
Clinton, W. J. Remarks on the completion of the first survey of the human genome. The American Presidency Project https://www.presidency.ucsb.edu/node/227458 (2000).
Amaral, P. et al. The status of the human gene catalogue. Nature 622, 41–47 (2023). This paper describes the efforts to characterize every transcript isoform for every human gene and the challenges involved in determining which isoforms are functional.
Rhie, A. et al. Towards complete and error-free genome assemblies of all vertebrate species. Nature 592, 737–746 (2021).
Robinson, G. E. et al. Creating a buzz about insect genomes. Science 331, 1386 (2011).
Cheng, S. et al. 10KP: a phylodiverse genome sequencing plan. GigaScience https://doi.org/10.1093/gigascience/giy013 (2018).
Lewin, H. A. et al. Earth biogenome project: sequencing life for the future of life. Proc. Natl Acad. Sci. USA 115, 4325–4333 (2018). This paper introduces a massive effort to sequence every eukaryotic species on the planet.
Blaxter, M. et al. The Earth Biogenome Project Phase II: illuminating the eukaryotic tree of life. Front. Sci. https://doi.org/10.3389/fsci.2025.1514835 (2025).
Koepfli, K.-P. & Paten, B. the Genome 10K Community of Scientists & O’Brien, S. J. The Genome 10K Project: a way forward. Annu. Rev. Anim. Biosci. 3, 57–111 (2015).
Borodovsky, M. & McIninch, J. GENMARK: parallel gene recognition for both DNA strands. Computers Chem. 17, 123–133 (1993).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78–94 (1997).
Stanke, M. & Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19, ii215–ii225 (2003).
Korf, I. Gene finding in novel genomes. BMC Bioinform. 5, 59 (2004).
Majoros, W., Pertea, M. & Salzberg, S. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997). This publication describes the second major upgrade to BLAST, which remains one of the most highly cited computational methods in the field.
Lister, R. et al. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 133, 523–536 (2008).
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L. & Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628 (2008). This landmark paper describes the invention of RNA-seq technology and applies it to mice.
Nagalakshmi, U. et al. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320, 1344–1349 (2008).
Shiraki, T. et al. Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. Proc. Natl Acad. Sci. USA 100, 15776–15781 (2003).
Jaganathan, K. et al. Predicting splicing from primary sequence with deep learning. Cell 176, 535–548.e524 (2019).
Zeng, T. & Li, Y. I. Predicting RNA splicing from DNA sequence using Pangolin. Genome Biol. 23, 103 (2022).
Chao, K.-H., Mao, A., Salzberg, S. L. & Pertea, M. Splam: a deep-learning-based splice site predictor that improves spliced alignments. Genome Biol. 25, 243 (2024).
Scalzitti, N. et al. Spliceator: multi-species splice site prediction using convolutional neural networks. BMC Bioinform. 22, 561 (2021).
Wang, R., Wang, Z., Wang, J. & Li, S. SpliceFinder: ab initio prediction of splice sites using convolutional neural network. BMC Bioinform. 20, 652 (2019).
Ji, Y., Zhou, Z., Liu, H. & Davuluri, R. V. DNABERT: pre-trained bidirectional encoder representations from transformers model for DNA-language in genome. Bioinformatics 37, 2112–2120 (2021).
Dehghannasiri, R., Olivieri, J. E., Damljanovic, A. & Salzman, J. Specific splice junction detection in single cells with SICILIAN. Genome Biol. 22, 219 (2021).
Rentzsch, P., Schubach, M., Shendure, J. & Kircher, M. CADD-Splice — improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 13, 31 (2021).
Cheng, J. et al. MMSplice: modular modeling improves the predictions of genetic variant effects on splicing. Genome Biol. 20, 48 (2019).
Ingolia, N. T., Ghaemmaghami, S., Newman, J. R. S. & Weissman, J. S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223 (2009). This paper introduces ribosome profiling and its use to study translated regions.
