Introduction
Mulberry (Morus spp. L.), belonging to the Moraceae family, has a long being cultivated in Asia for its economic significance in sericulture, food and pharmaceutical industries worldwide1. The rhizosphere of mulberry, a complex ecosystem harboring diverse microbial communities, remains an unexplored frontier for discovering novel plant-growth-promoting rhizobacteria (PGPR). Over the years, the PGPR has gained global importance and acceptance for crops and is considered as an alternative to chemical fertilizer. Thus, undoubtedly, PGPR deserves great attention in biological control. The concept of PGPR, introduced by Kloepper and Schroth (1978)2, refers to soil bacteria capable of colonizing plant roots and exerting beneficial effects. These bacteria employ multiple mode of action to facilitate plant growth, including: (i) improving nutrient access (solubilizing phosphate, potassium, oxidizing sulfur, fixing nitrogen, and binding iron and copper); (ii) helps in suppression of soil-borne pathogens by releasing antimicrobials, hydrogen cyanide and siderophores; (iii) improving plant stress tolerance to environmental stresses such as drought, metal toxicity and salinity; and (iv) promoting growth through phytohormone production like indole-3-acetic acid (IAA)3. IAA, the most common plant hormone in the auxin class, plays a vital role in regulating key physiological processes viz., cell growth, division, and tissue differentiation. Certain bacteria capable of producing IAA are particularly beneficial in agriculture, as they promote plant growth by stimulating root development, enhancing nutrient absorption, and strengthening overall plant resilience4,5. Several soil bacterial genera identified as PGPRs, these include Azospirillum, Bacillus, Beijerinckia, Clostridium, Klebsiella, Phyllobacterium Pseudomonas, Vario–vovax, and Xanthomonas6.
The predominant bacterial genera present in healthy mulberry rhizosphere soil include Bacillus, Pseudomonas, Proteobacteria, and Actinobacteria7. Among PGPR, the Bacillus genus stands out as effective rhizobacteria with multiple beneficial traits. In addition to promoting plant growth and nutrient uptake they also play a crucial role in biocontrol as well by outcompeting harmful microbes, inducing systemic resistance (ISR), and producing antimicrobial compounds8. Furthermore, Bacillus species synthesize various enzymes that help plants cope with biotic stresses, making them valuable allies in sustainable agriculture. Their capability to survive in diverse soil conditions, form resilient endospores, and produce bioactive compounds makes Bacillus a promising candidate for enhancing plant growth under environmental stressors9,10,11. This study thus aimed at isolating and characterizing PGPR strains with strong PGP traits from the mulberry rhizosphere. The plant-growth-enhancing features, along with the whole genome analysis of the most potent PGPR strain depicted as “RMG6T,” were also initiated to explore its potential applications as biocontrol agent. Our study presents a comprehensive genome sequence analysis of RMG6T alongside its phylogenetically related species, aiming to identify potential biosynthetic gene clusters (BGCs), predict their putative metabolic products, and classify protein-coding genes based on their functional attributes. Thus, our findings not only validate the use of Bacillus RMG6T as a PGPR but also provide a strong foundation for future research into its wider applications in sustainable agriculture.
Materials and methods
Sampling and isolation
The mulberry rhizosphere soil samples were taken in replicates in a sterile vial from a depth of 6 inches (15 cm) using a sterilized sampler and preserved at 4 °C for transferring to lab from the mulberry garden (25.6072° N, 88.1303° E), of Raiganj University, West Bengal, India. For each replicate, three to five soil samples were collected, thoroughly mixed, and a 10–25 g portion was selected as a representative sample and heat treated at 65 °C for 2 h to facilitate the selective enrichment of spore-forming bacteria. After that, the samples were suspended in 90 mL sterile 0.9% NaCl solutions, serially diluted, spread on nutrient agar (NA) (Himedia) medium, and incubated overnight at 35 °C12. NA plates without any inoculum were used as a negative control to check any spontaneous growth. The bacterial colonies with distinct morphological traits were selected for further purifications. Repeated sub-culturing was performed to ensure the purity of the isolates and consistency in its characteristics. The purified colonies were picked and preserved on freshly prepared NA slants for future use.
Screening of Bacillus like isolates with biocontrol potential against Gram-positive and Gram-negative bacteria
Antimicrobial assay was performed using agar-well diffusion method on Mueller–Hinton (MH) agar. By this method, two days old liquid culture of different Bacillus like isolates were centrifuged and the supernatants were collected and added on the MH agar plates wells (6 mm diameter) having the lawn of the test organism (Escherichia coli MTCC 43 & Enterococcus faecalis MTCC 439) to screen the most potent antimicrobial producing isolates. After 24 h the zone of inhibitions was recorded and the strain exhibiting highest antimicrobial activity was selected and designated as RMG6T. Further, RMG6T cells were kept on NA plates consecutively for 12 generations prior to preservation at − 80 °C in 20% glycerol. To determine the changes in cellular morphology of the test organisms were examined via scanning electron microscopy (SEM; Hitachi S-3400) analysis as described earlier13. Briefly, the bacterial cells were fixed with a 2.5% glutaraldehyde solution, followed by dehydration through a progressive series of ethanol treatments, and subjected to examination under SEM with an accelerating voltage of 15–20 kV. Multiple fields of vision were viewed at different magnifications13,14. Moreover, The SEM micrographs of RMG6T cells were also captured following the previously reported protocol13.
Physiology and chemotaxonomy
Morphological, cultural, physiological, and biochemical characteristics were analyzed as previously described15. Colonies grown on NA plates was examined for their cultural and morphological characteristic; that includes the cell shape, colonial appearance, and endospore formation after 48 h of incubation at 35 °C. The Gram staining was done using a Gram staining kit (NICE chemicals G21950) as per manufacturer’s protocol. The pure culture of RMG6T was biochemically characterized using Vitek-2 Compact Automated system as described earlier14. Growth in various salt concentrations (0–14% w/v), at different pH values (3–11) and at different temperatures (4, 15, 20, 25, 30, 37, 40, and 45 °C). All the tests were carried out incubating the cultures at 35 °C, except for investigations into the effect of temperature upon growth. After confirming the optimum growth conditions of RMG6T, the doubling time (TD) and specific growth rate (k) was also determined16. Furthermore, RMG6T was identified using a Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) Microflex LT Mass Spectrometer (Bruker Daltonics) equipped with MALDI Biotyper 3.0 software (Bruker Daltonics). The samples were precisely applied onto the target plate and overlaid with 1 μL of alpha-cyano-4-hydroxycinnamic acid (HCCA) matrix before undergoing MALDI-TOF MS analysis. Identification was considered reliable at the species level for log scores of ≥ 2.0, at the genus level for scores of ≥ 1.7, while scores below 1.7 were deemed insufficient for accurate classification. Moreover, the total content of cellular fatty acid in RMG6T was determined following the MIDI protocol (Microbial Identification). Extraction and methylation of fatty acids were done to form fatty acid methyl esters (FAME) and analyzed using Gas Chromatography (Agilent 6850 gas chromatograph) with the help of MIDI Sherlock software (MIDI Sherlock™ Microbial Identification System v.6.02 USA; http://www.midi-inc.com/) for FAME. Aerobic library (RTSBA6 6.21) was referred for the analysis17,18.
Detection and quantification of IAA
Spectrophotometric method
The bacteria RMG6T were incubated in L-tryptophan (2 mg/mL) containing Nutrient broth in 100 mL Erlenmeyer flasks in triplicates at 35 °C on a BOD incubator and shaker for 48 h. Initially, centrifugation of the RMG6T culture was done at 10,000 g for 10 min, and the supernatant was mixed with Salkowski reagent (50 mL, 35% of perchloric acid, 1 mL 0.5 M FeCl3 solution) in a 1:1 ratio. A pink color indicated IAA production, and its absorbance was measured at 530 nm. The IAA concentration was determined using a standard curve (10–100 μg/mL) from HiMedia.
Extraction of indolic derivatives
The extraction of indolic derivatives was performed from the bacterial culture supernatants obtained after 48 h of incubation in nutrient broth with tryptophan (Trp; 2 mg/mL) at 35 ± 2 °C. Following acidification to pH 2.5 with 1 N HCl, the culture supernatants underwent three sequential extractions with equal volume of C4H8O2. A fraction of the C4H8O2 phase was then air-dried and reconstituted in one-tenth of its original volume using methanol, in accordance with previously described earlier19. The methanolic extracts were then used for HPTLC analysis.
High-performance thin-layer chromatography (HPTLC) analysis
Thin-layer chromatography (TLC) analysis was conducted using the spray-on technique for sample loading onto TLC foils (20 × 10 cm) with a Linomat 5 applicator (CAMAG, Muttenz, Germany) equipped with a Hamilton 100 μL syringe. The application parameters included an 8 mm band length, a 150 nL/s application rate, a table speed of 10 mm/s, and an automatically set band spacing. The first track was positioned at 15 mm (X), with 11.3 mm between tracks, while scanning positions ranged from 5 to 70 mm (Y). The solvent front was set at 70 mm. A methanolic solution containing standard IAA, tryptamine, and IAM (250 µg/mL) was applied, with loaded volumes of 2 μL (500 ng), 3 μL (750 ng), 4 μL (1000 ng), and 5 μL (1250 ng), forming four tracks per standard. The TLC plate was developed using isopropanol: ammonia: water (8:1.4:0.6, v/v) as a mobile phase and pre-saturated with the same mobile phase for 10 min at 26 °C. After development of the TLC plates, those were air-dried, and visualized under a UV chamber at 254 nm. The mobile phase took 90 min to reach the solvent front (70 mm). For quantitative analysis, densitometric measurements were performed at 254 nm using a CAMAG TLC scanner 3 (D2 lamp) with a scanning speed of 20 mm/s. The slit dimensions were set at 5 × 0.45 mm, with a data resolution of 100 µm per step, using the Savitzky-Golay 7 filter for smoothing. Baseline correction was applied at the lowest slope, and display scaling was adjusted automatically. The limit of detection (LOD) and limit of quantification (LOQ) were measured via plotting a scatter graph of peak area versus the amount of standard loaded per band. These values were calculated using the formula LOD = 3.3 σ/S, where σ represents the standard deviation (SD) and S is the slope of the calibration curve. The SD (Y-axis) and slope were obtained from the standard curve (sample amount in µg on X-axis vs. peak area on Y-axis) using the LINEST function in MS Excel 2017. The recovery percentage of each standard was calculated comparing the measured quantity to the initially loaded amount, considering 100% as the reference20,21,22,23.
