
Introduction
Despite its inherent fragility, mRNA is one of the most prolific substances in the context of therapeutic gene delivery1. To ensure sufficient biological activity of therapeutic mRNA, it is necessary to protect it from premature cleavage by numerous RNA-targeting enzymes, particularly exonucleases2. Circularization is one of the most promising modifications of mRNA in this context, as it prevents exonucleolytic digestion3,4. Consequently, circRNAs have shown higher stability than corresponding linear RNAs and exhibited significantly longer protein expression profiles5,6,7. Unfortunately, the circularization of macromolecular RNA still poses a significant challenge.
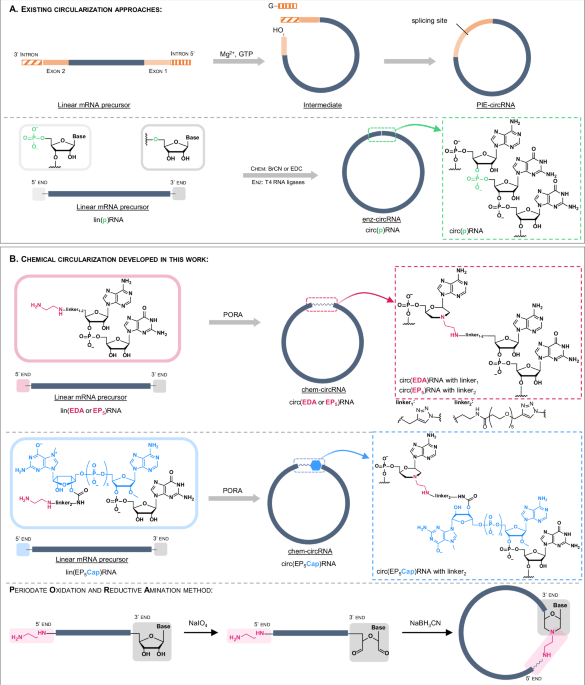
Currently established methods of RNA circularization are based on chemical reactions, enzymatic reactions, or the activity of autocatalytic RNA sequences8. The chemical circularization methods rely on short precursor RNA sequences, synthesized via phosphoramidite chemistry9,10,11. In consequence, the so-far chemically synthesized circRNAs encoded relatively short sequences and have limited use in the context of therapeutic gene delivery. Alternatively, RNA circularization can be carried out with ligases, such as T4 RNA ligase I and T4 RNA ligase II (Fig. 1A)12,13,14. Both enzymes require 5′ monophosphorylated precursors that are first adenylated and subsequently ligated with the 3′ OH of the ribose at the 3′ end RNA. The ligases are strongly dependent on the target RNA length and sequence5,6,14,15. As a result, it is difficult to predict or improve the performance of such enzymes. Other methods of RNA circularization involve catalytic nucleic acids (Fig. 1A)5,6,16,17,18,19. Wesselhoeft et al. have circularized RNA in vitro by using the permuted intron-exon (PIE) system and optimized self-splicing intronic sequences5,6. Litke et al. have shown the Twister-optimized RNA durable overexpression (Tornado) system for RNA circularization in cells16. These methods are based on sequence-dependent activity and can be hindered by methylated nucleobases [e.g. N1-methylpseudouridine (m1Ψ) or N6-methyladenosine] and other chemical modifications6. Moreover, the translation of currently known circRNAs relies on internal ribosome entry site (IRES) sequences20,21. It is well-established that, for linear mRNAs, translation driven by an IRES sequence is less efficient than the main pathways of eukaryotic translation involving cap-and-poly(A)-dependent initiation22,23,24,25. Hence, robust and scalable methods of RNA circularization that are compatible with mRNA body modifications or cap-dependent translation mechanisms are strongly desired.

A Overview of existing circularization approaches. B Chemical circularization developed in this work. The 5′ modified linear RNA precursor is obtained from a 5′ initiator (EDA-AG, EP5AG or EP5Cap) during an in vitro transcription (IVT) reaction and is subsequently circularized by PORA reaction. The produced circRNA contains a single chemically modified linkage.
