the Animal Experimentation Ethics Committee of Shenzhen Third People’s Hospital (Approval No: 2024-066-01).
Construction, expression and purification of recombinant proteins
Based on Mtb H37Rv (GenBank: AL123456.3), five clinically evaluated antigens were selected: Ag85A (Rv3804c, GenBank: CCP46633.1), ESAT-6 (Rv3875, GenBank: CCP46704.1), CFP10 (Rv3874, GenBank: CCP46703.1), Rv2660c(GenBank: CCP45458.1) and TB10.4 (Rv0288, GenBank: CCP43018.1). ESAT-6 and CFP10, as well as Rv2660c and TB10.4, were tandemly expressed using a GSGGSG linker, while Ag85A was truncated and expressed as △Ag85A (43A-338A) individually (Fig. 1a). Additionally, the cysteine at position 66 of Rv2660c was mutated to alanine to prevent the formation of intermolecular disulfide bond dimers in the Rv2660c and TB10.4 fusion protein. The 3D structures of the three designed fusion proteins were predicted online using the AlphaFold3 Server (https://alphafoldserver.com), as shown in Fig. 1d. Each protein was engineered with an AHIVMVDAYKPTK tag followed by a 6His tag at the C-terminus via a GSGGSG linker to facilitate affinity purification. The recombinant genes were cloned into the pET28a vector, resulting in plasmids pET28a-△Ag85A (43A-338A), pET28a-ESAT-6-CFP10 and pET28a-Rv2660c-TB10.4, corresponding to the proteins 85 A, EC and RT, respectively (Fig. 1a, sequences in Supplementary Sequence. 1a–c). To generate the M72-like construct (Fig. 1a), the M72 sequence disclosed in GSK’s patent was cloned into the pET28a vector to express a 6×His-tagged M72-like protein (sequence provided in Supplementary Sequence 1d), comprising the fusion components Mtb32a (GenBank: CCP42850.1) and Mtb39a (GenBank: CCP43952.1). Gene fragments encoding each recombinant antigen were synthesized by GenScript (Nanjing, China), codon-optimized for E. coli, and the resulting plasmid constructs were verified by Sanger sequencing at the same facility.
Recombinant vectors were transformed into Escherichia coli BL21 (DE3) cells (TransGen Biotech, China), and protein expression was induced with 0.5 mM isopropyl β-D-thiogalactopyranoside (IPTG; Sangon Biotech, China) at 18 °C in shaking flasks for 18 h. The bacterial pellets were harvested and resuspended in lysis buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl; Sigma-Aldrich, USA). Cells were lysed using a high-pressure homogenizer (ATS Engineering, Canada), and the supernatant was collected by centrifugation at 12,000 × g for 30 min at 4 °C. Proteins were sequentially purified using a 5 mL HisTrap™ Excel column (Cytiva, USA) and a Superdex™ 200 Increase 10/300 GL column (Cytiva, USA) on an ÄKTA pure chromatography system (Cytiva, USA), followed by buffer exchange into 20 mM Tris-HCl, 150 mM NaCl, pH 7.4. Purified proteins were stored at –80 °C until further use. SDS-PAGE analysis was conducted using NuPAGE™ 4–12% Bis-Tris Midi Gels (Thermo Fisher Scientific, USA). Protein samples were mixed with NuPAGE™ LDS Sample Buffer (Thermo Fisher Scientific, USA) containing 5 mM dithiothreitol (DTT; Sigma-Aldrich, USA) and denatured at 90 °C for 10 min before being separated by electrophoresis at 150 V for 1 h in the XCell4 SureLock™ Midi-Cell system (Thermo Fisher Scientific, USA) with 1× NuPAGE™ MES SDS Running Buffer (Thermo Fisher Scientific, USA). Gels were subsequently stained with InstaBlue™ Protein Stain Solution (Expedeon, UK) and visualized using the Amersham™ ImageQuant 800 imaging system (Cytiva, USA).
After SDS-PAGE analysis, purified 85 A, EC, RT and M72-like antigens were subjected to Western blotting. Proteins were transferred onto membranes using the Trans-Blot Turbo Transfer System and Transfer Pack (Bio-Rad, USA). Membranes were incubated with a mouse anti-His monoclonal antibody (Proteintech, China; Cat# 66005-1), followed by an AP-conjugated goat anti-mouse secondary antibody (Proteintech, China; Cat# SA00002-1) using the iBind FLEX system (Invitrogen, USA). Signal detection was performed using SIGMAFAST™ BCIP/NBT substrate tablets (Sigma-Aldrich, USA; Cat# B5655).
To further assess the integrity of the purified proteins, intact mass analysis was performed using a Vanquish™ Flex UHPLC system coupled with a Q Exactive™ Plus high-resolution mass spectrometer (Thermo Fisher Scientific, USA) operated in full MS and positive ion mode. The raw data were processed using Thermo BioPharma Finder software (version 5.1) to determine the accurate molecular weights of the four target proteins.
Preparation of conjugated nanoparticles
As previously described29,49, the isopeptide bond-based conjugation system was employed to prepare nanoparticles using 2-Keto-3-deoxy-6-phosphogluconate (KDPG) aldolase-derived mi3 as the nanoparticle scaffold. Based on protein structural considerations, the Catcher component of the isopeptide linkage system was fused to the subunit of the selected nanoparticle carriers via a flexible linker, and these were designated as NP (Fig. 1a, sequence in Supplementary Sequence. 1e). The gene encoding NP was cloned into the pET30a vector and transformed into Escherichia coli BL21 (DE3) for expression in a 5 L bioreactor. Following the expression, the harvested cells were lysed using an AH-PILOT high-pressure homogenizer (ATS Engineering Ltd., Canada), followed by centrifugation, and the resulting lysate underwent a series of purification steps according to a previously established protocol compliant with Good Manufacturing Practice (GMP) standards29. Western blot (WB) identification of NP was carried out using a mouse monoclonal antibody specifically generated in-house (GenScript, Nanjing, China), following the same procedure as described above. In addition, intact mass analysis of NP was conducted by LC-MS using the same method as previously described.
As shown in Fig. 1b, conjugated nanoparticles were prepared through covalent conjugation between the Tags on the antigens and the Catcher on the nanoparticles (NPs). The recombinant proteins 85 A, EC and RT were individually mixed with NPs at a 1:6 molar ratio and incubated at 22 °C for 24 h to facilitate isopeptide bond formation. Following incubation, unbound 85A, EC and RT were removed from the conjugated products via size-exclusion chromatography using a Superdex™ 200 Increase 10/300 GL column (Cytiva, USA) on an ÄKTA pure chromatography system (Cytiva, USA). The purified conjugated NPs (85A-NP, EC-NP and RT-NP) were analyzed by SDS-PAGE using NuPAGE™ 4–12% Bis-Tris Midi Gels, NuPAGE™ LDS Sample Buffer, and NuPAGE™ MES SDS Running Buffer (Thermo Fisher Scientific, USA), as described above, to assess coupling efficiency. Endotoxin levels were determined using the Nexgen-PTS™ chromogenic endotoxin detection system (Charles River, USA) and were confirmed to be below 10 EU/mg29,49.