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). This paper describes AlphaFold2, the first program ever to predict the 3D structure of proteins with accuracy comparable with that achieved by crystallography experiments.
Baek, M. et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871–876 (2021).
Sommer, M. J. et al. Structure-guided isoform identification for the human transcriptome. eLife 11, e82556 (2022). This paper describes how to use AlphaFold2 or ColabFold to determine the functional and non-functional transcript isoforms of a gene.
Adiconis, X. et al. Comparative analysis of RNA sequencing methods for degraded or low-input samples. Nat. Methods 10, 623–629 (2013).
Singh, A. et al. A scalable and cost-efficient rRNA depletion approach to enrich RNAs for molecular biology investigations. RNA 30, 728–738 (2024).
Hafner, M. et al. RNA-ligase-dependent biases in miRNA representation in deep-sequenced small RNA cDNA libraries. RNA 17, 1697–1712 (2011).
Faridani, O. R. et al. Single-cell sequencing of the small-RNA transcriptome. Nat. Biotechnol. 34, 1264–1266 (2016).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Mudge, J. M. et al. GENCODE 2025: reference gene annotation for human and mouse. Nucleic Acids Res. 53, D966–D975 (2025).
Varabyou, A. et al. CHESS 3: an improved, comprehensive catalog of human genes and transcripts based on large-scale expression data, phylogenetic analysis, and protein structure. Genome Biol. 24, 249 (2023).
Goldfarb, T. et al. NCBI RefSeq: reference sequence standards through 25 years of curation and annotation. Nucleic Acids Res. 53, D243–D257 (2025).
Bult, C. J., Blake, J. A., Smith, C. L., Kadin, J. A. & Richardson, J. E. Mouse genome database (MGD) 2019. Nucleic Acids Res. 47, D801–d806 (2019).
Öztürk-Çolak, A. et al. FlyBase: updates to the Drosophila genes and genomes database. Genetics https://doi.org/10.1093/genetics/iyad211 (2024).
Cheng, C. Y. et al. Araport11: a complete reannotation of the Arabidopsis thaliana reference genome. Plant. J. 89, 789–804 (2017).
Lamesch, P. et al. The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res. 40, D1202–D1210 (2012).
RNAcentral Consortium. RNAcentral 2021: secondary structure integration, improved sequence search and new member databases. Nucleic Acids Res. 49, D212–D220 (2021).
Pan, Q., Shai, O., Lee, L. J., Frey, B. J. & Blencowe, B. J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 40, 1413–1415 (2008).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015). The paper describes StringTie, a maximum-flow-based algorithm for assembling transcripts into gene structures and for quantifying expression.
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 20, 278 (2019).
Shumate, A., Wong, B., Pertea, G. & Pertea, M. Improved transcriptome assembly using a hybrid of long and short reads with StringTie. PLoS Comput. Biol. 18, e1009730 (2022).
Shao, M. & Kingsford, C. Accurate assembly of transcripts through phase-preserving graph decomposition. Nat. Biotechnol. 35, 1167–1169 (2017).
Zhang, Q., Shi, Q. & Shao, M. Accurate assembly of multi-end RNA-seq data with Scallop2. Nat. Comput. Sci. 2, 148–152 (2022).
Swarbreck, D. et al. The Arabidopsis Information Resource (TAIR): gene structure and function annotation. Nucleic Acids Res. 36, D1009–D1014 (2008).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Burdick, J. T. et al. Nanopore-based direct sequencing of RNA transcripts with 10 different modified nucleotides reveals gaps in existing technology. G3 Genes Genomes Genet. 13, jkad200 (2023).
Chen, Y. et al. A systematic benchmark of nanopore long-read RNA sequencing for transcript-level analysis in human cell lines. Nat. Methods https://doi.org/10.1038/s41592-025-02623-4 (2025).