In this work, we develop a chemical circularization method that expands circ-mRNA design strategies beyond IRES-dependent translation. Taking advantage of the unique reactivity of the ethylenediamine moiety towards oxidized RNA 3′ ends, we establish a post-transcriptional circularization protocol relying on a one-pot, two-step chemical reaction (Fig. 1B)26,27,28,29. The protocol is based on affordable, readily available, non-invasive reagents and can be applied to RNA of virtually any size and sequence (Fig. 1B). This approach, combined with improved purification and isolation techniques, provides access to chem-circRNAs of up to 4000 nt. mRNAs generated using our approach are evaluated in living cells to determine their relative stability and translational activity. We show that our circularization method is compatible with incorporating chemical modifications such as N7-methylguanosine cap and m1Ψ. Our findings offer an alternative to chemoenzymatic methods of RNA circularization and promote circ-mRNA optimization.
Results
Development of the chemical circularization method
Our approach towards circRNAs was designed to minimize the number of steps necessary to functionalize and circularize the target RNA, maintaining the process as close as possible to the existing mRNA production pipelines. In the first step, we generate an in vitro transcribed (IVT) pre-circRNA that is 5′ functionalized with ethylenediamine by incorporating a properly designed transcription initiator (primer; Fig. 1B). Such pre-circRNA is then post-transcriptionally circularized by one-pot periodate oxidation and reductive amination (PORA) reaction, resulting in the formation of a morpholine-derived inter-nucleotide linkage (Fig. 1B). As transcription initiators, we designed two types of AG dinucleotides equipped with ethylenediamine linkers (EDA-linkers) of different lengths— EDA-AG (EDA) and EDA-PEG5-AG (EP5) (Fig. 1B). We also designed an EDA-functionalized cap 1 analog – EDA-PEG5-m7GpppAmG (EP5Cap)—to provide unprecedented access to circular mRNAs containing an endocyclic 5′ cap structure (Fig. 1B). The presence of circular RNAs was verified by polyacrylamide gel electrophoresis (PAGE) and compared to analogous circRNA obtained by enzymatic ligation (Supplementary Fig. 1).
The synthesis of the primers was performed in a single synthetic step using click chemistry (NHS-based amidation and CuAAC) from an azido-modified dinucleotide synthesized on solid-support (N3-AG, Supplementary Fig. 2).
We began our attempts at chemical RNA circularization using two RNA oligonucleotide models: (EDA)RNA01 and (EDA)RNA02, (35 nucleotide-long sequences, Table 1, Fig. 2), transcribed in the presence of EDA-AG initiator (Fig. 1B, Supplementary Fig. 2). The transcription products were obtained in reasonable yields and quality, suggesting successful generation of RNA with functionalized 5′ end (the heterogeneity observed as multiple bands on the PAGE gel is typical for in vitro transcription, which generates mRNA products of various sizes, primarily due to non-templated nucleotide additions at the 3′ and 5′ ends)30.

A PAGE analysis of the circularization reaction of (EDA)RNA01 and (EDA)RNA02. RNAs were circularized at concentrations of 0.3, 1.2, or 12 µM, at 4 or 20 °C, with (+ΔT) or without (−ΔT) thermal refolding (rapid heating and subsequent slow cooling) prior to circularization. S refers to an untreated RNA substrate. Data presented is representative of one independent experiment. B–D Minimum-free-energy (MFE) secondary structure (2D) models (predicted with RNAfold web server) and tertiary structure (3D) of predicted models (RNA Composer web server) for RNA01 (B), RNA02 (C), and RNA02 dimer (D). Content of mRNA species was quantified using CLIQS (Core Laboratory Image Quantification Software).
The subsequent step of chemical circularization was based on our previous findings that oxidized RNA 3′ ends show particularly high reactivity in PORA reactions toward compounds containing ethylenediamine26. To execute the PORA reaction, (EDA)RNA01 and (EDA)RNA02 were incubated in a sodium periodate (NaIO4) solution, leading to the formation of an acyclic 2′,3′-dialdehyde at the 3′ terminal nucleotide. This derivative spontaneously reacted with the ethylenediamine motif to form a cyclic intermediate. The subsequent addition of sodium cyanoborohydride (NaBH3CN) led to the reduction of the cyclic intermediate, ensuring the irreversibility of the reaction. As a result, a morpholine linkage was formed between the former 3′ terminal nucleotide and the 5′ terminus of the transcription initiator (Fig. 2).