Characterization of nanoparticles
Dynamic light scattering (DLS) measurements were carried out using a Zetasizer Lab system (Malvern Panalytical Ltd., UK) equipped with a 633 nm He-Ne laser. Particle size analysis was conducted at a fixed scattering angle of 90°, with data acquisition and interpretation performed using ZS XPLORER software (Malvern Panalytical Ltd., UK). Each sample, at a concentration of 0.2 mg/mL, was dispensed into a DTS0012 disposable polystyrene cuvette (Malvern Panalytical Ltd., UK) and maintained at a precise temperature of 25 °C. The viscosity and refractive index were calibrated for measurements at 25 °C, and each sample was measured in triplicate to ensure reproducibility.
For electron microscopy, 5 μL of 85A-NP, EC-NP, RT-NP and NP samples (each at 100 ng/mL) were applied to glow-discharged carbon-coated copper grids (Zhongjingkeyi Technology Co., Ltd., China) for 1 min. The grids were rinsed with Milli-Q water (Millipore, USA), blotted dry, and stained with 0.75% uranyl formate (Electron Microscopy Sciences, USA) for 1 min. Images were acquired using a Tecnai 12 transmission electron microscope (Thermo Fisher Scientific, USA) equipped with an Orius SC200 CCD camera (Gatan, USA) at an accelerating voltage of 120 kV.
Production of AS01E-biosimilar adjuvant
Based on its robust stimulation of both cellular and humoral immunity, the AS01 adjuvant, which contains monophosphoryl lipid A (MPLA) and QS-21 (a purified saponin fraction), has been successfully approved for use in the shingles vaccine SHINGRIX140 and in the malaria vaccine RTS,S/AS01 (Mosquirix). In this study, traditional MPLA (Sigma-Aldrich, USA) and QS-21 (Desert King International, USA), derived from Salmonella enterica serovar Minnesota and the bark of Quillaja saponaria Molina, respectively, were used as the primary active ingredients in the adjuvant preparation. A specified amount of cholesterol (Avanti Polar Lipids, USA), DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine; Avanti Polar Lipids, USA), and MPLA were dissolved in isopropanol (Sinopharm Chemical Reagent Co., Ltd., China) and subjected to rotary evaporation using a RE-52AA rotary evaporator (Yarong Biochemical Instrument Factory, China) to form a lipid film. The lipid film was then hydrated in sterile Milli-Q water (Millipore, USA) to obtain a coarse liposome suspension, which was subsequently reduced in size using an Avestin EmulsiFlex-C3 high-pressure homogenizer (Avestin Inc., Canada) to produce homogeneous liposomes. The final adjuvant formulation, designated AS01E-bio, was prepared by adding QS-21 to the liposome suspension. The composition of AS01E-bio consists of 25 μg of QS-21, 25 μg of MPLA, 125 μg of cholesterol and 500 μg of DOPC per 0.5 mL, with the key components comparable to those in the AS01E formulation.
Vaccine preparation and vaccination schedule
Recombinant proteins 85A, EC, RT, M72-like and conjugated nanoparticles (85A-NP, EC-NP and RT-NP) were diluted in buffer (20 mM Tris-HCl, 25% sucrose, 150 mM NaCl, pH 7.4; Sigma-Aldrich, USA) to a final volume of 50 μL, then emulsified with 50 μL of AS01E-bio adjuvant to produce a 100 μL vaccine dose. Protein concentrations were determined by bicinchoninic acid (BCA) assay using the Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, USA).
For the immunogenicity study, 66 female C57BL/6 mice, aged 5–6 weeks, were selected for immunization and stratified into 11 groups (Vac1–Vac11, 6 mice per group). Vac1 (85A-NP: EC-NP: RT-NP) contains a mixture of 85A-NP, EC-NP and RT-NP, while Vac2 (85A: EC: RT) consists of recombinant proteins 85A, EC and RT, each at 0.2 μg, totaling 0.6 μg per dose. Vac3 to Vac5 represent individual 85A-NP, EC-NP and RT-NP groups (each containing 0.6 μg per dose), and Vac6 to Vac8 contain individual recombinant proteins 85A, EC and RT (0.6 μg per dose). The Vac9, Vac10 and Vac11 groups received M72-like control (0.6 μg per dose), BCG (Chengdu Institute of Biological Products Co., Ltd.) and saline, respectively. Mice in groups Vac1 through Vac9 were administered 100 μL intramuscular injections (50 μL per hind leg) twice, with a 3-week interval between doses. The saline control group received an equal volume of sterile saline via the same route. Group Vac10 (BCG) received a single 100 μL subcutaneous injection (multi-point immunization at 4–6 sites on the dorsal surface) containing 5 × 10⁴ CFU of BCG. Six weeks post-initial immunization, mice were anesthetized using a mixture of 2% isoflurane and 98% air until deep anesthesia was achieved, followed by blood collection. Euthanasia was then performed by administering a mixture of 5% isoflurane and 95% air until cessation of circulation was confirmed, after which spleens were harvested for further analysis. Details of group allocations and vaccination schedules are provided in Supplementary Table 1 and Fig. 3a.
Female C57BL/6 mice (5–6 weeks old) were housed in a biosafety level 2 laboratory and randomly assigned to the designated experimental groups for the Mtb H37Rv challenge experiments. In the high-dose challenge, 48 female C57BL/6 mice (aged 5–6 weeks) were assigned to 8 groups (Vac1–Vac8, 6 mice per group). Vac1 (85A-NP: EC-NP: RT-NP) contains a mixture of 85A-NP, EC-NP and RT-NP, while Vac2 (85A: EC: RT) consists of recombinant proteins 85A, EC and RT, each at 0.2 μg, totaling 0.6 μg per dose. Vac3 to Vac6 represent individual 85A-NP, EC-NP and RT-NP, and M72-like groups (0.6 μg per dose). Vac7 and Vac8 serve as the BCG and saline groups, respectively. Mice in Vac1 to Vac6 received 100 μL intramuscular injections (50 μL per hind leg), administered twice with a 3-week interval, and the saline group received an equal volume of saline, while Vac7 (BCG) received a single 100 μL subcutaneous injection (multi-point immunization at 4–6 sites on the dorsal surface) containing 5 × 10⁴ CFU of BCG. Detailed group allocations and immunization schedules are provided in Supplementary Table 2 and Fig. 6a.
In the low-dose challenge, 24 female C57BL/6 mice (aged 5–6 weeks) were assigned to 4 groups (Vac1–Vac4, 6 mice per group). Vac1 (85A-NP: EC-NP: RT-NP) contains a mixture of 85A-NP, EC-NP and RT-NP, each at 0.2 μg, totaling 0.6 μg per dose. Vac2 represents the individual M72-like group (0.6 μg per dose), and Vac3 and Vac4 serve as the BCG and saline groups, respectively. Mice in Vac1 and Vac2 received 100 μL intramuscular injections (50 μL per hind leg), administered twice with a 3-week interval, and the Vac4 (saline) group received an equal volume of saline. The immunization protocol for the Vac3 (BCG) group was consistent with the high-dose challenge experiment. Detailed group allocations and immunization schedules are provided in Supplementary Table 3 and Fig. 6a.