Pardo-Palacios, F. J. et al. Systematic assessment of long-read RNA-seq methods for transcript identification and quantification. Nat. Methods 21, 1349–1363 (2024).
Wu, T. D. & Watanabe, C. K. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21, 1859–1875 (2005).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018). This publication describes minimap2, one of the fastest and most accurate tools for aligning both short and long DNA sequencing reads to a genome.
Tung, L. H., Shao, M. & Kingsford, C. Quantifying the benefit offered by transcript assembly with Scallop-LR on single-molecule long reads. Genome Biol. 20, 287 (2019).
Prjibelski, A. D. et al. Accurate isoform discovery with IsoQuant using long reads. Nat. Biotechnol. 41, 915–918 (2023).
Gao, Y. et al. ESPRESSO: robust discovery and quantification of transcript isoforms from error-prone long-read RNA-seq data. Sci. Adv. 9, eabq5072 (2023).
Chen, Y. et al. Context-aware transcript quantification from long-read RNA-seq data with Bambu. Nat. Methods 20, 1187–1195 (2023).
Han, S. W., Jewell, S., Thomas-Tikhonenko, A. & Barash, Y. Contrasting and combining transcriptome complexity captured by short and long RNA sequencing reads. Genome Res. 34, 1624–1635 (2024).
Nip, K. M. et al. Reference-free assembly of long-read transcriptome sequencing data with RNA-Bloom2. Nat. Commun. 14, 2940 (2023).
Tang, F. et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 6, 377–382 (2009).
Vento-Tormo, R. et al. Single-cell reconstruction of the early maternal–fetal interface in humans. Nature 563, 347–353 (2018).
Montoro, D. T. et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 560, 319–324 (2018).
Vieira Braga, F. A. et al. A cellular census of human lungs identifies novel cell states in health and in asthma. Nat. Med. 25, 1153–1163 (2019).
Park, J.-E. et al. A cell atlas of human thymic development defines T cell repertoire formation. Science 367, eaay3224 (2020).
HuBMAP Consortium. The human body at cellular resolution: the NIH Human Biomolecular Atlas Program. Nature 574, 187–192 (2019).
Regev, A. et al. The human cell atlas. eLife https://doi.org/10.7554/eLife.27041 (2017).
Lähnemann, D. et al. Eleven grand challenges in single-cell data science. Genome Biol. 21, 31 (2020).
Nip, K. M. et al. RNA-Bloom enables reference-free and reference-guided sequence assembly for single-cell transcriptomes. Genome Res. 30, 1191–1200 (2020).
Liu, J., Liu, X., Ren, X. & Li, G. scRNAss: a single-cell RNA-seq assembler via imputing dropouts and combing junctions. Bioinformatics 35, 4264–4271 (2019).
Shi, Q., Zhang, Q. & Shao, M. Transcriptome assembly at single-cell resolution with Beaver. Bioinformatics 41, i323–i331 (2025).
Picelli, S. et al. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 9, 171–181 (2014).
Hagemann-Jensen, M. et al. Single-cell RNA counting at allele and isoform resolution using Smart-seq3. Nat. Biotechnol. 38, 708–714 (2020).
Ramsköld, D. et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat. Biotechnol. 30, 777–782 (2012).
Healey, H. M., Bassham, S. & Cresko, W. A. Single-cell Iso-Sequencing enables rapid genome annotation for scRNAseq analysis. Genetics https://doi.org/10.1093/genetics/iyac017 (2022).
Tian, L. et al. Comprehensive characterization of single-cell full-length isoforms in human and mouse with long-read sequencing. Genome Biol. 22, 310 (2021).
Dondi, A. et al. Detection of isoforms and genomic alterations by high-throughput full-length single-cell RNA sequencing in ovarian cancer. Nat. Commun. 14, 7780 (2023).
Li, Z. et al. An isoform-resolution transcriptomic atlas of colorectal cancer from long-read single-cell sequencing. Cell Genom. https://doi.org/10.1016/j.xgen.2024.100641 (2024).