To test the scope and limitations of this reaction, we first investigated the basic factors affecting chemical circularization. To that end, we tested the influence of pH, temperature, and RNA concentration on the circularization yield.
The circularization efficiencies were estimated by PAGE combined with densitometric analyses (Fig. 2 and Supplementary Fig. 3). To minimize bias arising from the arbitrary selection of main band(s) (for product or substrate) and to reduce errors from incorrect intensity calculations (caused by overlapping peaks or challenges in baseline assignment), the entire regions corresponding to the substrates and products of circularization (sum of the six main peaks) were used in our calculations. We observed that the conversion of linear precursor (~50%) was not significantly affected by pH (5.6–8.0) (Supplementary Fig. 3), time, temperature (2 h at 20 °C or 16 h at 4 °C), or the concentration of the precursor (0.3–12 µM) (Fig. 2A). However, both RNA sequence and concentration were crucial for circularization selectivity. The circularization of (EDA)RNA01 was, on average, more selective (50–70%) than that of (EDA)RNA02 (30–55%), which also produced linear and circular dimers. Notably, the circularization of (EDA)RNA01 did not lead to any linear and circular polymeric by-products (dimers and concatemers), especially at lower concentrations of RNA (Fig. 2A).
To gain insight into the sequence-dependence of RNAs circularization, secondary and tertiary structural models of the RNA01 and RNA02 sequences were generated using automated structure predictors (Fig. 2B–D)31,32,33. Computational predictions suggested that both RNA01 and RNA02 can form distinct secondary and tertiary structures. The structure of RNA01 features a stable hairpin (free energy of the thermodynamic ensemble −8.2 kcal/mol) with an open loop motif within which the 3ʹ and 5ʹ ends spatially converge. The predicted average distance between the 5ʹ and 3ʹ ends in the RNA01 model is shorter (2.3 nm), making the molecule prone to circularization (Fig. 2B). In contrast, the secondary structure of RNA02 formed a low stability hairpin (−5.1 kcal/mol). The 3ʹ and 5ʹ ends reside within a bigger open loop, with an increased end-to-end distance (4.9 nm). Moreover, the tertiary structure model of RNA02 suggested that the 3ʹ and 5ʹ ends could point in opposite directions (Fig. 2C). The spatial arrangement of these termini probably enhances the likelihood of intermolecular interactions rather than intramolecular ones, ultimately leading to the preferential formation of stable RNA dimers (−18.3 kcal/mol), especially at higher concentrations (Fig. 2A, D). Overall, the chemical circularization method worked well on the oligonucleotide models, and its selectivity and efficiency could be rationalized with the help of computational methods. Considering that the majority of long RNA sequences adopt tertiary structures with termini separated by no more than 10 nm, these results encouraged us to test circularization on longer RNAs34.
To that end, we performed chemical circularization of macromolecular RNAs (RNA03–RNA08, 276–760 nt), which included various protein-coding RNAs or their 5′ terminal fragments (Table 1). Here, we took advantage of a dinucleotide initiator with a longer linker (EP5AG; Supplementary Fig 2). The chemical circularization process proceeded quite efficiently for most of the studied sequences, independent of their length, yielding circularization efficiencies ranging from 47% to 60% (Table 1). The chemical circularization yield was generally lower for RNAs longer than 900 nt compared to shorter RNAs (Table 1; compare RNA04 versus RNA09). Furthermore, the introduction of a poly(A) sequence (A30) at the 3′ end of the precursor resulted in a dramatic reduction in circularization yield (<5%). For instance, the circularization efficiency of RNA10, which lacks the poly(A) (A30), was 33%, whereas for the same sequence with the poly(A) (RNA11), it was only 2%. A similar result was obtained for another sequence containing an A30 tract (RNA09) (Table 1, Supplementary Fig. 4). The low efficiency of circularization for these sequences is likely due to the unstructured nature of the poly(A) region, which increases the 5′−3′ end-to-end distance in the precursor34. Since our overall goal was to develop a circularization method applicable to all RNA sequences and lengths, we attempted to overcome this issue by applying a splint-mediated approach. To decrease the distances between the 5′- and 3′-terminal moieties in longer and