Endpoint ELISA for IgG measurement and IgG avidity determination
Blood samples were collected from all groups of mice six weeks after the initial immunization via retro-orbital sinus under anesthesia, followed by euthanasia. After clotting, the samples were centrifuged at 4000 rpm for 10 min, and the supernatant was stored at −20 °C. 96-well ELISA plates (Thermo Fisher Scientific, USA) were coated with 85A, EC, RT and M72-like proteins (2 μg/mL, 100 ng/50 μL per well) and incubated overnight at 4 °C. After washing twice with PBST (0.05% Tween 20), 200 μL of blocking buffer (Thermo Fisher Scientific, USA) was added per well. The plates were blocked at room temperature (25 °C ± 3 °C) for 1–4 h, washed twice with PBST, and subsequently incubated for 1 h with immune sera. Sera were diluted 1:100 and serially diluted 3-fold for vaccine groups, while a 1:3 initial dilution was applied to saline controls. After four washes, 50 μL of 1:5000 diluted HRP-conjugated anti-IgG, anti-IgG1 and anti-IgG2c secondary antibodies (Abcam, UK) were added to each well. After a 1-h incubation at room temperature and six washes, 100 μL of substrate solution was added for 10 min of color development in the dark. The reaction was stopped with 100 μL of 1 M HCl, and absorbance was measured at 450 nm (reference 620 nm). Measurements were completed within 5 min of stopping the reaction. The endpoint titer is determined as the dilution at which the absorbance value exceeds the optical density (OD) of naïve serum by at least three standard deviations or 0.15, whichever is greater, as extrapolated from the X-axis intercept of the dilution curve.
Avidity index (AI) was determined by an ELISA method as previously described141. Briefly, 96-well ELISA plates (Thermo Fisher Scientific, USA) were coated with 85A, EC, RT, M72-like or inactivated H37Rv lysate protein (Shanghai Gene-Optimal Science & Technology Co., Ltd., China) and a mixed protein (85 A/EC/RT) at 1 μg/mL (180 ng per well, with 60 ng of each protein in the mixed protein) and incubated overnight at 4 °C. After washing with PBST (0.05% Tween 20), the plates were blocked with 200 μL blocking buffer for 1 h. Sera were diluted 1:100 in PBS containing 0.1% Tween-80, and three-fold serial dilutions were applied and incubated for 90 min at 37 °C. Following three washes, 100 μL of 1.0 M sodium thiocyanate (NaSCN) solution or PBS was added to half of the wells for 15 min. After washing, the plates were incubated with 1:5000 HRP-conjugated rabbit anti-human IgG for 90 min at 37 °C. Substrate solution was added, and the reaction was developed in the dark for 10 min. The reaction was stopped with 1 N HCl, and absorbance was measured at 450 nm (reference at 620 nm). The determination of endpoint titers follows the same method described above for ELISA. The AI, representing the percentage of antibodies remaining bound to the antigen after sodium thiocyanate treatment, was calculated by comparing the titer with NaSCN to that without, expressed as a percentage: AI = (titer with NaSCN)/(titer without NaSCN) × 100.
Quantification of cytokine secretion by ELISPOT
Six weeks after the initial immunization, mice were euthanized, and spleens were aseptically harvested. Splenic lymphocytes were isolated via mechanical dissociation using the GentleMACS™ Octo Dissociator (Miltenyi Biotec, Germany)142,143, followed by filtration through a sterile mesh. Red blood cells (RBCs) were subsequently lysed using ammonium-chloride-potassium (ACK) lysis buffer. Following lysis and washing, viable lymphocytes were enumerated and utilized for ELISPOT and flow cytometry analyses. Following the manufacturer’s instructions for the commercial kit (Mabtech, Nacka Strand, Sweden), IFN-γ, IL-2, TNFα and IL-4 were assayed. Plates were washed four times with sterile PBS, followed by blocking with serum-containing medium for at least 30 min. After discarding the medium, splenocytes isolated from mice three weeks post-final immunization were seeded at 1.0 × 10⁶ cells per well. Splenic lymphocytes from the 85A-NP, EC-NP and RT-NP, 85A, EC, RT, and M72-like groups were stimulated with their respective specific antigens at a concentration of 2 μg/mL. Cytokine assays for specific antigens in the recombinant protein mixture group (85A:EC:RT) and nanoparticle mixture group (85A-NP:EC-NP:RT-NP) were conducted by stimulating with the respective antigens at 2 μg/mL, while total cytokine levels were assessed by stimulating with a mixture of 85A, EC and RT, each at a concentration of 2 μg/mL. The plates were incubated at 37 °C in a 5% CO₂ atmosphere for 24 h. Subsequently, the plates were washed with PBS, and biotinylated antibodies specific to mouse IFN-γ, IL-2, TNFα and IL-4 were added, followed by a 2-h incubation at room temperature. After further washing, streptavidin-alkaline phosphatase (Mabtech, Nacka Strand, Sweden) was applied, and the plates were incubated for 1 h at room temperature. For color development, BCIP/NBT-plus substrate solution was added, and the reaction proceeded for 10 min before being stopped by washing with water. Finally, spot counting was performed using an ELISPOT reader (Bioreader® 7000–E, BIOSYS, Germany), and the data were subjected to analysis.
Intracellular cytokine staining for immune profiling
In the mouse intracellular cytokine staining (ICS) assay, isolated splenocytes (1 × 10⁶ cells per well) were stimulated in vitro with the corresponding individual antigens or antigen mixtures, following the same stimulation protocol used for the ELISPOT assay. During stimulation, cells were incubated with co-stimulatory antibodies against CD28 and CD49d (BD Biosciences, USA; Cat# 347690) for 6 h. Subsequently, protein transport was inhibited by adding Brefeldin A (BD Biosciences, USA; Cat# 555029) for an additional 4-h incubation prior to staining. After washing with PBS, cells were stained with Fixable Viability Stain 780 (1:1000 dilution; BD Biosciences, USA; Cat# 565388) and mouse Fc Block (BD Biosciences, USA; Cat# 553141) for 15 min at room temperature. Following a wash with staining buffer (BD Biosciences, USA; Cat# 554656)—composed of phosphate-buffered saline (PBS) supplemented with 2% fetal bovine serum (FBS) and 0.1% sodium azide (NaN₃)—surface staining was performed using a cocktail of PE-Cyanine7 anti-mouse CD3 (1:50; BD Biosciences, USA; Cat# 552774), PE anti-mouse CD4 (1:50; BD Biosciences, USA; Cat# 553730), and PerCP-Cyanine5.5 anti-mouse CD8 (1:50; BD Biosciences, USA; Cat# 551162) for 15 min. Cells were then fixed and permeabilized using the Fixation/Permeabilization Kit (BD Biosciences, USA; Cat# 554714), washed twice with Perm/Wash buffer, and subjected to intracellular staining with APC anti-mouse IFN-γ (BD Biosciences, USA; Cat# 554413), FITC anti-mouse IL-2 (BD Biosciences, USA; Cat# 554427), PE anti-mouse IL-4 (BD Biosciences, USA; Cat# 562450), and R718 anti-mouse TNF-α (BD Biosciences, USA; Cat# 567041), all at a dilution of 1:50. After final washing, cells were resuspended and analyzed using a DxFLEX flow cytometer (Beckman Coulter, USA), and data were processed using CytExpert software. Results were expressed as background-subtracted frequencies of CD4⁺ and CD8⁺ T cells producing IFN-γ, TNF-α, IL-4 or IL-2.