Seed germination assay
Efficacy of RMG6T was tested for any stimulating activity on the seed germination of Vigna radiata. Five viable seeds were collected and surface sterilized using a 0.1% HgCl₂ solution, followed by through rinsing in sterile distilled water to remove any residual HgCl2. The sterilized seeds were then soaked in sterile distilled water for one hour and placed on a sterile, moistened tissue paper at the bottom of a Petri dish. The bacteria were cultured in a L-tryptophan (0.2%) containing Nutrient Broth at 35 °C for 24 h. Bacterial culture was centrifuge at 10,000 rpm for 10 min. Supernatant (containing IAA) 2 mL was utilised for seed germination assay. For the control, only sterile nutrient broth was used. The experiment was conducted in triplicate. Seed germination was observed after 24–48 h in each experimental set, following a previously reported method24.
Antibiotic susceptibility testing
The Kirby-Bauer disc diffusion assay was conducted to assess the antibiotic susceptibility profile of Bacillus sp. strain RMG6T25. A total of 43 antibiotics, commonly used in human and veterinary medicine, were obtained from HiMedia Laboratories Pvt. Ltd., India. The strain was cultured overnight spreading uniformly onto MH agar (HiMedia Laboratories Pvt. Ltd., India) plates. The antibiotic discs were placed on agar surface, and incubated at 37 °C for 24 h. Based on the formation of inhibition zones, the antibiotic sensitivity was determined; the presence of a halo indicated susceptibility, while the absence of a halo indicated resistance. The results were interpreted according to the European Committee on Antimicrobial Susceptibility Testing (EUCAST) clinical breakpoints (version 14.0, https://www.eucast.org/clinical_breakpoints). The strain was classified as resistant (R), intermediate (I), or susceptible (S) based on EUCAST guidelines for Bacillus sp.
Molecular identification of isolated bacteria
The genomic DNA of the isolated bacteria was extracted using the phenol–chloroform extraction method26. The purity of the extracted DNA was assessed spectrophotometrically using a NanoDrop 1000. Its quality was also verified through electrophoresis on a 1% agarose gel. Briefly, 2 µL of isolated DNA was mixed with 1 µL of 2 × loading dye (Invitrogen) and then subjected to electrophoresis at 120 V for 60 min. The results were documented using a BioRad ChemiDoc MP Imager (Model No. 12003154). For molecular identification, the 16S rDNA was amplified using universal primers 27F, 704F, and 907R27. The PCR reaction (25 µL total volume) contained 12.5 µL of PCR Master Mix (Sigma Aldrich, St. Louis, MO), 2 µL of template DNA, 1 µL of each primer, and 8.5 µL of nuclease-free distilled water. The thermal cycling conditions included denaturation at 95 °C for 5 min, followed by 35 cycles of denaturation at 95 °C for 30 s and annealing at 55 °C for 30 s and elongation at 72 °C for 1 min, and then cycling was completed by a final elongation step for 10 min at 72 °C. A negative control with water was included in each PCR reaction. The amplified PCR products were analyzed by electrophoresis on a 1% agarose gel. The purified 16S rDNA was sequenced using the Sanger sequencing method. Both strands of the nucleotide sequences were aligned using BioEdit software (BioEdit Sequence Alignment Editor v.7.2.5; http://www.mbio.ncsu.edu/BioEdit/bioedit.html) to obtain the partial or complete sequence of the 16S rDNA. The sequence was then submitted to the EZBioCloud server for identification of the closest type strains. Multiple sequence alignment of the retrieved type strains was performed using the ClustalW program built in MEGA11 software. The neighbor-joining (NJ) method was adopted to construct the phylogenetic trees. The evolutionary distances were calculated using the Kimura 2-parameter model. Bootstrap analysis was performed with 1000 replications to validate the clades of the phylogenetic tree. The NCBI accession number for the 16S rDNA sequence of strain RMG6T is PP494329.
Multi-locus DNA sequence based phylogenetic position analysis
Multi-Locus Sequence Analysis (MLSA) is a method that can be used for determining the evolutionary ancestry of species within a genus. In order to differentiate species from their close relatives, a phylogenetic tree based on multiple genes generates more information than one that relies exclusively on the 16S rRNA gene. The housekeeping gene sequences were used for construction of a phylogenetic tree of MLSA, and phylogenies are subsequently generated28. The whole genome sequence was used to extract nine genes for the MLSA of RMG6T, including gyrB (DNA gyrase subunit B), atpD (F0-F1 ATP synthase subunit β), dnaK (molecular chaperone protein), gmk (guanylate kinase), ilvD (dihydroxy-acid dehydratase), purH (bifunctional phosphoribosylaminoimidazolecarboxamide formyltransferase), pycA (pyruvate carboxylase), rpoB (DNA-directed RNA polymerase subunit beta), rpoD (RNA polymerase sigma factor) from the whole genome sequence (accession number-JBEFNT000000000). The housekeeping gene sequences and their closest relatives (according to 16S rRNA gene sequence-based analysis) were downloaded from GenBank. Following alignment using MEGA 11 software, the nucleotide sequences associated to the corresponding genes were manually trimmed at identical positions. The nine-housekeeping gene sequences were concatenated head to tail as gyrB-atpD-dnaK-gmk-ilvD-purH-pycA-rpoB-rpoD for RMG6T and its nearest relatives. Pairwise distances were computed using the Kimura 2-parameter model29. MEGA 11 software was used to generate the best model for phylogenetic analysis, and phylogenetic analysis took place using the the best model parameters. The Tamura nei (TN93) model by Gamma distributed with invariant sites (G + I) was determined to be the best fit for maximum likelihood30.
Draft genome sequencing and assembly
The genome of Bacillus sp. strain RMG6T was sequenced to identify genes involved in the biosynthesis of antimicrobial compounds and production of different types of plant growth-promoting traits like siderophore, indole acetic acid, etc.
DNA extraction was performed using the DNeasy Ultraclean Microbial Kit., followed by quantity confirmation using Nanodrop 1000. Construction of paired-end libraries was done utilizing Ultra II DNA Library Prep Kit (NEBNext #E7645S/L) and Illumina NovaSeq 6000 platform (Neuberg Diagnostics Pvt. Ltd., Ahmedabad, India) used for sequencing, producing 13,625,248 reads having 2 × 159 bp paired-end read length. The quantification of final DNA libraries was performed using a Qubit 4.0 fluorometer (Thermo Fisher #Q33238) with a DNA HS assay kit (Thermo Fisher #Q32851), as per manufacturer’s guidelines. The library insert size was analyzed using the TapeStation 4150 system (Agilent) with highly sensitive D1000 ScreenTapes (Agilent #5067–5582).
Quality assessment of the samples raw fastq reads was done and processed using FastQC v.0.11.9 (default parameters)31 and Fastp v.0.20.132 (parameters: –length_required 50 –correction –trim_poly_g –qualified_quality_phred 30 –unqualified_percent_limit 30 –average_qual 30) respectively. Thereafter, the processed data were re-assessed using FastQC. The processed reads were de novo assembled using Unicycler v.0.4.4,33 Megahit v1.2.934 and Spades v4.035 with default parameters. The completeness and contamination rate of the assembled genome were finally determined using CheckM2 v.1.0.136.
Genome sequence annotation and genomic data analysis
Prokaryotic Genome Annotation Pipeline (PGAP) v6.7 in NCBI was used to carry out the genome sequence annotation utilizing the Best-Placed Reference Protein Set and GeneMarkS-2 + methods37 and later it was also compared with another annotation platform viz. BV-BRC v3.46.3. The completed genome assembly sequence was submitted to the DSMZ Type Strain Genome Server (TYGS v.391; https://tygs.dsmz.de/user_requests/new), and analyzed using the Genome to Genome Distance Calculator (GGDC v3.0; https://ggdc.dsmz.de/ggdc.php#)38. Phylogenetic trees produced by TYGS were visualized using the interactive tree of life (iTOL V6; iTOL: Interactive Tree Of Life (embl.de)39 with Escherichia coli DSM 30,083 selected as an outgroup40. Average nucleotide identity (ANI) was calculated on the Ezbiocloud server (https://www.ezbiocloud.net/tools/ani) using the OrthoANIu algorithm41 against the closest relative identified by the Ezbiocloud server. Finally, a genome map of Bacillus sp. strain RMG6T was constructed using CGView Server (https://cgview.ca/)42 to visualize distinct contigs, CDSs (in both forward and reverse orientations), GC content and skew. Furthermore, the assembled genomic data underwent annotation and comparative analysis using the Subsystem Technology tool kit (RASTtk). This process facilitated the prediction of genes associated with risk assessment, including virulence factors, antibiotic resistance genes (ARGs), potential drug targets, and human homologs)43. Moreover, the AutoMLST web platform (autoMLST: Automated Multi-Locus Species Tree (ziemertlab.com)44 was used to evaluate the presence BGCs and the potential of the isolate RMG6T for secondary metabolite production. The antiSMASH v 7.1.0 (https://antismash.secondarymetabolites.org/#!/start) was used for the genome mining of secondary metabolite BGCs in strain RMG6T45. The metabolic pathways of strain RMG6T and other closely related Bacillus species were characterized and compared with the METABOLIC software v4.046. In addition, the metabolic pathway annotation and completeness of the pathway modules of strain RMG6T and other closely related Bacillus species were determined using GhostKOALA47,48,49,50. Heatmap analysis of differentiated pathways was performed with the complex heatmap package in the R program51. Genomes were aligned and compared with BLAST Ring Image Generator (BRIG) to generate a circular map of closely related species52.
Results and discussion
Isolation, screening, 16S rDNA, and Multilocus DNA sequence based phylogenetic position analysis of RMG6T
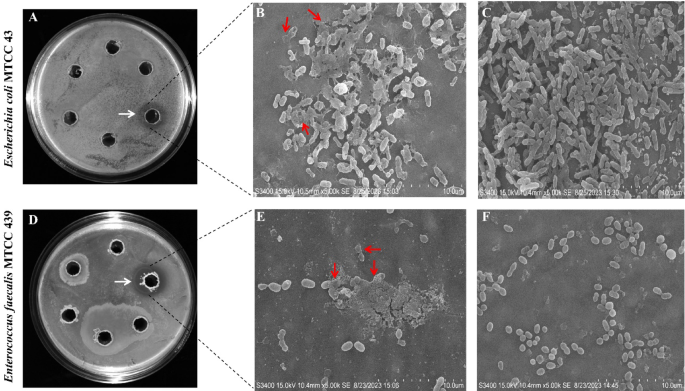
A total of 48 bacterial colonies were obtained from the rhizosphere soil of mulberry. Among them, only one isolate, RMG6T exhibited significant biocontrol potential and demonstrated in vitro antagonism against Escherichia coli MTCC 43 and Enterococcus faecalis MTCC 439 Our results revealed that the culture supernatant (50 µL) of RMG6T showed a larger zone of inhibition against Enterococcus faecalis MTCC 439 (1.92 cm) compared to Escherichia coli MTCC 43 (1.62 cm) (Fig. 1A,D; as highlighted with white color arrow). SEM micrographs also revealed RMG6T supernatant mediated cell-lysis of the tested bacteria as evidenced by cytoplasmic leakage (as highlighted with red color arrow) (Fig. 1B,E) compared to the control (Fig. 1C,F). This study aligned with an earlier study that reported the antimicrobial activity of cell-free supernatant of Bacillus sp. against E. faecalis53.