unstructured RNAs, we annealed the linear precursors with oligonucleotide DNA splints (short DNA sequences that are complementary to the 5′ and 3′ flanking regions of the precursor RNA; Fig. 3, Supplementary Table 2). To that end, we applied computational modeling (RNAfold web server) to RNA11 (952 nt) containing human beta globin (HBB) 5′ and 3′ UTRs, Gaussia luciferase (GLuc) ORF, and A30 tract, to design four DNA sequences differing in length and targeting specific structural features of the precursor RNAs (ON1-ON4; 10–67 nt; Fig. 3A, Supplementary Table 2). Next, we synthesized a molecular probe consisting of RNA11 labeled with a fluorescent FRET pair, Cy5 and Cy3, at the 5ʹ and 3′ ends, respectively (Fig. 3B)26. Measuring and analyzing the fluorescence spectra recorded at different temperatures, before and after splint annealing with the probe, led to the selection of an optimal splint sequence (ON2, 28 nt; Fig. 3). For RNA11, the circularization efficiency in the presence of the optimal DNA splint (ON2, 28 nt, 50%) was over twenty-fold higher than that in the absence of DNA (2%) or in the presence of sub-optimal splints (2–40%, Fig. 3B, C). The use of longer DNA splints, ON3 (48 nt) and ON4 (67 nt), resulted in reduced circularization selectivity and increased linear RNA polymers formation (Fig. 3C), whereas the shorter splint (ON1, 10 nt) did not improve circularization efficiency at all. The experiment revealed that the optimal splint has to form a sufficiently stable RNA-DNA duplex (melting temperature ~50 °C) to unwind the less stable RNA-RNA secondary structure (melting temperature ~20–30 °C), but should not be too long as it shifts the equilibrium toward dimer formation (Fig. 3A). The addition of the splint improved circularization of RNAs longer than 900 nt but had no effect for shorter (760 nt) sequences (Table 1; see data for RNA04 or RNA18).

A The predicted MFE secondary structure of RNA11 aligned with the tested DNA splints (ON1–4) and tertiary structure model of a selected fragment (predicted with RNAfold web server). Numbers 1–4 mark the regions of hypothetical interactions between RNA and the splints (Supplementary Table 2). B Fluorescence emission spectra (ex. 500 nm) of a FRET probe (Cy5-RNA11-Cy3) after hybridization with oligonucleotide splints (ON1–4, Supplementary Table 2) or without splint (w/o), recorded at 4 °C or 20 °C. C PAGE analysis of the chemical products of RNA11 circularization, preformed after annealing the precursor with (ON1–4, Supplementary Table 2) or without (w/o) an oligonucleotide splint. S refers to the untreated precursor. Content of mRNA species was quantified densitometrically using CLIQS (Core Laboratory Image Quantification Software). Data presented is representative of one independent experiment.
To verify the universality of this splint-based approach for longer RNAs, we applied it to RNA sequences: RNA09, RNA10 RNA12-RNA19. Our observations indicated that the efficiency of chemical circularization, particularly for sequences possessing a poly(A) tract, increased significantly in the presence of a suitably designed splint (Table 1). The longest RNA we attempted to circularize was 3893 nt long mRNA (RNA17) encoding the BNT162b anti-COVID vaccine29, achieving successful chemical circularization also in this case, albeit with lower yield compared to shorter RNAs (35%).
Furthermore, during the circularization reaction, we observed little to no formation of polymeric side-products or nicking of the desired product. Overall, we found that splint-aided chemical circularization is equally efficient (44–63%) as the PIE methodology (54%, Supplementary Fig 5). Ligation of the respective mRNAs with T4 RNA ligase II yielded circularization efficiencies from 40 to 60% and were comparable to chemical circularization (Table 1). However, we noted that enzymatic ligation was significantly more sequence-dependent than chemical circularization. Since the circularization efficiency may be limited by the efficiency of co-transcriptional incorporation of the modified initiators, EP5AG or EP5Cap, we evaluated the incorporation (capping) efficiencies for one of the model RNAs. To that end, we performed ribozyme-mediated cleavage of (EP5)RNA11 and (EP5Cap)RNA11, and compared the results to uncapped (triphosphorylated) RNA11 [(p3)RNA11)]. The incorporation efficiency of the initiators was 94% and 87% for EP5AG and EP5Cap (Supplementary Fig 6), respectively, which may suggest that the circularization yield can be slightly improved by decreasing the content of uncapped RNA.