Aerosol challenge with Mtb in mice
The Mycobacterium tuberculosis (Mtb) H37Rv strain (provided by the biosafety level 3 laboratory at the Third People’s Hospital of Shenzhen, China) was cultured in a shaking incubator at 37 °C in Middlebrook 7H9 medium (Becton, Dickinson and Company, USA) until reaching an optical density at 600 nm (OD600) of 0.8–1.0. The culture was then diluted 1:100, aliquoted into cryogenic vials, and stored at −80 °C. Prior to infection, an aliquot was thawed, sonicated for 1 min using a cup-horn sonicator (Branson Ultrasonics Corporation, Danbury, CT, USA) to disperse clumps, and diluted in PBS containing 0.01% Tween-80 to a final volume of 5 mL. The suspension was titrated in a pilot study by assessing lung CFU 24 h post-exposure, allowing adjustment of the inoculum concentration. Four weeks following the final immunization, mice subjected to high-dose challenge were exposed to an aerosolized suspension delivering approximately 1500 colony-forming units (CFU) per lung using a Glas-Col Inhalation Exposure System (Glas-Col LLC, Terre Haute, IN, USA) for 30 min141. Four weeks post-challenge, mice were anesthetized with a 2% isoflurane-98% air mixture until deep anesthesia was achieved. Euthanasia was then performed by inhaling a 5% isoflurane-95% air mixture until circulatory cessation was confirmed. Following euthanasia, lungs were harvested for further analysis: the middle lobe of the left lung was fixed in 4% paraformaldehyde for histopathological analysis (H&E staining), while the remaining lung tissue was homogenized in 1 mL phosphate-buffered saline (PBS) and plated onto Middlebrook 7H10 agar (Becton, Dickinson and Company) for colony-forming unit (CFU) enumeration after four weeks of incubation at 37 °C (Fig. 6a). Similarly, mice exposed to a low-dose challenge were administered an infectious dose of 100 CFU per lung and euthanized as described above, followed by lung tissue homogenization and bacterial culture. The bacterial load in lung tissues following H37Rv challenge was quantified as CFU, and the results were log₁₀-transformed for statistical analysis to ensure normalization and facilitate comparisons between groups.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 10.0, with p-values < 0.05 considered statistically significant. One-way and two-way ANOVA, Tukey’s multiple comparisons post-test were used for comparisons of multiplex groups. Statistical significance thresholds were set as follows: *(0.01 < P < 0.05), **(0.001 < P < 0.01), ***(0.0001 < P < 0.001) and ****(P < 0.0001). ns indicates no statistical significance (P > 0.05).
Data availability
All data supporting the findings of this study are available within the article and its Supplementary Information. Protein sequences referenced in the study are publicly available in the NCBI GenBank database under the following accession numbers: Ag85A (GenBank: CCP46633.1), ESAT-6 (GenBank: CCP46704.1), CFP10 (GenBank: CCP46703.1), Rv2660c (GenBank: CCP45458.1), TB10.4 (GenBank: CCP43018.1) and Catcher-NPM (GenBank: AXF54357.1). The M72-like fusion protein comprises Mtb32a (GenBank: CCP42850.1) and Mtb39a (GenBank: CCP43952.1).
References
-
Hosseinpoor, A. R. et al. Monitoring inequalities is a key part of the efforts to end AIDS, tuberculosis, and malaria. Lancet 399, 1208–1210 (2022).
-
Fernandes, G. F. S., Thompson, A. M., Castagnolo, D., Denny, W. A. & Dos Santos, J. L. Tuberculosis drug discovery: challenges and new horizons. J. Med. Chem. 65, 7489–7531 (2022).
-
Lange, C. et al. Drug-resistant tuberculosis: An update on disease burden, diagnosis and treatment. Respirology 23, 656–673 (2018).
-
Wang, H. et al. Enhancing TB vaccine efficacy: current progress on vaccines, adjuvants and immunization strategies. Vaccines 12, 38 (2023).
-
Zhuang, L., Ye, Z., Li, L., Yang, L. & Gong, W. Next-generation TB vaccines: progress, challenges, and prospects. Vaccines 11, 1304 (2023).
-
Andersen, P. & Doherty, T. M. The success and failure of BCG – implications for a novel tuberculosis vaccine. Nat. Rev. Microbiol 3, 656–662 (2005).
-
Fine, P. E. The BCG story: lessons from the past and implications for the future. Rev. Infect. Dis. 11, S353–S359 (1989).
-
Poyntz, H. C. et al. Non-tuberculous Mycobacteria have diverse effects on BCG efficacy against Mycobacterium tuberculosis. Tuberculosis 94, 226–237 (2014).
-
Fatima, S., Kumari, A., Das, G. & Dwivedi, V. P. Tuberculosis vaccine: a journey from BCG to present. Life Sci. 252, 117594 (2020).
-
Brandt, L. et al. Failure of the Mycobacterium bovis BCG vaccine: some species of environmental Mycobacteria block multiplication of BCG and induction of protective immunity to tuberculosis. Infect. Immun. 70, 672–678 (2002).
-
Scriba, T. J., Netea, M. G. & Ginsberg, A. M. Key recent advances in TB vaccine development and understanding of protective immune responses against Mycobacterium tuberculosis. Semin Immunol. 50, 101431 (2020).
-
Tait, D. R. et al. Final analysis of a trial of M72/AS01(E) vaccine to prevent tuberculosis. N. Engl. J. Med. 381, 2429–2439 (2019).
-
Schrager, L. K., Vekemens, J., Drager, N., Lewinsohn, D. M. & Olesen, O. F. The status of tuberculosis vaccine development. Lancet Infect. Dis. 20, e28–e37 (2020).
-
Lu, L. L. et al. A functional role for antibodies in tuberculosis. Cell 167, 433–443.e414 (2016).
-
Urdahl, K. B., Shafiani, S. & Ernst, J. D. Initiation and regulation of T-cell responses in tuberculosis. Mucosal Immunol. 4, 288–293 (2011).
-
Luabeya, A. K. et al. First-in-human trial of the post-exposure tuberculosis vaccine H56:IC31 in Mycobacterium tuberculosis infected and non-infected healthy adults. Vaccine 33, 4130–4140 (2015).