Evaluation of biocontrol potential of RMG6T. (A) Zone of inhibition study through agar-well diffusion assay of RMG6T against E. coli MTCC 43, (B) SEM micrograph of E. coli MTCC 43 treated with cell-free supernatant of RMG6T, (C) SEM micrograph of E. coli MTCC 43 as control, (D) Zone of inhibition study through agar-well diffusion assay of RMG6T against E. faecalis MTCC 439, (E) SEM micrograph of E. faecalis MTCC 439 treated with cell-free supernatant of RMG6T, and (F) SEM micrograph of E. faecalis MTCC 439 as control.
To determine the taxonomic position of the isolate, a 16S rDNA-based phylogenetic analysis was conducted. The short (1545 bp) 16S rDNA-based phylogenetic analysis of closely related type strains revealed that the isolate RMG6T was a member of the Bacillus genus and the closest relative to the isolate was the Bacillus siamensis KCTC 13,613 (Fig. 2) with 99.93% similarity and 1 nt variation was present.

16S rDNA sequence phylogeny of Bacillus sp. strain RMG6T. Only > 50% of bootstrap values (expressed as % of 1000 replications) are shown at the nodes.
Further, The MLSA pairwise distance, calculated by employing Kimura-2 parameter model, between strain RMG6T and its phylogenetically associated species exceeded the threshold value of 0.007, supporting its designation as a new species, as recommended by Rong and Huang (2010)54 (Table 1).
The ML tree generated using the best fit model parameters for the MLSA analysis revealed that RMG6T, Bacillus siamensis KCTC 13,613 and Bacillus amyloliquefaciens DSM 7, all descended from the same ancestor indicating close relativeness (Fig. 3).

Maximum-likelihood (ML) tree of RMG6T showing position of the strain amongst its phylogenetic neighbors. Maximum likelihood tree using Tamura nei (TN93) model by Gamma distributed with invariant sites (G + I) method based on the house-keeping gene (gyrB-atpD-dnaK-gmk-ilvD-purH-pycA-rpoB-rpoD).
Furthermore, the digital DNA-DNA hybridization (dDDH) similarity of RMG6T with Bacillus siamensis KCTC 13,613 and Bacillus amyloliquefaciens DSM 7 was only 56.80% and 55.20%, respectively, which is much lower than the suggested threshold of 70% for species delineation55. Thus, RMG6T was considered as a novel species within the Bacillus genus and designated as Bacillus ayatagriensis sp. nov. RMG6T (= MCM-B-1537).
Physiology and chemotaxonomy
The RMG6T colonies exhibited milky white, wrinkled protuberances at the center and was opaque with an irregular edge when observed after 48 h of incubation on NA medium at 35 °C (Fig. 4A). Morphology evaluation of colonies derived from Bacillus siamensis on NA plates exhibited small, irregular, round yellowish white colonies on NA medium56. Another study reported that the colonies of Bacillus siamensis on NA plates were round, greyish, flat and sticky with smoother edges57. Thus, the differences in the morphology of RMG6T colony pattern suggesting an initial clues about the bacterial classification of different species. When observed under light microscope, RMG6T cells belong to the Gram-positive class (Fig. 4B). SEM micrograph of RMG6T vegetative cells revealed that the isolated strain RMG-6 was rod-shaped cells with a diameter of 2.45 µm, chains or clusters with a relatively smooth surface (Fig. 4C,D); the most notable feature is the presence of endospore, which can appear as intact and cylindrical with bumpy, rough, wrinkled surfaces that possess numerous folds and creases (Fig. 4E,F), consistent with previously reported findings58.

Morphology and microscopic view of RMG6T. (A) RMG6T colony on NA agar plate, (B) Light microscopic view of RMG6T at 40 × magnification, (C) SEM micrograph of RMG6T at 5000 × magnification, (D) Diameter measurement of RMG6T cells at 5000 × magnification, (E) SEM micrograph of RMG6T spores at 20,000 × magnification, and (F) at 15,000 × magnification.
To further characterize the isolate, biochemical profiling of RMG6T was conducted using Vitek-2 automated technology. Based on the biochemical characteristics, the Vitek-2 automated system was unable to identify the strain RMG6T (Supplementary Fig. 1). Table 2 shows the main features that distinguished Bacillus sp. RMG6T from other phenotypical and phylogenetic taxa.
The strain exhibited a broad growth range, thriving at temperatures between 20 °C and 50 °C (Fig. 5A; Table 2), with an optimum growth temperature of 35 °C (Fig. 5B; Table 2). It also demonstrated adaptability to a pH range of 4–9, with optimal growth at pH 6 and showed remarkable salt tolerance, growing in NaCl concentrations of up to 14% (Fig. 5C; Table 2). Furthermore, the growth pattern of the strain RMG6T up to 16 h in a nutrient broth medium was also recorded and presented in Fig. 5D. The growth kinetics of RMG6T revealed that the TD (= doubling time) and of k (= growth rate constant) of RMG6T cells, was 76.17 ± 1.71 min (mean ± standard deviation) and k was 0.0085 ± 0.0001 doubling times per min, respectively. These findings are in agreement with previously reported results64,65.