Analytical methods for identifying and isolating chem-circRNA
The purity and homogeneity of circular RNA are crucial for mitigating an innate immune response and ensuring sustained protein production. Depending on the circularization method used, the resulting mixture may contain remnants of unreacted linear precursor, polymerized linear and circular side products such as dimers and concatemers, excised intron fragments, nicked circRNA, or splint templates that can activate the innate immune response6,35. To establish optimal methods for identifying and purifying chem-circRNA, we first reviewed established methods from the literature. Then, we conducted a detailed examination and optimization of selected methods, leading to significant improvements and insightful findings.
The yields of RNA circularization and the homogeneity of circRNAs were determined based on electrophoretic analyses. Initially, we attempted to resolve linear and circRNAs on denaturing agarose gel. However, we typically observed poor resolution or overlap between linear and circular species of corresponding weight, which has also been reported previously5,6,14,36. Consequently, we turned our attention to denaturing polyacrylamide gel electrophoresis (PAGE). By varying both the degree of cross-linking of the polyacrylamide (PAA) and the final concentration of the AA/MBAA (acrylamide/N, N’-methylenebisacrylamide) blend, we determined the optimal density and cross-linking degree of polyacrylamide gels for the best migration and separation of linear and circRNA (Fig. 4, Supplementary Table 3). In all studied cases, the migration speed of the circular RNA macromolecules in polyacrylamide gel was significantly slower than that of linear precursors of corresponding weight (Fig. 4A) or high-molecular-weight (HMW, 3000–6000 nt) linear RNA molecules (Fig. 4B, C). A higher gel density (20% PAA) with a cross-linking degree of 5% (19:1 AA/MBAA) favored the separation of short linear mRNAs and their circularization products (RNA01, 35 nt) (Fig. 4A). A lower gel percentage, corresponding to an increase in pore size, enhanced the migration rate of nucleic acids, as demonstrated for RNA03 (276 nt) and RNA04 (760 nt) on a 15% and 6% PAA gel, respectively. Conversely, a higher degree of cross-linking (19:1 AA/MBAA) significantly inhibited the migration of circRNA03 (276 nt) and circRNA04 (760 nt). Reducing the degree of cross-linking to 2.3% (38:1 AA/MBAA) increased the migration rate of circRNA while maintaining substantial separation between linear and circular RNA topologies. Such pronounced differences in nucleic acid migration are primarily attributed to variations in secondary structure rather than molecular weight. Through these experiments, we demonstrated that by manipulating gel density (from 20 to 4% PAA) and cross-linking degree (from 19:1 to 99:1 AA/MBAA ratio), one can achieve separation of linear and circular RNA species up to 4000 nt (Fig. 4; Supplementary Fig 7, Supplementary Table 3). High cross-linking degrees and high gel density enable the separation of short RNAs, whereas lower density and cross-linking degree gels are required for the efficient separation of long RNAs (for details, see Supplementary Table 3). Using these conditions, we could access the composition of the reaction mixtures in a simple, precise, and reproducible manner.

A–C Resolution of circular RNAs and their linear precursors based on molecular size (A RNA01 35 nt, B RNA03 276 nt, C RNA04 760 nt), gel density, and cross-linking degree of denaturing polyacrylamide. D, E Treatment of linear and circular mRNA with endo- and exonucleases (D RNase H—RH and E RNase R—RR, respectively) to confirm the circular topology of the reaction products. A–E Data presented is representative of one independent experiment.