Westoby, J., Artemov, P., Hemberg, M. & Ferguson-Smith, A. Obstacles to detecting isoforms using full-length scRNA-seq data. Genome Biol. 21, 74 (2020).
Deng, E. et al. Systematic evaluation of single-cell RNA-seq analyses performance based on long-read sequencing platforms. J. Adv. Res. 71, 141–153 (2025).
Carninci, P. et al. Genome-wide analysis of mammalian promoter architecture and evolution. Nat. Genet. 38, 626–635 (2006).
Valen, E. et al. Genome-wide detection and analysis of hippocampus core promoters using DeepCAGE. Genome Res. 19, 255–265 (2009).
Noguchi, S. et al. FANTOM5 CAGE profiles of human and mouse samples. Sci. Data 4, 170112 (2017).
Harrow, J. et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774 (2012).
Ozsolak, F. et al. Comprehensive polyadenylation site maps in yeast and human reveal pervasive alternative polyadenylation. Cell 143, 1018–1029 (2010).
Beck, A. H. et al. 3′-End sequencing for expression quantification (3SEQ) from archival tumor samples. PLoS One 5, e8768 (2010).
Shepard, P. J. et al. Complex and dynamic landscape of RNA polyadenylation revealed by PAS-Seq. RNA 17, 761–772 (2011).
Martin, G., Gruber, A. R., Keller, W. & Zavolan, M. Genome-wide analysis of pre-mRNA 3′ end processing reveals a decisive role of human cleavage factor I in the regulation of 3′ UTR length. Cell Rep. 1, 753–763 (2012).
Tian, B., Hu, J., Zhang, H. & Lutz, C. S. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res. 33, 201–212 (2005).
Lianoglou, S., Garg, V., Yang, J. L., Leslie, C. S. & Mayr, C. Ubiquitously transcribed genes use alternative polyadenylation to achieve tissue-specific expression. Genes Dev. 27, 2380–2396 (2013).
Gruber, A. J. et al. A comprehensive analysis of 3′ end sequencing data sets reveals novel polyadenylation signals and the repressive role of heterogeneous ribonucleoprotein C on cleavage and polyadenylation. Genome Res. 26, 1145–1159 (2016).
You, L. et al. APASdb: a database describing alternative poly(A) sites and selection of heterogeneous cleavage sites downstream of poly(A) signals. Nucleic Acids Res. 43, D59–D67 (2015).
Herrmann, C. J. et al. PolyASite 2.0: a consolidated atlas of polyadenylation sites from 3′ end sequencing. Nucleic Acids Res. 48, D174–D179 (2020).
Moon, Y., Herrmann, C. J., Mironov, A. & Zavolan, M. PolyASite v3. 0: a multi-species atlas of polyadenylation sites inferred from single-cell RNA-sequencing data. Nucleic Acids Res. 53, D197–D204 (2025).
Crappé, J. et al. PROTEOFORMER: deep proteome coverage through ribosome profiling and MS integration. Nucleic Acids Res. 43, e29–e29 (2015).
Chun, S. Y., Rodriguez, C. M., Todd, P. K. & Mills, R. E. SPECtre: a spectral coherence-based classifier of actively translated transcripts from ribosome profiling sequence data. BMC Bioinform. 17, 482 (2016).
Fields, A. P. et al. A regression-based analysis of ribosome-profiling data reveals a conserved complexity to mammalian translation. Mol. Cell 60, 816–827 (2015).
Erhard, F. et al. Improved Ribo-seq enables identification of cryptic translation events. Nat. Methods 15, 363–366 (2018).
Swirski, M. I. et al. Translon: a single term for translated regions. Nat. Methods 22, 2002–2006 (2025).
Lin, M. F., Jungreis, I. & Kellis, M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics 27, i275–i282 (2011). This paper describes PhyloCSF, a powerful method for determining patterns of sequence conservation that indicate whether a genomic region encodes a protein.