Effect of temperature (A), pH, (B) and NaCl concentration (C) on the growth of Bacillus sp. strain RMG6T, and growth kinetics of Bacillus sp. strain RMG6T (D).
Further identification of RMG6T using MALDI-TOF MS yielded a log score of ≥ 2.0 or ≥ 1.9, confirming its classification at the genus level. One Bacillus isolate was identified with a log score of 2.04 using the Bruker system. While MALDI-TOF MS is widely used to identify Bacillus species, its accuracy can be affected by the presence of endospores, which alter the protein composition of samples. The extent of endospore formation can influence MALDI spectra, depending on culture conditions. Due to these limitations, a more precise identification of RMG6T was pursued through additional fatty-acid profiling and genome-based analyses.
The total cellular fatty acid profile of RMG6T revealed a high proportion of branched fatty acids; the major components (> 5.0%) were anteiso-C15:0 (28.13%), iso-C15:0 (20.72%), C18:0 (10.72%), iso-C17:0 (7.00%), iso-C16:0 (5.88%), and anteiso-C17:0 (5.87%) (Table 3 & Supplementary Fig. 2).
FAME analysis suggested that strain RMG6T have highest diversity of fatty acids from the other closest related species of Bacillus. For example, C11:0, anteiso-C17:1ω9c, C18:3ω6c (6, 9, 12), C18:0, and C20:0 exclusively present in the RMG6T strain. However, fatty acids such as C16:1ω7c (present only in Bacillus subtilis NCIB 3610), C16:1ω11c (present in Bacillus velezensis NRRL B-41580, Bacillus subtilis NCIB 3610, Bacillus amyloliquefaciens DSM 7, Bacillus nakamurai NRRL B-41091), and iso-C17:1ω10c (present only in Bacillus subtilis NCIB 3610) were not detected in the RMG6T strain, but they were identified in other related strains. These differences further support the taxonomic uniqueness of RMG6T.
Auxin production and plant growth-promoting potential of Bacillus ayatagriensis sp. nov. RMG6T
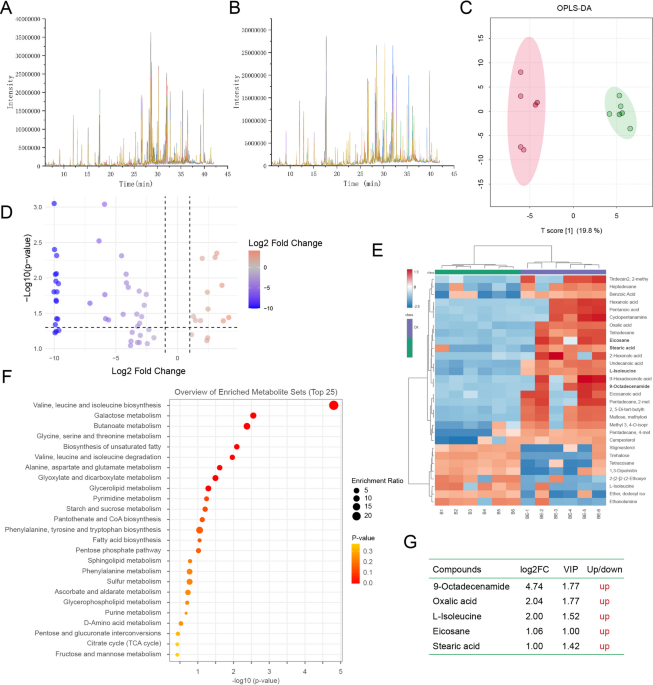
Auxin plays a crucial role in plant root development, making its production an important trait for plant-associated bacteria. To further investigate if RMG6T produces auxin, the total content of auxin was evaluated in the cell-free culture supernatant of RMG6T. The indole-3-pyruvic acid pathway (IPA) is one of the main pathways for the biosynthesis of IAA in bacteria. The synthesis is completed in three steps. In the first step tryptophan is converted to the indole-3-pyruvic acid by aminotransferases and then indole-3-acetaldehyde is formed by pyruvate decarboxylase. In final step, aldehyde dehydrogenases form indole-3-acetic acid67. Strain RMG6T uses IPA pathway for the biosynthesis of IAA from tryptophan. MEQ7682189.1 protein has 83% similarity to aromatic-amino-acid aminotransferase of Bacillus subtilis. Strain RMG6T has three pyruvate decarboxylase; MEQ7680847.1 (pdhA), MEQ7680848.1 (pdhB) and MEQ7680849.1 (pdhC) similar to Bacillus subtilis counterparts. Finally Strain RMG6T has three aldehyde dehydrogenases protein; MEQ7681358.1, MEQ7682689.1 and MEQ7683083.1, further supporting its ability to synthesize IAA (Supplementary Table 1). The IAA production capability of the strain RMG6T was experimentally validated in the L-tryptophan-supplemented nutrient broth. It was found that the strain could produce a maximum IAA of 73.4 μg/mL in 0.2% optimum concentration of L-tryptophan (Fig. 6B,C,E,F). HPTLC confirmed the IAA production (Fig. 6D), and the results were compared with the standard IAA (Fig. 6G–I). Moreover, the spectra comparison confirmed the presence of IAA in the strain RMG6T culture (Fig. 6A), reinforcing its ability to produce this essential phytohormone.

Spectra comparison (A) of ethyl acetate fraction extracted (Rf 0.48, 254 nm) from the strain RMG6T culture with different amounts (500 ng, 750 ng, 1000 ng and 1250 ng) of standard (Rf 0.49, 254 nm). (Inset Pic- Left: IAA production by RMG6T (B) with Control (C); Right: HPTLC plate (D) (Lanes 1, 2, 3, and 4 denote 500, 750, 1000, and 1250 ng mixture of standard IAA, Tryptamine and IAM respectively, and Lane 5 denotes RMG6T supernatant), (E) & (F) denotes estimation and Standard curve generation of IAA through spectrophotometrically analysis and (G–I) show Standard curve of IAA (G), Tryp. (H), IAM (I) in HPTLC.
Beyond its role in bacterial taxonomy, IAA production serves as an important ecological indicator, highlighting the potential interaction of RMG6T with mulberry plants. Since IAA is critical for plant growth and development, the significant levels produced by RMG6T suggest its potential as an effective plant growth-promoting bacterium (PGPB). Notably, RMG6T exhibited higher IAA production compared to previously reported Bacillus species, further distinguishing it within its taxonomic group (Table 4).
We anticipate that the growth-promoting effects on other crops could be further enhanced through RMG6T treatment, which facilitates increased IAA production. To substantiate this, Vigna radiata seed germination and rooting assays were conducted. The results demonstrated that treatment with RMG6T cell-free supernatant, containing bacterial-derived auxin, supported 100% seed germination and promoted the development of healthy radicles with an average length of 5.4 cm, significantly longer than the 3.06 cm observed in the untreated control group (Fig. 7). These findings are in accordance with earlier studies, reinforcing the potential of RMG6T for cross-species bioaugmentation72.

Vigna radiata seed germination effect in control (A) and cell-free bacterial supernatant (B).
Antibiotic susceptibility testing
The antibiogram profiling of RMG6T was tested against different clinically relevant antibiotics to determine its resistance profile. The antibiogram results revealed that the strain RMG6T was mainly resistant to cell wall inhibitors like penicillin, ampicillin, and ceftazidime and protein synthesis inhibitors like piperacillin (Supplementary Table 2). Genome annotation further supported these findings, revealing the presence of a Beta-lactamase class A (penP) gene, which likely contributes to its resistance against penicillin, ampicillin, and ceftazidime. Additionally, DeepARG analysis identified ARGs related to multidrug resistance, including those for bacitracin and phenicol, highlighting the genetic basis of RMG6T resistance (Supplementary Table 3).
Genome sequencing and biosynthetic gene clusters analysis
The genome assembly resulted in a sequence spanning 3,818,111 base pairs, distributed across 37 contigs. The assembly exhibited an N50 value of 995,572 base pairs and an L50 count of 2. The estimated GC content was determined to be 46.62%, with a sequencing coverage of 496.06 × . A total of 3,723 coding sequences were annotated, which include 3 rRNA genes (one 5S, one 16S, and one 23S) along with 76 tRNA genes. Additionally, according to BV-BRC v3.46.3 annotation platform, it was observed that the genome length and G + C content of RMG6T was 3,821,630 bp long and 46.63%, respectively. N50 length, L50 count, number of t-RNA, and r-RNA was found to be consistent with the statistics of NCBI-PGAP (Table 5).
The completeness and contamination of all genome assemblies produced by Unicycler, Megahit and Spades were 100% and 0%, respectively. However, Unicycler genome assembly was produced less contigs (37 contigs) compared to Megahit (49 contigs) and Spades (288 contigs). Thus, Unicycler genome assembly was selected for further analysis (Supplementary Table 4). Figure 8 shows the genome map generated in the CGView server.