-
Coccia, M. et al. Cellular and molecular synergy in AS01-adjuvanted vaccines results in an early IFN gamma response promoting vaccine immunogenicity. NPJ Vaccines 2, 25 (2017).
-
Schellack, C. et al. IC31, a novel adjuvant signaling via TLR9, induces potent cellular and humoral immune responses. Vaccine 24, 5461–5472 (2006).
-
Seydoux, E. et al. Effective combination adjuvants engage both TLR and inflammasome pathways to promote potent adaptive immune responses. J. Immunol. 201, 98–112 (2018).
-
Gillard, P. et al. Safety and immunogenicity of the M72/AS01E candidate tuberculosis vaccine in adults with tuberculosis: a phase II randomised study. Tuberculosis 100, 118–127 (2016).
-
Penn-Nicholson, A. et al. Safety and immunogenicity of the novel tuberculosis vaccine ID93 + GLA-SE in BCG-vaccinated healthy adults in South Africa: a randomised, double-blind, placebo-controlled phase 1 trial. Lancet Respir. Med. 6, 287–298 (2018).
-
Tkachuk, A. P. et al. Multi-subunit BCG booster vaccine GamTBvac: assessment of immunogenicity and protective efficacy in murine and guinea pig TB models. PLoS One 12, e0176784 (2017).
-
Zhao, L. et al. Nanoparticle vaccines. Vaccine 32, 327–337 (2014).
-
Gomes, A. C., Mohsen, M. & Bachmann, M. F. Harnessing nanoparticles for immunomodulation and vaccines. Vaccines 5, 6 (2017).
-
Engeroff, P. & Bachmann, M. F. The 5th virus-like particle and nano-particle vaccines (VLPNPV) conference. Expert Rev. Vaccines 18, 1–3 (2019).
-
Nooraei, S. et al. Virus-like particles: preparation, immunogenicity and their roles as nanovaccines and drug nanocarriers. J. Nanobiotechnol. 19, 59 (2021).
-
Kanekiyo, M. et al. Rational design of an Epstein-Barr virus vaccine targeting the receptor-binding site. Cell 162, 1090–1100 (2015).
-
Marcandalli, J. et al. Induction of potent neutralizing antibody responses by a designed protein nanoparticle vaccine for respiratory syncytial virus. Cell 176, 1420–1431.e1417 (2019).
-
Li, Y. et al. An RBD virus-like particle vaccine for SARS-CoV-2 induces cross-variant antibody responses in mice and macaques. Signal. Transduct. Target Ther. 8, 173 (2023).
-
Huzair, F. & Sturdy, S. Biotechnology and the transformation of vaccine innovation: the case of the hepatitis B vaccines 1968-2000. Stud. Hist. Philos. Biol. Biomed. Sci. 64, 11–21 (2017).
-
Tsang, S. H. et al. Durability of cross-protection by different schedules of the bivalent HPV vaccine: the CVT trial. J. Natl. Cancer Inst. 112, 1030–1037 (2020).
-
Chowdhury, F. et al. A non-inferiority trial comparing two recombinant vaccines (Hepa-B vs. Engerix-B) for hepatitis B among adults in Dhaka, Bangladesh. Vaccine 39, 6385–6390 (2021).
-
Joura, E. A. et al. A 9-valent HPV vaccine against infection and intraepithelial neoplasia in women. N. Engl. J. Med. 372, 711–723 (2015).
-
Gordon, D. M. et al. Safety, immunogenicity, and efficacy of a recombinantly produced Plasmodium falciparum circumsporozoite protein-hepatitis B surface antigen subunit vaccine. J. Infect. Dis. 171, 1576–1585 (1995).
-
RTS,S Clinical Trials Partnership et al. First results of phase 3 trial of RTS,S/AS01 malaria vaccine in African children. N. Engl. J. Med. 365, 1863–1875 (2011).
-
Datoo, M. S. et al. Efficacy of a low-dose candidate malaria vaccine, R21 in adjuvant Matrix-M, with seasonal administration to children in Burkina Faso: a randomised controlled trial. Lancet 397, 1809–1818 (2021).
-
Fuenmayor, J., Godia, F. & Cervera, L. Production of virus-like particles for vaccines. N. Biotechnol. 39, 174–180 (2017).
-
Pascha, M. N. et al. Nanoparticle display of neuraminidase elicits enhanced antibody responses and protection against influenza A virus challenge. NPJ Vaccines 9, 97 (2024).
-
Wuertz, K. M. et al. A SARS-CoV-2 spike ferritin nanoparticle vaccine protects hamsters against Alpha and Beta virus variant challenge. NPJ Vaccines 6, 129 (2021).
-
Arunachalam, P. S. et al. Adjuvanting a subunit COVID-19 vaccine to induce protective immunity. Nature 594, 253–258 (2021).
-
Zakeri, B. & Howarth, M. Spontaneous intermolecular amide bond formation between side chains for irreversible peptide targeting. J. Am. Chem. Soc. 132, 4526–4527 (2010).
-
Song, J. Y. et al. Immunogenicity and safety of SARS-CoV-2 recombinant protein nanoparticle vaccine GBP510 adjuvanted with AS03: interim results of a randomised, active-controlled, observer-blinded, phase 3 trial. EClinicalMedicine 64, 102140 (2023).
-
Tang, R. et al. Safety and immunogenicity of the SARS-CoV-2 LYB001 RBD-based VLP vaccine (CHO cell) phase 1 in Chinese adults: a randomized, double-blind, positive-parallel-controlled study. Expert Rev. Vaccines 23, 498–509 (2024).
-
Yin, Y. et al. Hepatitis B virus core particles displaying Mycobacterium tuberculosis antigen ESAT-6 enhance ESAT-6-specific immune responses. Vaccine 29, 5645–5651 (2011).
-
Dhanasooraj, D., Kumar, R. A. & Mundayoor, S. Vaccine delivery system for tuberculosis based on nano-sized hepatitis B virus core protein particles. Int. J. Nanomed. 8, 835–843 (2013).
-
Zhou, F. & Zhang, D. Nano-sized chimeric human papillomavirus-16 L1 virus-like particles displaying Mycobacterium tuberculosis antigen Ag85B enhance Ag85B-specific immune responses in female C57BL/c mice. Viruses 15, 2123 (2023).
-
Wang, J. et al. Correction: Wang et al. A VLP-based vaccine displaying HBHA and MTP antigens of Mycobacterium tuberculosis induces potentially protective immune responses in M. tuberculosis H37Ra infected mice. Vaccines 11, 941 (2023).
-
Kang, Y. F. et al. Rapid development of SARS-CoV-2 spike protein receptor-binding domain self-assembled nanoparticle vaccine candidates. ACS Nano 15, 2738–2752 (2021).
-
Li, Y. et al. A nanoparticle vaccine displaying varicella-zoster virus gE antigen induces a superior cellular immune response than a licensed vaccine in mice and non-human primates. Front. Immunol. 15, 1419634 (2024).