Menschaert, G. et al. Deep proteome coverage based on ribosome profiling aids mass spectrometry-based protein and peptide discovery and provides evidence of alternative translation products and near-cognate translation initiation events. Mol. Cell. Proteom. 12, 1780–1790 (2013).
Chong, C. et al. Integrated proteogenomic deep sequencing and analytics accurately identify non-canonical peptides in tumor immunopeptidomes. Nat. Commun. 11, 1293 (2020).
Ouspenskaia, T. et al. Unannotated proteins expand the MHC-I-restricted immunopeptidome in cancer. Nat. Biotechnol. 40, 209–217 (2022).
Mudge, J. M. et al. Standardized annotation of translated open reading frames. Nat. Biotechnol. 40, 994–999 (2022).
Ji, H. & Salzberg, S. L. Upstream open reading frames may contain hundreds of novel human exons. PLoS Comput. Biol. 20, e1012543 (2024).
Calviello, L., Hirsekorn, A. & Ohler, U. Quantification of translation uncovers the functions of the alternative transcriptome. Nat. Struct. Mol. Biol. 27, 717–725 (2020).
Pertea, M., Lin, X. Y. & Salzberg, S. L. GeneSplicer: a new computational method for splice site prediction. Nucleic Acids Res. 29, 1185–1190 (2001).
Yeo, G. & Burge, C. B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 11, 377–394 (2004).
The UniProt Consortium. UniProt: the universal protein knowledgebase in 2025. Nucleic Acids Res. 53, D609–D617 (2025).
Omer, S., Harlow, T. J. & Gogarten, J. P. Does sequence conservation provide evidence for biological function? Trends Microbiol. 25, 11–18 (2017).
Platt, A., Ross, H. C., Hankin, S. & Reece, R. J. The insertion of two amino acids into a transcriptional inducer converts it into a galactokinase. Proc. Natl Acad. Sci. USA 97, 3154–3159 (2000).
Balmer, P. et al. A curated catalog of canine and equine keratin genes. PLoS One 12, e0180359 (2017).
van Dam, S., Võsa, U., van der Graaf, A., Franke, L. & de Magalhães, J. P. Gene co-expression analysis for functional classification and gene–disease predictions. Brief. Bioinform. 19, 575–592 (2018).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 9, 559 (2008).
Zhang, R. et al. A CRISPR screen defines a signal peptide processing pathway required by flaviviruses. Nature 535, 164–168 (2016).
Li, W. et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554 (2014).
Berns, K. et al. A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature 428, 431–437 (2004).
Moffat, J. et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 124, 1283–1298 (2006).
Paddison, P. J. et al. A resource for large-scale RNA-interference-based screens in mammals. Nature 428, 427–431 (2004).
Westbrook, T. F. et al. A genetic screen for candidate tumor suppressors identifies REST. Cell 121, 837–848 (2005).
Boutros, M. et al. Genome-wide RNAi analysis of growth and viability in Drosophila cells. Science 303, 832–835 (2004).
Xu, Y. et al. CRISPR screens in Drosophila cells identify Vsg as a Tc toxin receptor. Nature 610, 349–355 (2022).
van Kempen, M. et al. Fast and accurate protein structure search with Foldseek. Nat. Biotechnol. https://doi.org/10.1038/s41587-023-01773-0 (2024).
Zhang, Y. & Skolnick, J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 33, 2302–2309 (2005).
Holm, L. & Sander, C. Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 233, 123–138 (1993).
Berman, H. M. et al. The Protein Data Dank. Nucleic Acids Res. 28, 235–242 (2000).
Orengo, C. A. et al. CATH — a hierarchic classification of protein domain structures. Structure 5, 1093–1109 (1997).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Pratt, O. S. et al. AlphaFold 2, but not AlphaFold 3, predicts confident but unrealistic β-solenoid structures for repeat proteins. Comput. Struct. Biotechnol. J. 27, 467–477 (2025).