Bacillus ayatagriensis sp. nov. strain RMG6T genome map generated using the CGView server, showing a full view of the genome. The blue arrows represent the CDSs and the gray arrows represent the contigs. The black plot shows GC content, while the green and magenta plot shows CG skew + and − , respectively.
The circular map generated by comparing the genome of Bacillus ayatagriensis sp. nov. RMG6T with other closely related Bacillus genomes showed a large number of variations with gaps or regions of low similarity (Fig. 9).

Circular map of the whole genome of Bacillus ayatagriensis sp. nov. and comparative genome maps of genome sequences of closely related Bacillus species. Gaps in the circles represent regions of low or no similarity.
GGDC v3.0 calculated the dDDH values. In this analysis, Formula 2 was used because its calculations are independent of genome length and are specifically recommended for incompletely sequenced genomes. The distance among the genomes of strain RMG6T and closely related Bacillus siamensis KCTC 13,613 was 56.80%. According to the previous reports, the dDDH values not exceeding 70% indicate that the tested organisms belong to different species73,74. OrthoANIu was employed to determine the ANI between the isolated strain and its phylogenetically closest type strains. Based on this algorithm, strain RMG6T showed a maximum average value of 94.44% with closely related Bacillus siamensis KCTC 13,613. Typically, the ANI values between genomes of the same species are above 95%75.
Comparative metabolic analyses were performed METABOLIC and GhostKOALA. Detailed metabolic pathway annotations were presented in supplementary Table 5. Heatmap of differentiated pathways shown that B. ayatagriensis sp. nov. RMG6T clustered with B. methylotrophicus and B. velezensis (Fig. 10).

Functional classification of protein-coding genes present in Bacillus ayatagriensis sp. nov genomes and closely related Bacillus species by the completeness of Kyoto Encyclopedia of Genes and Genomes (KEGG). Permission has been obtained from Kanehisa laboratories for using KEGG pathway database48,49,50. The color scale represents the completeness of pathway.
Furthermore, a whole-genome-based phylogenetic tree was constructed via the Type (Strain) Genome Server (TYGS) to elucidate the phylogenetic placement of the strains. The constructed tree showed that the strain RMG6T formed an individual clade in comparison with highly related Bacillus species (Fig. 11). Thus, as per the phylogenomic analysis, dDDH and ANI scores between Bacillus sp. strain RMG6T and closely related strains reveal that the strain RMG6T should be considered a distinct species.

Whole-genome phylogenetic tree produced with TYGS. 14 strains were automatically chosen using TYGS with GGDC. E. coli DSM 30,083 was included as an outgroup and the tree is rooted on this branch. Mean branch support is 65.8%, and bootstrap data is shown as a percent for each branch. δ statistics = 0.039–0.13. Excluding the outgroup: G + C 43.12–46.63 mol%, genome size 3.6–4.5 Mb, number of proteins 3622–4511. The branch lengths are scaled in terms of the GBDP distance formula d5.
The whole-genome sequence (WGS) of strain RMG6T was submitted to the RAST server (https://rast.nmpdr.org/) for comprehensive functional annotation76. Default parameters were applied across all software tools. The annotation process identified a total of 3,899 coding sequences and 79 predicted RNAs. RAST analysis further revealed the presence of siderophore gene clusters, as well as genes associated with the biosynthesis of indole acetic acid (IAA) and the metabolism of sulfur, phosphate, and nitrogen, key functions linked to plant growth promotion and biocontrol. A summary of the subsystem categories assigned to the genome of Bacillus ayatagriensis sp. nov. RMG6T is presented in Fig. 12.

An overview of the subsystem categories assigned to the genome of Bacillus sp. strain RMG6T. The WGS sequence of the strain RMG6T was annotated using the RAST server.
A phylogenetic tree of 49 Bacillus strains and representative species was also generated through In silico AutoMLST analysis using Bacillus ayatagriensis sp. nov. RMG6T as a query sequence (Fig. 13). The biosynthetic potentials (number of BGCs detected by antiSMASH) of each Bacillus strain are reflected in the color code. Among the 49 analyzed Bacillus strains, 1 had no BGCs (grey), 34 had between 7 and 13 BGCs (blue), and 13 strains had between 14 and 20 BGCs (green). This shows that Bacillus has a high potential for producing secondary metabolites.

Bacillus phylogenetic tree generated with autoMLST. Color code represents the number of BGCs identified by antiSMASH.
In silico analysis using antiSMASH v.7.1.0 (default parameters were used for all software unless otherwise noted) revealed that genome of the isolate RMG6T harbor putatively 20 BGCs (Table 6). The most structurally related compounds (≥ 40%) were characterized chemically and cataloged in the MiBIG (Minimum Information about a Biosynthetic Gene Cluster) database, as identified directly through antiSMASH analysis.
Analysis of cluster 1.3: The genome of the strain RMG6T was found to harbor a Trans-acyltransferase polyketide synthase (transAT-PKS) cluster which presented 100% similarity with a biosynthetic gene cluster known to code for macrolactin H (polyketide) in Bacillus velezensis FZB4279,80,81. Known cluster BLAST using MIBiG 3.1 showed highest similarity with BGC0000181 (macrolactin H biosynthetic gene cluster from Bacillus velezensis FZB42) this particular BGC shown in Fig. 14A. This particular cluster consist of 88,213 nucleotides comprising of a total 44 ORFs out of which 7 are for the core biosynthetic genes, 11 are for additional biosynthetic genes, 1 transport-related gene, 1 regulatory gene, and 23 other genes. The structure of these 7 core genes (1 to 7) is consisting of 11 modules with domain (Fig. 15A). The polymer prediction by the above-mentioned cluster is (ohmal–ccmal–ccmal) + (ccmal) + (ccmal–mal) + (ccmal–mal) + (ohmal) + (ohmal–mal); and the predicted core structure for this particular cluster is shown in Fig. 15B.

Highly similar biosynthetic gene clusters of Bacillus sp. RMG6T strain compared with known clusters in the antiSMASH database. Gene clusters for Macrolactin H (A), Bacillaene (B), Fengycin (C), Baciilysin (D), Bacillibactin (E), Difficidin (F and G), Bacinapeptin (H), Surfactin (I).