-
Bruun, T. U. J., Andersson, A. C., Draper, S. J. & Howarth, M. Engineering a rugged nanoscaffold to enhance plug-and-display vaccination. ACS Nano 12, 8855–8866 (2018).
-
Ma, X. et al. Nanoparticle vaccines based on the receptor binding domain (RBD) and heptad repeat (HR) of SARS-CoV-2 elicit robust protective immune responses. Immunity 53, 1315–1330 e1319 (2020).
-
Boyoglu-Barnum, S. et al. Quadrivalent influenza nanoparticle vaccines induce broad protection. Nature 592, 623–628 (2021).
-
Walls, A. C. et al. Elicitation of potent neutralizing antibody responses by designed protein nanoparticle vaccines for SARS-CoV-2. Cell 183, 1367–1382 e1317 (2020).
-
Brune, K. D. et al. Plug-and-display: decoration of virus-like particles via isopeptide bonds for modular immunization. Sci. Rep. 6, 19234 (2016).
-
Putim, C. et al. Secretome profile analysis of multidrug-resistant, monodrug-resistant and drug-susceptible Mycobacterium tuberculosis. Arch. Microbiol. 200, 299–309 (2018).
-
Karbalaei Zadeh Babaki, M., Soleimanpour, S. & Rezaee, S. A. Antigen 85 complex as a powerful Mycobacterium tuberculosis immunogene: biology, immune-pathogenicity, applications in diagnosis, and vaccine design. Micro Pathog. 112, 20–29 (2017).
-
Lindestam Arlehamn, C. S., Lewinsohn, D., Sette, A. & Lewinsohn, D. Antigens for CD4 and CD8 T cells in tuberculosis. Cold Spring Harb. Perspect. Med. 4, a018465 (2014).
-
Kremer, L., Maughan, W. N., Wilson, R. A., Dover, L. G. & Besra, G. S. The M. tuberculosis antigen 85 complex and mycolyltransferase activity. Lett. Appl. Microbiol. 34, 233–237 (2002).
-
Veerapandian, R., Gadad, S. S., Jagannath, C. & Dhandayuthapani, S. Live attenuated vaccines against tuberculosis: targeting the disruption of genes encoding the secretory proteins of Mycobacteria. Vaccines 12, 530 (2024).
-
Singh, S., Saavedra-Avila, N. A., Tiwari, S. & Porcelli, S. A. A century of BCG vaccination: immune mechanisms, animal models, non-traditional routes and implications for COVID-19. Front. Immunol. 13, 959656 (2022).
-
Aghababa, H., Mobarez, A. M., Behmanesh, M., Khoramabadi, N. & Mobarhan, M. Production and purification of mycolyl transferase B of Mycobacterium tuberculosis. Tanaffos 10, 23–30 (2011).
-
Dang, S. et al. Ag85a-S2 activates cGAS-STING signaling pathway in intestinal mucosal cells. Vaccines 10, 2170 (2022).
-
Pathan, A. A. et al. Boosting BCG with recombinant modified vaccinia Ankara expressing antigen 85A: different boosting intervals and implications for efficacy trials. PLoS ONE 2, e1052 (2007).
-
Chen, Z. et al. A multistage protein subunit vaccine as BCG-booster confers protection against Mycobacterium tuberculosis infection in murine models. Int. Immunopharmacol. 139, 112811 (2024).
-
Jeyanathan, M. et al. Aerosol delivery, but not intramuscular injection, of adenovirus-vectored tuberculosis vaccine induces respiratory-mucosal immunity in humans. JCI Insight 7, e155655 (2022).
-
Huygen, K. The immunodominant T-cell epitopes of the mycolyl-transferases of the antigen 85 complex of M. tuberculosis. Front. Immunol. 5, 321 (2014).
-
Abdallah, A. M. et al. Type VII secretion-Mycobacteria show the way. Nat. Rev. Microbiol. 5, 883–891 (2007).
-
Yu, X. & Xie, J. Roles and underlying mechanisms of ESAT-6 in the context of Mycobacterium tuberculosis-host interaction from a systems biology perspective. Cell Signal. 24, 1841–1846 (2012).
-
Liu, L. et al. Exploration of novel cellular and serological antigen biomarkers in the ORFeome of Mycobacterium tuberculosis. Mol. Cell Proteom. 13, 897–906 (2014).
-
Rueda, C. M., Marin, N. D., Garcia, L. F. & Rojas, M. Characterization of CD4 and CD8 T cells producing IFN-gamma in human latent and active tuberculosis. Tuberculosis 90, 346–353 (2010).
-
Petruccioli, E. et al. Characterization of the CD4 and CD8 T-cell response in the Quanti FERON-TB Gold Plus kit. Int. J. Mycobacteriol. 5, S25–S26 (2016).
-
Arlehamn, C. S. et al. Dissecting mechanisms of immunodominance to the common tuberculosis antigens ESAT-6, CFP10, Rv2031c (hspX), Rv2654c (TB7.7), and Rv1038c (EsxJ). J. Immunol. 188, 5020–5031 (2012).
-
Serafini, A., Pisu, D., Palu, G., Rodriguez, G. M. & Manganelli, R. The ESX-3 secretion system is necessary for iron and zinc homeostasis in Mycobacterium tuberculosis. PLoS One 8, e78351 (2013).
-
Griffin, J. E. et al. High-resolution phenotypic profiling defines genes essential for Mycobacterial growth and cholesterol catabolism. PLoS Pathog. 7, e1002251 (2011).
-
Woodworth, J. S. et al. Mycobacterium tuberculosis directs immunofocusing of CD8+ T cell responses despite vaccination. J. Immunol. 186, 1627–1637 (2011).
-
Staff, P. P. Correction: Human and murine clonal CD8+ T cell expansions arise during tuberculosis because of TCR selection. PLoS Pathog. 11, e1005144 (2015).
-
Skjot, R. L. et al. Epitope mapping of the immunodominant antigen TB10.4 and the two homologous proteins TB10.3 and TB12.9, which constitute a subfamily of the esat-6 gene family. Infect. Immun. 70, 5446–5453 (2002).
-
Skjot, R. L. et al. Comparative evaluation of low-molecular-mass proteins from Mycobacterium tuberculosis identifies members of the ESAT-6 family as immunodominant T-cell antigens. Infect. Immun. 68, 214–220 (2000).
-
Lewinsohn, D. A. et al. Comprehensive definition of human immunodominant CD8 antigens in tuberculosis. NPJ Vaccines 2, 8 (2017).
-
Betts, J. C., Lukey, P. T., Robb, L. C., McAdam, R. A. & Duncan, K. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol. Microbiol. 43, 717–731 (2002).
-
Govender, L. et al. Higher human CD4 T cell response to novel Mycobacterium tuberculosis latency associated antigens Rv2660 and Rv2659 in latent infection compared with tuberculosis disease. Vaccine 29, 51–57 (2010).
-
Zakeri, B. et al. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc. Natl. Acad. Sci. USA 109, E690–E697 (2012).