Aubel, M., Eicholt, L. & Bornberg-Bauer, E. Assessing structure and disorder prediction tools for de novo emerged proteins in the age of machine learning. F1000Research 12, 347 (2023).
Hashimoto, K. et al. CAGE profiling of ncRNAs in hepatocellular carcinoma reveals widespread activation of retroviral LTR promoters in virus-induced tumors. Genome Res. 25, 1812–1824 (2015).
Zhou, K.-R. et al. ChIPBase v2.0: decoding transcriptional regulatory networks of non-coding RNAs and protein-coding genes from ChIP-seq data. Nucleic Acids Res. 45, D43–D50 (2017).
Ganot, P., Bortolin, M.-L. & Kiss, T. Site-specific pseudouridine formation in preribosomal RNA is guided by small nucleolar RNAs. Cell 89, 799–809 (1997).
Kiss-László, Z., Henry, Y., Bachellerie, J.-P., Caizergues-Ferrer, M. & Kiss, T. Site-specific ribose methylation of preribosomal RNA: a novel function for small nucleolar RNAs. Cell 85, 1077–1088 (1996).
Hammond, S. M., Bernstein, E., Beach, D. & Hannon, G. J. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature 404, 293–296 (2000).
Elbashir, S. M. et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411, 494–498 (2001).
Pillai, R. S. et al. Inhibition of translational initiation by Let-7 microRNA in human cells. Science 309, 1573–1576 (2005).
Chendrimada, T. P. et al. MicroRNA silencing through RISC recruitment of eIF6. Nature 447, 823–828 (2007).
Derrien, T. et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 22, 1775–1789 (2012).
Quek, X. C. et al. lncRNAdb v2.0: expanding the reference database for functional long noncoding RNAs. Nucleic Acids Res. 43, D168–D173 (2015).
Volders, P.-J. et al. LNCipedia: a database for annotated human lncRNA transcript sequences and structures. Nucleic Acids Res. 41, D246–D251 (2013).
Ma, L. et al. LncBook: a curated knowledgebase of human long non-coding RNAs. Nucleic Acids Res. 47, D128–D134 (2019).
Hon, C.-C. et al. An atlas of human long non-coding RNAs with accurate 5′ ends. Nature 543, 199–204 (2017).
Iyer, M. K. et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 47, 199–208 (2015).
Li, J. et al. Long noncoding RNA XIST: Mechanisms for X chromosome inactivation, roles in sex-biased diseases, and therapeutic opportunities. Genes Dis. 9, 1478–1492 (2022).
Rinn, J. L. et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129, 1311–1323 (2007).
Tripathi, V. et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 39, 925–938 (2010).
Clemson, C. M. et al. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol. Cell 33, 717–726 (2009).
Berghoff, E. G. et al. Evf2 (Dlx6as) lncRNA regulates ultraconserved enhancer methylation and the differential transcriptional control of adjacent genes. Development 140, 4407–4416 (2013).
Samson, J., Cronin, S. & Dean, K. BC200 (BCYRN1) — the shortest, long, non-coding RNA associated with cancer. Non-coding RNA Res. 3, 131–143 (2018).
Glažar, P., Papavasileiou, P. & Rajewsky, N. circBase: a database for circular RNAs. RNA 20, 1666–1670 (2014).
Kozomara, A., Birgaoanu, M. & Griffiths-Jones, S. miRBase: from microRNA sequences to function. Nucleic Acids Res. 47, D155–D162 (2019).
Cheetham, S. W., Faulkner, G. J. & Dinger, M. E. Overcoming challenges and dogmas to understand the functions of pseudogenes. Nat. Rev. Genet. 21, 191–201 (2020).