Schematic representation of the NRPS/PKS modules of gene clusters (A, C, E, G, I, K, M, O, Q, S) and the putative compounds produced by these clusters (B, D, F, H, J, L, N, P, R, T).
Analysis of cluster 1.4: This cluster was found to be a hybrid (NRPS, T3PKS, transAT-PKS) cluster exhibiting 100% similarity with the biosynthetic gene cluster known to code for bacillaene (polyketide + NRP) in Bacillus velezensis FZB4279,80,82,83,84. Known cluster BLAST using MIBiG 3.1 showed highest similarity with BGC0001089 (bacillaene biosynthetic gene cluster from Bacillus velezensis FZB42) this particular BGC shown in Fig. 14B. This particular BGCs is of 110,118 nucleotides length comprising 52 ORFs. Out of the 9 core biosynthetic genes, the NRPS/PKS modules analysis reveals that rest 4 of the core genes (5 to 9) is made up of 16 modules with domains (Fig. 15C). Polymer prediction by this particular cluster is (Gly–ccmal–ohmal –mal) + (ccmal–mal–ohmal–mal) + (Me–ccmal–ohmal) + (Ala–ccmal–ccmal–Me–ccmal) + (mal–mal); and the predicted core structure for this particular cluster is shown in Fig. 15D.
Analysis of cluster 1.5: This cluster was found to be hybrid (NRPS, transAT-PKS, betalactone) cluster which exhibited 80% similarity with the BGC known to code for the fengycin (NRP) in Bacillus velezensis FZB4280,85,86. Known cluster BLAST using MIBiG 3.1 showed highest similarity with BGC0001095 (fengycin biosynthetic gene cluster from Bacillus velezensis FZB42) this particular BGCs shown in Fig. 14C. This particular cluster contains 87,643 nucleotides comprising of 46 ORFs. It has been observed that this cluster presents three candidate BGCs within it [i.e., Candidate Cluster 1.5.1 (location 985,466–1,073,109): neighbouring NRPS-transAT-PKS-betalactone; Candidate Cluster 1.5.2 (location 985,466–1,062,712): chemical hybrid NRPS-transAT-PKS; and Candidate Cluster 1.5.3 (location 1,045,626–1,073,109): chemical hybrid NRPS-betalactone]. The 5 core biosynthetic genes for the candidate cluster 1.5.1 is made up of 10 modules, the 3 core biosynthetic genes for the candidate cluster 1.5.2 is made up of 3 modules, and 2 core biosynthetic genes for the candidate cluster is made up of 2 modules (Fig. 15E,G,I). Polymer prediction for the cluster 1.5.1 is (D-Tyr) + (Ile) + (mal–Asn) + (D-Tyr–D-Asn–Gln–Pro) + (D-Asn–Ser); for cluster 1.5.2 is (mal–Asn) + (D-Tyr–D-Asn–Gln–Pro) + (D-Asn–Ser); and for the cluster 1.5.3 is (D-Tyr) + (Ile). The predicted core structures for these three particular candidate clusters are shown in Fig. 15F,H,J.
Analysis of cluster 2.1: BGCs was predicted to encode a protein associated with secondary metabolism that does not align with any established secondary metabolite categories identified in antiSMASH. Nonetheless, this cluster exhibited 100% similarity with the BGC of Bacillus velezensis FZB42 which has been found to encode bacilysin87,88,89. Known cluster BLAST using MIBiG 3.1 showed highest similarity with BGC0001184 (bacilysin biosynthetic gene cluster from Bacillus velezensis FZB42) this particular BGC shown in Fig. 14D. This cluster was found to be of 41,419 nucleotides, having 42 ORFs, with one core biosynthetic gene, 12 additional biosynthetic genes, 4 transport-related genes, 2 regulatory genes and rest other genes. The NRPS/PKS modules, polymer structure predictions were not determined for this cluster.
Analysis of cluster 2.2: This cluster is a hybrid (RiPP-like, NRP-metallophore, NRPS) cluster which presented 100% similarity with the biosynthetic gene cluster known to encode bacillibactin (NRP) in Bacillus subtilis subsp. subtilis str. 16890,91,92. Known cluster BLAST using MIBiG 3.1 showed highest similarity with BGC0000309 (bacillibactin biosynthetic gene cluster from Bacillus subtilis subsp. subtilis str. 168) this particular BGC shown in Fig. 14E. This particular cluster consists of 51,794 nucleotides comprising of 46 ORFs. It has been observed that this cluster presents three candidate BGCs within it [i.e., Candidate Cluster 2.2.1 (location 866,688–918,482): neighbouring RiPP-like-NRP-metallophore-NRPS; and Candidate Cluster 2.2.2 (location 866,746–918,482): chemical hybrid NRP-metallophore-NRPS]. Interestingly, both the candidate clusters are identically made of 2 domains (Fig. 15K,M), with both of their polymer prediction being (Gly–Thr) and their polymer structure shown in Fig. 15L,N.
Analysis of cluster 3.1: This Trans-acyltransferase polyketide synthase (transAT-PKS) cluster presented 46% similarity according to MIBiG 3.1, with a biosynthetic gene cluster known to code for BGC0000176 (difficidin biosynthetic gene cluster from Bacillus velezensis FZB42)79, this particular BGC shown in Fig. 14F. This cluster consists of 33,927 nucleotides, comprising of 26 ORFs among which 2 is for the core biosynthetic gene, 9 are for the additional biosynthetic genes, 2 is for transport-related gene, 1 regulatory gene, and rest 12 other genes. The structure of these 2 core genes is made up of 1 module with domains (Fig. 15O). The polymer prediction by the aforementioned cluster is (Me-ccmal); and the predicted core structure for this particular cluster is shown in Fig. 15P.
Analysis of cluster 4.1: This Trans-acyltransferase polyketide synthase like (transAT-PKS-like) cluster which presented 53% similarity with a biosynthetic gene cluster known to code for BGC0000176 (difficidin biosynthetic gene cluster from Bacillus velezensis FZB42)79, this particular BGCs shown in Fig. 14G. This cluster consists of 45,721 nucleotides, comprising of 30 ORFs among which 4 is for the core biosynthetic gene, 8 are for the additional biosynthetic genes, and rest 18 other genes. The structure of these 4 core genes comprising 6 modules with domains (Fig. 15Q). The prediction of polymers by the aforementioned cluster is (ohmal–ccmal) + (ohmal–mal) + (Me-ccmal) + (mal); and the predicted core structure for this particular cluster is shown in Fig. 15R.
Analysis of cluster 6.1: This cluster exhibits 100% similarity with the BGCs known to code for bacinapeptin in Bacillus nakamurai. Known cluster BLAST using MIBiG 3.1 showed highest similarity with BGC0002700 (bacinapeptin biosynthetic gene cluster from Bacillus nakamurai)94, this particular BGCs shown in Fig. 14H. This cluster consists of 22,616 nucleotides, comprising of 21 ORFs among which 1 is for the core biosynthetic gene, 5 are for the additional biosynthetic genes, 1 is for transport-related gene, 4 regulatory genes, and rest 8 other genes.
Analysis of cluster 6.2: This Non-ribosomal peptide synthetase (NRPS) cluster which presented 43% similarity with a biosynthetic gene cluster known to code for BGC0000433 (surfactin biosynthetic gene cluster from Bacillus velezensis FZB42)86, this particular BGC shown in Fig. 14I. This cluster consists of 27,706 nucleotides, comprising of 19 ORFs among which 1 is for the core biosynthetic gene, 6 are for the additional biosynthetic genes, 1 is for regulatory gene, and rest 11 other genes. The structure of this 1 core genes is made up of 2 modules with domains (Fig. 15S). The polymer prediction by the aforementioned cluster is (Glu–Leu); and the predicted core structure for this particular cluster is shown in Fig. 15T.
The genome sequence of Bacillus ayatagriensis sp. nov. RMG6T provides information on unique biosynthetic gene clusters for secondary metabolite production suggesting that the bacterial strain may be a promising source for the discovery of new antimicrobial compounds. Moreover, deciphering the complete genome of RMG6T will unlock deep insights into the molecular foundations of plant growth promotion and biocontrol, paving the way for groundbreaking research and transformative biotechnological advancements.
Conclusion
The isolation and characterization of Bacillus ayatagriensis sp. nov. RMG6T highlight its potential as a novel biocontrol agent with plant growth-promoting properties. The strain’s antimicrobial activity, phylogenetic novelty, and ability to enhance seed germination suggest its applicability in sustainable agriculture. However, further research is necessary to fully explore its capabilities and practical applications. Future studies should focus on experimental validation of plant–microbe interactions under field conditions to assess the effectiveness of RMG6T in enhancing crop yield and suppressing plant pathogens. Metabolomic and proteomic analyses can help elucidate the biochemical pathways underlying its biocontrol mechanisms. Additionally, transcriptomic studies could provide insights into gene expression dynamics in response to environmental stimuli. To establish its viability as a commercial biocontrol agent, long-term ecological impact assessments and biosafety evaluations are essential. Investigations into formulation techniques, such as bio-encapsulation or consortia-based applications, could further enhance its stability and efficacy. Finally, regulatory approvals and large-scale field trials will be crucial for integrating Bacillus ayatagriensis sp. nov. RMG6T into agricultural practices, ensuring its effectiveness in real-world farming systems.