-
Aves, K. L. & Sander, A. F. Design and purification of Tag/Catcher AP205-based capsid virus-like particle vaccines. Methods Mol. Biol. 2720, 127–141 (2024).
-
Ahlborg, N., Ling, I. T., Holder, A. A. & Riley, E. M. Linkage of exogenous T-cell epitopes to the 19-kilodalton region of Plasmodium yoelii merozoite surface protein 1 (MSP1(19)) can enhance protective immunity against malaria and modulate the immunoglobulin subclass response to MSP1(19). Infect. Immun. 68, 2102–2109 (2000).
-
Nazeri, S., Zakeri, S., Mehrizi, A. A., Sardari, S. & Djadid, N. D. Measuring of IgG2c isotype instead of IgG2a in immunized C57BL/6 mice with Plasmodium vivax TRAP as a subunit vaccine candidate in order to correct interpretation of Th1 versus Th2 immune response. Exp. Parasitol. 216, 107944 (2020).
-
Nazeri, S., Zakeri, S., Mehrizi, A. A. & Djadid, N. D. Naturally acquired immune responses to thrombospondin-related adhesion protein (TRAP) of Plasmodium vivax in patients from areas of unstable malaria transmission. Acta Trop. 173, 45–54 (2017).
-
Baldwin, S. L. et al. Therapeutic efficacy against Mycobacterium tuberculosis using ID93 and liposomal adjuvant formulations. Front. Microbiol.13, 935444 (2022).
-
Du, X. et al. A new poly(I:C)-decorated PLGA-PEG nanoparticle promotes Mycobacterium tuberculosis fusion protein to induce comprehensive immune responses in mice intranasally. Micro Pathog. 162, 105335 (2022).
-
Peralta-Alvarez, M. P. et al. MTBVAC induces superior antibody titers and IgG avidity compared to BCG vaccination in non-human primates. NPJ Vaccines 9, 230 (2024).
-
Brandt, L. et al. The protective effect of the Mycobacterium bovis BCG vaccine is increased by coadministration with the Mycobacterium tuberculosis 72-kilodalton fusion polyprotein Mtb72F in M. tuberculosis-infected guinea pigs. Infect. Immun. 72, 6622–6632 (2004).
-
Fan, X. et al. A novel multistage antigens ERA005f confer protection against Mycobacterium tuberculosis by driving Th-1 and Th-17 type T cell immune responses. Front. Immunol. 14, 1276887 (2023).
-
Li, W. et al. A recombinant adenovirus expressing CFP10, ESAT6, Ag85A and Ag85B of Mycobacterium tuberculosis elicits strong antigen-specific immune responses in mice. Mol. Immunol. 62, 86–95 (2014).
-
Tkachuk, A. P. et al. Safety and immunogenicity of the GamTBvac, the recombinant subunit tuberculosis vaccine candidate: a phase II, multi-center, double-blind, randomized, placebo-controlled study. Vaccines 8, 652 (2020).
-
Chen, S. et al. Particulate Mycobacterial vaccines induce protective immunity against tuberculosis in mice. Nanomaterials 11, 2060 (2021).
-
Mpande, C. A. M. et al. Immune profiling of Mycobacterium tuberculosis-specific T cells in recent and remote infection. EBioMedicine 64, 103233 (2021).
-
Nemes, E. et al. Prevention of M. tuberculosis Infection with H4:IC31 vaccine or BCG revaccination. N. Engl. J. Med. 379, 138–149 (2018).
-
Jenum, S. et al. A phase I/II randomized trial of H56:IC31 vaccination and adjunctive cyclooxygenase-2-inhibitor treatment in tuberculosis patients. Nat. Commun. 12, 6774 (2021).
-
Zuniga, J. et al. Cellular and humoral mechanisms involved in the control of tuberculosis. Clin. Dev. Immunol. 2012, 193923 (2012).
-
Ma, J. et al. A multistage subunit vaccine effectively protects mice against primary progressive tuberculosis, latency and reactivation. EBioMedicine 22, 143–154 (2017).
-
Cooper, A. M. Cell-mediated immune responses in tuberculosis. Annu. Rev. Immunol. 27, 393–422 (2009).
-
Chackerian, A. A., Perera, T. V. & Behar, S. M. Gamma interferon-producing CD4+ T lymphocytes in the lung correlate with resistance to infection with Mycobacterium tuberculosis. Infect. Immun. 69, 2666–2674 (2001).
-
Spreng, R. L. et al. Identification of RTS,S/AS01 vaccine-induced humoral biomarkers predictive of protection against controlled human malaria infection. JCI Insight. 9, e178801 (2024).
-
Day, C. L. et al. Induction and regulation of T-cell immunity by the novel tuberculosis vaccine M72/AS01 in South African adults. Am. J. Respir. Crit. Care Med. 188, 492–502 (2013).
-
Rodo, M. J. et al. A comparison of antigen-specific T cell responses induced by six novel tuberculosis vaccine candidates. PLoS Pathog. 15, e1007643 (2019).
-
Cao, X., Fu, Y. X. & Peng, H. Promising cytokine adjuvants for enhancing tuberculosis vaccine immunity. Vaccines 12, 477 (2024).
-
Urdahl, K. B. Understanding and overcoming the barriers to T cell-mediated immunity against tuberculosis. Semin. Immunol. 26, 578–587 (2014).
-
Hu, Z. et al. The profile of T Cell responses in Bacille Calmette-Guerin-Primed mice boosted by a novel Sendai virus vectored anti-tuberculosis vaccine. Front. Immunol. 9, 1796 (2018).
-
Thacher, E. G. et al. Safety and immunogenicity of the M72/AS01 candidate tuberculosis vaccine in HIV-infected adults on combination antiretroviral therapy: a phase I/II, randomized trial. AIDS 28, 1769–1781 (2014).
-
Geldenhuys, H. et al. The tuberculosis vaccine H4:IC31 is safe and induces a persistent polyfunctional CD4 T cell response in South African adults: a randomized controlled trial. Vaccine 33, 3592–3599 (2015).
-
Sander, C. R. et al. Safety and immunogenicity of a new tuberculosis vaccine, MVA85A, in Mycobacterium tuberculosis-infected individuals. Am. J. Respir. Crit. Care Med. 179, 724–733 (2009).
-
Kirk, N. M. et al. Recombinant Pichinde viral vector expressing tuberculosis antigens elicits strong T cell responses and protection in mice. Front. Immunol. 14, 1127515 (2023).
-
Baldwin, S. L. et al. Protection and long-lived immunity induced by the ID93/GLA-SE vaccine candidate against a clinical Mycobacterium tuberculosis isolate. Clin. Vaccin. Immunol. 23, 137–147 (2016).
-
Woodworth, J. S. et al. A Mycobacterium tuberculosis-specific subunit vaccine that provides synergistic immunity upon co-administration with Bacillus Calmette-Guerin. Nat. Commun. 12, 6658 (2021).
-
Aagaard, C. et al. Immunization with Mycobacterium tuberculosis-specific antigens bypasses T cell differentiation from prior Bacillus Calmette-Guerin vaccination and improves protection in mice. J. Immunol. 205, 2146–2155 (2020).