Sisu, C. et al. Comparative analysis of pseudogenes across three phyla. Proc. Natl Acad. Sci. USA 111, 13361–13366 (2014). This paper summarizes the processes that create and degrade pseudogenes across the human, nematode and fruit fly genomes.
Esnault, C., Maestre, J. & Heidmann, T. Human LINE retrotransposons generate processed pseudogenes. Nat. Genet. 24, 363–367 (2000).
Wagner, A. The fate of duplicated genes: loss or new function? Bioessays 20, 785–788 (1998).
Suzuki, I. K. et al. Human-specific NOTCH2NL genes expand cortical neurogenesis through delta/notch regulation. Cell 173, 1370–1384 e1316 (2018).
Fiddes, I. T. et al. Human-specific NOTCH2NL genes affect notch signaling and cortical neurogenesis. Cell 173, 1356–1369 e1322 (2018).
Pei, B. et al. The GENCODE pseudogene resource. Genome Biol. 13, R51 (2012).
Gabriel, L. et al. BRAKER3: Fully automated genome annotation using RNA-seq and protein evidence with GeneMark-ETP, AUGUSTUS, and TSEBRA. Genome Res. 34, 769–777 (2024). This paper describes BRAKER3, an automated annotation pipeline that includes both ab initio prediction and RNA-seq evidence.
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinform. 12, 491 (2011).
Souvorov, A. et al. Gnomon–NCBI eukaryotic gene prediction tool. National Center for Biotechnology Information https://www.ncbi.nlm.nih.gov/core/assets/genome/files/Gnomon-description.pdf (2010).
Aken, B. L. et al. The Ensembl gene annotation system. Database https://doi.org/10.1093/database/baw093 (2016).
Banerjee, S. et al. FINDER: an automated software package to annotate eukaryotic genes from RNA-Seq data and associated protein sequences. BMC Bioinform. 22, 205 (2021).
Brůna, T. et al. Galba: genome annotation with miniprot and AUGUSTUS. BMC Bioinform. 24, 327 (2023).
Keilwagen, J., Hartung, F., Paulini, M., Twardziok, S. O. & Grau, J. Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinform. 19, 189 (2018).
Zimin, A. V., Puiu, D., Pertea, M., Yorke, J. A. & Salzberg, S. L. Efficient evidence-based genome annotation with EviAnn. Nat. Methods (in the press). This paper describes EviAnn, an automated annotation pipeline that combines RNA-seq evidence, protein-to-DNA alignment and transcripts from related species.
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 (2010).
Slater, G. S. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinforma. 6, 31 (2005).
Li, H. Protein-to-genome alignment with miniprot. Bioinformatics https://doi.org/10.1093/bioinformatics/btad014 (2023).
Gotoh, O. Spaln3: improvement in speed and accuracy of genome mapping and spliced alignment of protein query sequences. Bioinformatics https://doi.org/10.1093/bioinformatics/btae517 (2024).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644 (2008).
Brůna, T., Lomsadze, A. & Borodovsky, M. GeneMark-ETP significantly improves the accuracy of automatic annotation of large eukaryotic genomes. Genome Res. 34, 757–768 (2024).
Gabriel, L., Becker, F., Hoff, K. J. & Stanke, M. Tiberius: end-to-end deep learning with an HMM for gene prediction. Bioinformatics https://doi.org/10.1093/bioinformatics/btae685 (2024).
Shumate, A. & Salzberg, S. L. Liftoff: accurate mapping of gene annotations. Bioinformatics 37, 1639–1643 (2021). This publication describes Liftoff, the first standalone tool that could map annotation from one genome directly onto another.
Shumate, A. & Salzberg, S. LiftoffTools: a toolkit for comparing gene annotations mapped between genome assemblies. F1000Research 11, 1230 (2022).
Chao, K.-H. et al. Combining DNA and protein alignments to improve genome annotation with LiftOn. Genome Res. 35, 311–325 (2025).