Data availability
This whole-genome shotgun project was deposited at NCBI under the GenBank accession number JBEFNT000000000. The version described in this paper is the first version, JBEFNT010000000, and consists of sequences JBEFNT010000001-JBEFNT010000037. The BioProject and BioSample accession numbers are PRJNA1115086 and SAMN41506556, respectively. The raw data are available at the Sequence Read Archive (SRA) under the accession number SRR29330546.
References
-
Vijayan, K., Gnanesh, B. N. & Tikader, A. Botanical features and economic significance of mulberry. In The mulberry genome (eds Gnanesh, B. N. & Vijayan, K.) 1–11 (Springer International Publishing, 2023).
-
Kloepper, K. J. & Schroth, M. N. Plant growth-promoting rhizobacteria on radishes. In Proc. of the 4th Internet. Conf. on Plant Pathogenic Bacter, Station de Pathologie Vegetale et Phytobacteriologie, INRA, Angers, France, 1978 2, 879–882 (1978).
-
Kloepper, J. W., Ryu, C.-M. & Zhang, S. Induced systemic resistance and promotion of plant growth by Bacillus spp. Phytopathology® 94, 1259–1266 (2004).
-
Cleland, R. E. Auxin and cell elongation. In Plant hormones and their role in plant growth and development (ed. Davies, P. J.) 132–148 (Springer, 1987).
-
Zhao, Y. Auxin biosynthesis and its role in plant development. Annu. Rev. Plant Biol. 61, 49–64 (2010).
-
Fischer, S. E., Fischer, S. I., Magris, S. & Mori, G. B. Isolation and characterization of bacteria from the rhizosphere of wheat. World J. Microbiol. Biotechnol. 23, 895–903 (2007).
-
Yuan, T. et al. Analysis of changes in bacterial diversity in healthy and bacterial wilt mulberry samples using metagenomic sequencing and culture-dependent approaches. Front. Plant Sci. 14, 1206691 (2023).
-
Sheoran, A. R. et al. Enhancing plant disease resistance: Insights from biocontrol agent strategies. J. Plant. Growth Regul. 44, 436–459 (2025).
-
Jiao, W. et al. The biocontrol potentials of rhizospheric bacterium Bacillus velezensis K0T24 against mulberry bacterial wilt disease. Arch. Microbiol. 206, 213 (2024).
-
Li, N., Li, J., Zhang, S., Lan, X. & Zhou, H. Unveiling the microbial mysteries of mulberry rhizosphere in saline-alkaline soils. Rhizosphere 33, 101040 (2025).
-
Ou, T. et al. Study on the potential for stimulating mulberry growth and drought tolerance of plant growth-promoting fungi. IJMS 24, 4090 (2023).
-
Todorova, S. & Kozhuharova, L. Characteristics and antimicrobial activity of Bacillus subtilis strains isolated from soil. World J. Microbiol. Biotechnol. 26, 1207–1216 (2010).
-
Nesa, J. et al. Antimicrobial potential of a ponericin-like peptide isolated from Bombyx mori L. hemolymph in response to Pseudomonas aeruginosa infection. Sci. Rep. 12, 15493 (2022).
-
Mondal, R. et al. Development of aptamer-functionalized gold nanoparticles as probes in point-of-care diagnostic device for rapid detection of multidrug-resistant bacteria in Bombyx mori L.. ACS Appl. Bio Mater. 7, 5740–5753 (2024).
-
Sumpavapol, P. et al. Bacillus siamensis sp. Nov., isolated from salted crab (poo-khem) in Thailand. Int. J. Systemat. Evolut. Microbiol. 60, 2364–2370 (2010).
-
Williams, S. E. et al. Discovery and biosynthetic assessment of ‘Streptomyces ortus’ sp. nov. isolated from a deep-sea sponge. Microbial Genom. 9, 000996 (2023).
-
Surve, V. V., Patil, M. U. & Dharmadhikari, S. M. FAME and 16srDNA sequence analysis of halophilic bacteria from solar salterns of Goa: A comparative study. Int. J. Sci. Res. Publ. 209, (2012).
-
Dunlap, C. A. et al. Bacillus nakamurai sp. nov., a black-pigment-producing strain. Int. J. Syst. Evolut. Microbiol. 66, 2987–2991 (2016).
-
Goswami, D., Thakker, J. N. & Dhandhukia, P. C. Simultaneous detection and quantification of indole-3-acetic acid (IAA) and indole-3-butyric acid (IBA) produced by rhizobacteria from l-tryptophan (Trp) using HPTLC. J. Microbiol. Methods 110, 7–14 (2015).
-
Dhandhukia, P. C. & Thakkar, V. R. Separation and quantitation of Jasmonic acid using HPTLC. J. Chromatogr. Sci. 46, 320–324 (2008).
-
Dhandhukia, P. C. & Thakker, J. N. Quantitative analysis and validation of method using HPTLC. In High-performance thin-layer chromatography (HPTLC) (ed. Srivastava, M.) 203–221 (Springer, 2011).
-
El-Koussi, W. M., Atia, N. N., Mahmoud, A. M. & El-Shabouri, S. R. HPTLC method for direct determination of gemifloxacin mesylate in human plasma. J. Chromatogr. B 967, 98–101 (2014).
-
Abou-Donia, A. H., Darwish, F. A., Toaima, S. M., Shawky, E. & Takla, S. S. A new approach to develop a standardized method for assessment of acetylcholinesterase inhibitory activity of different extracts using HPTLC and image analysis. J. Chromatogr. B 955–956, 50–57 (2014).
-
Xu, L., Li, S., Shabala, S., Jian, T. & Zhang, W. Plants grown in Parafilm-Wrapped petri dishes are stressed and possess altered gene expression profile. Front. Plant Sci. 10, 637 (2019).
-
Mondal, R. et al. Genomic dataset of a multiple-drug resistant Pseudomonas sp. strain RAC1 isolated from a flacherie infected Nistari race of Bombyx mori L. Data Brief 54, (2024).
-
Marmur, J. A procedure for the isolation of deoxyribonucleic acid from micro-organisms. J. Mol. Biol. 3, 208–218 (1961).
-
Mao, D.-P., Zhou, Q., Chen, C.-Y. & Quan, Z.-X. Coverage evaluation of universal bacterial primers using the metagenomic datasets. BMC Microbiol. 12, 66 (2012).
-
Glaeser, S. P. & Kämpfer, P. Multilocus sequence analysis (MLSA) in prokaryotic taxonomy. Syst. Appl. Microbiol. 38, 237–245 (2015).
-
Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120 (1980).
-
Nei, M. & Kumar, S. Molecular evolution and phylogenetics (Oxford University Press, 2000).
-
Brown, J., Pirrung, M. & McCue, L. A. FQC Dashboard: Integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics 33, 3137–3139 (2017).
-
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
-
Wick, R. R., Judd, L. M., Gorrie, C. L. & Holt, K. E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13, e1005595 (2017).
-
Li, D., Liu, C.-M., Luo, R., Sadakane, K. & Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676 (2015).
-
Prjibelski, A., Antipov, D., Meleshko, D., Lapidus, A. & Korobeynikov, A. Using SPAdes De novo assembler. CP Bioinf. 70, e102 (2020).
-
Chklovski, A., Parks, D. H., Woodcroft, B. J. & Tyson, G. W. CheckM2: a rapid, scalable and accurate tool for assessing microbial genome quality using machine learning. Nat. Methods 20, 1203–1212 (2023).
-
Tatusova, T. et al. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624 (2016).
-
Meier-Kolthoff, J. P. & Göker, M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 10, 2182 (2019).
-
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296 (2021).
-
Wang, A. & Ash, G. J. Whole genome phylogeny of Bacillus by feature frequency profiles (FFP). Sci. Rep. 5, 13644 (2015).
-
Yoon, S.-H., Ha, S., Lim, J., Kwon, S. & Chun, J. A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek 110, 1281–1286 (2017).
-
Stothard, P. & Wishart, D. S. Circular genome visualization and exploration using CGView. Bioinformatics 21, 537–539 (2005).
-
Brettin, T. et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 5, 1–6 (2015).
-
Hifnawy, M. S. et al. The genus Micromonospora as a model microorganism for bioactive natural product discovery. RSC Adv. 10, 20939–20959 (2020).
-
Blin, K. et al. antiSMASH 7.0: New and improved predictions for detection, regulation, chemical structures and visualisation. Nucleic Acids Res. 51, 46–50 (2023).
-
Zhou, Z. et al. METABOLIC: High-throughput profiling of microbial genomes for functional traits, metabolism, biogeochemistry, and community-scale functional networks. Microbiome 10, 33 (2022).
-
Kanehisa, M., Sato, Y. & Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731 (2016).
-
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: Biological systems database as a model of the real world. Nucleic Acids Res. 53, D672–D677 (2025).
-
Kanehisa, M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
-
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951 (2019).
-
Gu, Z., Eils, R. & Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849 (2016).
-
Alikhan, N.-F., Petty, N. K., Ben Zakour, N. L. & Beatson, S. A. BLAST ring image generator (BRIG): Simple prokaryote genome comparisons. BMC Genom. 12, 402 (2011).