-
Rungelrath, V. et al. Vaccination with mincle agonist UM-1098 and Mycobacterial antigens induces protective Th1 and Th17 responses. NPJ Vaccines 9, 100 (2024).
-
Tan, T. K. et al. A COVID-19 vaccine candidate using SpyCatcher multimerization of the SARS-CoV-2 spike protein receptor-binding domain induces potent neutralising antibody responses. Nat. Commun. 12, 542 (2021).
-
John, S. et al. Multi-antigenic human cytomegalovirus mRNA vaccines that elicit potent humoral and cell-mediated immunity. Vaccine 36, 1689–1699 (2018).
-
Kuang, B. et al. Different antigen ratio in bivalent vaccine can affect immunological activation and protection against Aeromonas salmonicida and Vibrio anguillarum in Atlantic salmon. Fish. Shellfish Immunol. 128, 644–650 (2022).
-
van Pinxteren, L. A., Cassidy, J. P., Smedegaard, B. H., Agger, E. M. & Andersen, P. Control of latent Mycobacterium tuberculosis infection is dependent on CD8 T cells. Eur. J. Immunol. 30, 3689–3698 (2000).
-
Villarreal-Ramos, B. et al. Investigation of the role of CD8+ T cells in bovine tuberculosis in vivo. Infect. Immun. 71, 4297–4303 (2003).
-
Jia, Q., Maslesa-Galic, S., Nava, S. & Horwitz, M. A. Listeria-vectored multi-antigenic tuberculosis vaccine protects C57BL/6 and BALB/c mice and guinea pigs against Mycobacterium tuberculosis challenge. Commun. Biol. 5, 1388 (2022).
-
Zhou, S. et al. The adjuvant effect of manganese on tuberculosis subunit vaccine Bfrb-GrpE. NPJ Vaccines 9, 248 (2024).
-
Chen, C. Y. et al. A critical role for CD8 T cells in a nonhuman primate model of tuberculosis. PLoS Pathog. 5, e1000392 (2009).
-
Woldemariam, T. et al. Zoonotic transmission of the Mycobacterium tuberculosis complex between cattle and humans in Central Ethiopia. Front. Vet. Sci. 12, 1527279 (2025).
-
Mohammed, T. et al. Epidemiology of Mycobacterium tuberculosis complex infections in cattle and humans in the remote pastoral settings of southern Ethiopia. Front. Vet. Sci. 12, 1551710 (2025).
-
Riopel, N. D. et al. Characterization of Mycobacterium orygis, Mycobacterium bovis, and Mycobacterium caprae infections in humans in western Canada. J. Infect. Dis. 230, e789–e797 (2024).
-
Rani, I. et al. Mycobacterium orygis and its unseen impact: re-evaluating zoonotic tuberculosis in animal and human populations. Front. Public Health 13, 1505967 (2025).
-
Alba, P. et al. Genomics insights into a Mycobacterium pinnipedii isolate causing tuberculosis in a captive South American sea lion (Otaria flavescens) from Italy. Front. Microbiol. 14, 1303682 (2023).
-
Kiers, A., Klarenbeek, A., Mendelts, B., Van Soolingen, D. & Koeter, G. Transmission of Mycobacterium pinnipedii to humans in a zoo with marine mammals. Int. J. Tuberc. Lung Dis. 12, 1469–1473 (2008).
-
Van Der Meeren, O. et al. Phase 2b controlled trial of M72/AS01(E) vaccine to prevent tuberculosis. N. Engl. J. Med. 379, 1621–1634 (2018).
-
Kwon, K. W. et al. BCG-booster vaccination with HSP90-ESAT-6-HspX-RipA multivalent subunit vaccine confers durable protection against hypervirulent Mtb in mice. NPJ Vaccines 9, 55 (2024).
-
Kwon, K. W. et al. Long-term protective efficacy with a BCG-prime ID93/GLA-SE boost regimen against the hyper-virulent Mycobacterium tuberculosis strain K in a mouse model. Sci. Rep. 9, 15560 (2019).
-
Chong, P. et al. Production of EV71 vaccine candidates. Hum. Vaccin. Immunother. 8, 1775–1783 (2012).
-
Velez-Suberbie, M. L. et al. Holistic process development to mitigate proteolysis of a subunit rotavirus vaccine candidate produced in Pichia pastoris by means of an acid pH pulse during fed-batch fermentation. Biotechnol. Prog. 36, e2966 (2020).
-
Teo, A. K. J., Singh, S. R., Prem, K., Hsu, L. Y. & Yi, S. Duration and determinants of delayed tuberculosis diagnosis and treatment in high-burden countries: a mixed-methods systematic review and meta-analysis. Respir. Res. 22, 251 (2021).
-
Lv, H. et al. Further analysis of tuberculosis in eight high-burden countries based on the global burden of disease study 2021 data. Infect. Dis. Poverty 13, 70 (2024).
-
Bonam, S. R., Partidos, C. D., Halmuthur, S. K. M. & Muller, S. An overview of novel adjuvants designed for improving vaccine efficacy. Trends Pharm. Sci. 38, 771–793 (2017).
-
Wang, Y. Q., Bazin-Lee, H., Evans, J. T., Casella, C. R. & Mitchell, T. C. MPL adjuvant contains competitive antagonists of human TLR4. Front. Immunol. 11, 577823 (2020).
-
Walkowicz, W. E. et al. Quillaja saponin variants with central glycosidic linkage modifications exhibit distinct conformations and adjuvant activities. Chem. Sci. 7, 2371–2380 (2016).
-
Stadtmauer, E. A. et al. A phase 1/2 study of an adjuvanted varicella-zoster virus subunit vaccine in autologous hematopoietic cell transplant recipients. Blood 124, 2921–2929 (2014).
-
Vermont, C. L. et al. Antibody avidity and immunoglobulin G isotype distribution following immunization with a monovalent meningococcal B outer membrane vesicle vaccine. Infect. Immun. 70, 584–590 (2002).
-
Lee, J. et al. CD11cHi monocyte-derived macrophages are a major cellular compartment infected by Mycobacterium tuberculosis. PLoS Pathog. 16, e1008621 (2020).
-
Hu, Z. et al. A multistage Sendai virus vaccine incorporating latency-associated antigens induces protection against acute and latent tuberculosis. Emerg. Microbes Infect. 13, 2300463 (2024).
Acknowledgements
Patronus Biotech Co. Ltd. funded the vaccine design, purification, formulation and immunogenicity study, and the challenge study was supported by the Science and Technology Planning Project of Guangdong Province, China (Grant No. 2021B1212030003).
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ding, Y., Li, Y., Wu, Z. et al. A novel nanoparticle vaccine displaying multistage tuberculosis antigens confers protection in mice infected with H37Rv. npj Vaccines 10, 173 (2025). https://doi.org/10.1038/s41541-025-01216-8
-
Received:
-
Accepted:
-
Published:
-
DOI: https://doi.org/10.1038/s41541-025-01216-8