Fiddes, I. T. et al. Comparative Annotation Toolkit (CAT)—simultaneous clade and personal genome annotation. Genome Res. 28, 1029–1038 (2018).
Shumate, A. et al. Assembly and annotation of an Ashkenazi human reference genome. Genome Biol. 21, 129 (2020).
Nurk, S. et al. The complete sequence of a human genome. Science 376, 44–53 (2022). This landmark paper describes the first ever complete human genome sequence, whose annotation includes 140 new protein-coding genes.
Alonge, M., Shumate, A., Puiu, D., Zimin, A. V. & Salzberg, S. L. Chromosome-scale assembly of the bread wheat genome reveals thousands of additional gene copies. Genetics 216, 599–608 (2020).
The International Wheat Genome Sequencing Consortium (IWGSC). Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 361, eaar7191 (2018).
Morales, J. et al. A joint NCBI and EMBL-EBI transcript set for clinical genomics and research. Nature 604, 310–315 (2022). This paper describes the MANE database, a very high-quality annotation containing a single splice isoform for nearly every human protein-coding gene.
Stelzer, G. et al. The GeneCards suite: from gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinform. 54, 1.30.31–31.30.33 (2016).
Laulederkind, S. J. F. et al. The rat genome database: genetic, genomic, and phenotypic data across multiple species. Curr. Protoc. 3, e804 (2023).
Yoshimura, J. et al. Recompleting the Caenorhabditis elegans genome. Genome Res. 29, 1009–1022 (2019).
Sakai, H. et al. Rice Annotation Project Database (RAP-DB): an integrative and interactive database for rice genomics. Plant Cell Physiol. 54, e6 (2013).
Dyer, S. C. et al. Ensembl 2025. Nucleic Acids Res. 53, D948–D957 (2025).
Salzberg, S. L. Genome re-annotation: a wiki solution? Genome Biol. 8, 102 (2007).
Sternberg, P. W. et al. WormBase 2024: status and transitioning to alliance infrastructure. Genetics https://doi.org/10.1093/genetics/iyae050 (2024).
Keilwagen, J. et al. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 44, e89–e89 (2016).
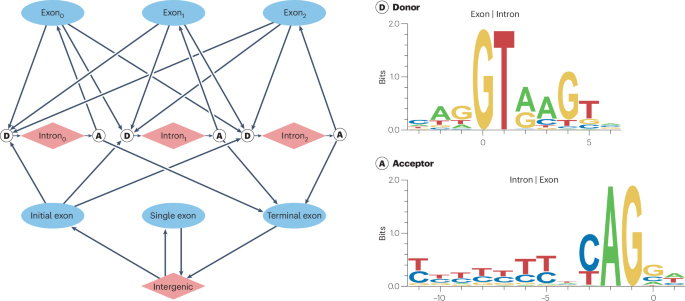
Crooks, G. E., Hon, G., Chandonia, J. M. & Brenner, S. E. WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190 (2004).
Goffeau, A. et al. Life with 6000 genes. Science 274, 546–567 (1996).
International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001).
Venter, J. C. et al. The sequence of the human genome. Science 291, 1304–1351 (2001).
Curwen, V. et al. The Ensembl automatic gene annotation system. Genome Res. 14, 942–950 (2004).
Cantarel, B. L. et al. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196 (2008).
Hubbard, T. et al. The Ensembl genome database project. Nucleic Acids Res. 30, 38–41 (2002).
Pruitt, K. D., Tatusova, T. & Maglott, D. R. NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 33, D501–D504 (2005).
The FlyBase Consortium. The FlyBase database of the Drosophila genome projects and community literature. Nucleic Acids Res. 31, 172–175 (2003).
Adams, M. D. et al. Complementary DNA sequencing: expressed sequence tags and human genome project. Science 252, 1651–1656 (1991).
Allen, J. E. & Salzberg, S. L. JIGSAW: integration of multiple sources of evidence for gene prediction. Bioinformatics 21, 3596–3603 (2005).