-
Pedretti, N. et al. Cell-free supernatant from a strain of bacillus siamensis isolated from the skin showed a broad spectrum of antimicrobial activity. Microorganisms 12, 718 (2024).
-
Rong, X. & Huang, Y. Taxonomic evaluation of the Streptomyces griseus clade using multilocus sequence analysis and DNA–DNA hybridization, with proposal to combine 29 species and three subspecies as 11 genomic species. Int. J. Syst. Evol. Microbiol. 60, 696–703 (2010).
-
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H.-P. & Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinf. 14, 60 (2013).
-
Islam, A., Kabir, S. & Khair, A. Characterization and evaluation of bacillus siamensis isolate for its growth promoting potential in tomato. Agriculture (Pol’nohospodárstvo) 65, 42–50 (2019).
-
Aranya Paul & Chinnamani PrasannaKumar. Isolation and characterization of Bacillus siamensis strain NCT39 from a marine crab. (2023) https://doi.org/10.13140/RG.2.2.10125.20960.
-
Lv, R. et al. Effect of ultrasonication and thermal and pressure treatments, individually and combined, on inactivation of Bacillus cereus spores. Appl. Microbiol. Biotechnol. 103, 2329–2338 (2019).
-
Ruiz-García, C., Béjar, V., Martínez-Checa, F., Llamas, I. & Quesada, E. Bacillus velezensis sp. nov., a surfactant-producing bacterium isolated from the river Vélez in Málaga, southern Spain. Int. J. Syst. Evolut. Microbiol. 55, 191–195 (2005).
-
Matloub, A. A., Gomaa, E. Z., Hassan, A. A., Elbatanony, M. M. & El-Senousy, W. M. Comparative chemical and bioactivity studies of intra- and extracellular metabolites of endophytic bacteria, bacillus subtilis NCIB 3610. Int. J. Pept. Res. Ther. 26, 497–511 (2020).
-
Morris, R. J., Stevenson, D., Sukhodub, T., Stanley-Wall, N. R. & MacPhee, C. E. Density and temperature controlled fluid extraction in a bacterial biofilm is determined by poly-γ-glutamic acid production. npj Biofilms Microbiomes 8, 98 (2022).
-
Borriss, R. et al. Relationship of Bacillus amyloliquefaciens clades associated with strains DSM 7T and FZB42T: A proposal for Bacillus amyloliquefaciens subsp. amyloliquefaciens subsp. nov. and Bacillus amyloliquefaciens subsp. plantarum subsp. nov. based on complete genome sequence comparisons. Int. J. Syst. Evolut. Microbiol. 61, 1786–1801 (2011).
-
Ngalimat, M. S. et al. A review on the biotechnological applications of the operational group bacillus amyloliquefaciens. Microorganisms 9, 614 (2021).
-
Dijkhuizen, L. & Arfman, N. Methanol metabolism in thermotolerant methylotrophic Bacillus species. FEMS Microbiol. Rev. 7, 215–219 (1990).
-
Burdett, I. D., Kirkwood, T. B. & Whalley, J. B. Growth kinetics of individual Bacillus subtilis cells and correlation with nucleoid extension. J. Bacteriol. 167, 219–230 (1986).
-
De Los SantosVillalobos, S. et al. Bacillus cabrialesii sp. nov., an endophytic plant growth promoting bacterium isolated from wheat (Triticum turgidum subsp. durum) in the Yaqui Valley, Mexico. Int. J. Syst. Evolut. Microbiol. 69, 3939–3945 (2019).
-
Spaepen, S. & Vanderleyden, J. Auxin and plant-microbe interactions. Cold Spring Harb. Perspect. Biol. 3, a001438 (2011).
-
Ambawade, M. S. & Pathade, G. R. Indole acetic acid (IAA) production by Bacillus siamensis BE 76 isolated from Musa balbisiana (banana). J. Chem. Pharm. Res. 10, 610 (2018).
-
Qian, S. et al. Bacillus amyloliquefaciens AK-12 helps rapeseed establish a protection against Brevicoryne brassicae. Int. J. Mol. Sci. 24, 15893 (2023).
-
Zhang, Y. et al. Growth promotion on maize and whole-genome sequence analysis of Bacillus velezensis D103. Microbiol. Spectrum 12, e01147-e1224 (2024).
-
Wagi, S. & Ahmed, A. Bacillus spp.: Potent microfactories of bacterial IAA. PeerJ 7, 7258 (2019).
-
Yadav, A. et al. Auxin biosynthesis by Microbacterium testaceum Y411 associated with orchid aerial roots and their efficacy in micropropagation. Front. Plant Sci. 13, 1037109 (2022).
-
Tindall, B. J., Rosselló-Móra, R., Busse, H.-J., Ludwig, W. & Kämpfer, P. Notes on the characterization of prokaryote strains for taxonomic purposes. Int. J. Syst. Evol. Microbiol. 60, 249–266 (2010).
-
Borriss, R. et al. Whole genome sequence comparisons in taxonomy. In Methods Microbiology vol. 38: 409–436 (Elsevier, 2011).
-
Rodriguez-R, L. M. et al. An ANI gap within bacterial species that advances the definitions of intra-species units. MBio 15, e02696-e2723 (2024).
-
Aziz, R. K. et al. The RAST server: Rapid annotations using subsystems technology. BMC Genom. 9, 75 (2008).
-
Ota, Y. et al. Butirosin-biosynthetic gene cluster from Bacillus circulans. J. Antibiot. 53, 1158–1167 (2000).
-
Kudo, F. et al. Extended sequence and functional analysis of the butirosin biosynthetic gene cluster in Bacillus circulans SANK 72073. J. Antibiot. 58, 373–379 (2005).
-
Chen, X.-H. et al. Structural and functional characterization of three polyketide synthase gene clusters in Bacillus amyloliquefaciens FZB 42. J. Bacteriol. 188, 4024–4036 (2006).
-
Chen, X. H. et al. Comparative analysis of the complete genome sequence of the plant growth–promoting bacterium Bacillus amyloliquefaciens FZB42. Nat. Biotechnol. 25, 1007–1014 (2007).
-
Schneider, K. et al. Macrolactin is the polyketide biosynthesis product of the pks2 cluster of bacillus amyloliquefaciens FZB42. J. Nat. Prod. 70, 1417–1423 (2007).
-
Butcher, R. A. et al. The identification of bacillaene, the product of the PksX megacomplex in Bacillus subtilis. Proc. Natl. Acad. Sci. U.S.A. 104, 1506–1509 (2007).
-
Moldenhauer, J., Chen, X., Borriss, R. & Piel, J. Biosynthesis of the antibiotic bacillaene, the product of a giant polyketide synthase complex of the trans -AT family. Angew. Chem. Int. Ed. 46, 8195–8197 (2007).
-
Moldenhauer, J. et al. The final steps of bacillaene biosynthesis in Bacillus amyloliquefaciens FZB42: Direct evidence for β, γ dehydration by a trans-acyltransferase polyketide synthase. Angew. Chem. 122, 1507–1509 (2010).
-
Yang, H., Li, X., Li, X., Yu, H. & Shen, Z. Identification of lipopeptide isoforms by MALDI-TOF-MS/MS based on the simultaneous purification of iturin, fengycin, and surfactin by RP-HPLC. Anal. Bioanal. Chem. 407, 2529–2542 (2015).
-
Koumoutsi, A. et al. Structural and functional characterization of gene clusters directing nonribosomal synthesis of bioactive cyclic lipopeptides in Bacillus amyloliquefaciens strain FZB42. J. Bacteriol. 186, 1084–1096 (2004).
-
Wu, L. et al. Difficidin and bacilysin from Bacillus amyloliquefaciens FZB42 have antibacterial activity against Xanthomonas oryzae rice pathogens. Sci. Rep. 5, 12975 (2015).
-
Chen, X.-H. et al. Difficidin and bacilysin produced by plant-associated Bacillus amyloliquefaciens are efficient in controlling fire blight disease. J. Biotechnol. 140, 38–44 (2009).
-
Wu, L. et al. Bacilysin from bacillus amyloliquefaciens FZB42 has specific bactericidal activity against harmful algal bloom species. Appl. Environ. Microbiol. 80, 7512–7520 (2014).
-
May, J. J., Wendrich, T. M. & Marahiel, M. A. The dhb operon of bacillus subtilisencodes the biosynthetic template for the catecholic siderophore 2,3-dihydroxybenzoate-glycine-threonine trimeric ester bacillibactin. J. Biol. Chem. 276, 7209–7217 (2001).
-
Barbe, V. et al. From a consortium sequence to a unified sequence: The Bacillus subtilis 168 reference genome a decade later. Microbiology 155, 1758–1775 (2009).
-
Kunst, F. J. et al. The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature 390, 249–256 (1997).
-
Li, X.-Y. et al. ESI LC-MS and MS/MS characterization of antifungal cyclic lipopeptides produced by Bacillus subtilis XF-1. J. Mol. Microbiol. Biotechnol. 22, 83–93 (2012).
-
Xue, D. et al. Correlational networking guides the discovery of unclustered lanthipeptide protease-encoding genes. Nat. Commun. 13, 1647 (2022).
Acknowledgements
Rittick Mondal would like to acknowledge the Department of Science & Technology, Government of India, for the DST-INSPIRE Ph.D. Fellowship (DST-INSPIRE-SRF; INSPIRE Code- IF190457). Pankaj Mandal would like to acknowledge Swami Vivekananda Merit-cum-Means Scholarship (WBP241720165040), Govt. of West Bengal. Amit Kumar Mandal acknowledges DST-SERB India for the financial assistance (sanction order number: EEQ/2021/000058).
Ethics declarations
Competing interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Das, S., Mondal, R., Mandal, P. et al. Bacillus ayatagriensis sp. nov., a novel plant growth-promoting rhizobacteria strain isolated from mulberry rhizosphere. Sci Rep 15, 26693 (2025). https://doi.org/10.1038/s41598-025-05508-w
-
Received:
-
Accepted:
-
Published:
-
DOI: https://doi.org/10.1038/s41598-025-05508-w