On the other hand, the topology of circRNAs can be readily verified by treating them with endonucleases and exonucleases, including RNase H and RNase R3,37,38. Digestion of intact chem-circRNA with oligonucleotide-guided RNase H predominantly produced a single band, in contrast to the nicking of its linear precursor, which yielded two split products of lower molecular weight (Fig. 4D). RNase R, with 3′ to 5′ exonuclease activity, eliminates linear contaminants from circRNA preparations3,4. Due to the unique closed-loop structure, circRNAs remain resistant to RNase R digestion, which has been used for circRNAs enrichment. We found that chem-circRNAs are essentially also resistant to RNase R, as the RNase-treated circularization reaction yielded solely the chem-circRNA band (Fig. 4E). However, when applying RNase R digestion for chem-circRNAs enrichment, we observed that both the quality of the circRNA sample and the presence of some sequence and structural elements may affect the selectivity of the reaction (Supplementary Fig. 8). Digestion of RP-HPLC-purified mixtures of circular and linear RNA significantly increased the selectivity of RNase R towards the linear RNA species (Supplementary Fig. 8A). To investigate the resistance of chemically circularized RNAs to RNase R, we conducted RNase R digestion on three chem-circRNA11 variants with different linker lengths [(EDA)RNA11, (EP5)RNA11 and (EP5Cap)RNA11], as well as an enz-circRNA11 [(p)RNA11] with a phosphodiester bond at the junction site. We established that introducing a chemical linker in chem-circRNA11 does not preclude the use of RNase R for chem-circRNA enrichment (Supplementary Fig. 8B). However, we also observed undesired degradation of chem-circRNAs11. This issue may be resolved by optimizing RNase R digestion conditions, such as enzyme concentration incubation times or buffer composition.
To obtain high-purity circRNA samples on a smaller scale (for in vitro studies), we employed PAGE separation of linear and circular RNA. The chem-circRNA molecules were resolved on PAA gel and extracted using either the crush-and-soak method or electroelution (Supplementary Fig. 9). We found that the UV spectra of the isolated material differed greatly, indicating that crush-and-soak isolation leads to contamination of RNA with PAA. In contrast, electroelution yielded chem-circRNA of high purity (>85% on PAGE) with an acceptable UV spectrum (Supplementary Fig. 9A–C). Given that RNAs separated on denaturing agarose gels are reportedly non-translatable due to interactions between denaturants (formaldehyde) and nucleotide monomers39, we investigated the translational activity of circRNAs isolated from polyacrylamide gels (Supplementary Fig 9D). The circ(EP5Cap)RNA12 and reference circular RNA obtained by PIE splicing strategy (circRNA16-PIE) were purified via PAGE/electroelution or RNase R treatment to compare their translational activity in cultured cells. The biological impact of circRNA isolation was assessed in three cell lines (A549, HEK293T, HepG2). Regardless of the circRNA sample, total protein levels were comparable between the isolation methods (Supplementary Fig 9D).
Another possible method for chem-circRNA purification is isolation from the reaction mixture by high-performance liquid chromatography (HPLC). We investigated this in three modes: ion-exchange chromatography (IEC), size-exclusion chromatography (SEC), and reversed-phase chromatography (RP) (Fig. 5). Previous studies utilized SEC for isolating circularization products from self-catalytic precursors using permuted intron-exon (PIE) methodology5,6,14. SEC can be effective in such a scenario because during the transesterification reaction, the linear precursor eliminates chain fragments corresponding to sequences of autocatalytic introns, contributing to a decrease in molecular mass. However, in our hands, SEC did not facilitate the separation of linear and circular RNA molecules of the same size and sequence (such as chem-circRNA and its precursor or the desired circRNA and nicked product). Successful separation of RNA molecules based on their topology was achieved only in RP mode in the presence of ion-pairing agents (IP-RP) (Fig. 5B). Optimal separations were achieved at 60 °C temperature on polystyrene divinylbenzene copolymer resin in the presence of n-hexylammonium acetate and acetonitrile (Fig. 5C). Notably, the IP-RP-HPLC method was effective only for RNAs shorter than 800 nt; for longer RNAs, the retention times for circular and linear forms were too similar (Supplementary Fig 10). The recovery rates ranged from 50% to 70%, with higher sample amounts leading to improved recovery. No correlation was observed between peak separation and the injected sample amount for the tested amounts ranging from 25 to 60 µg. The combination of IP-RP-HPLC and PAGE provided us with a set of tools to precisely analyze circularization reactions and isolate the desired circRNA products with high purity.

A Samples of linear (p)RNA04 and crude post-circularization mixture containing linear (p)RNA04 and circ(p)RNA04, (RNA04 760 nt) were analyzed using different HPLC methods: ion-exchange (IEC), size-exclusion (SEC), and reversed-phase (RP). B IP-RP-HPLC separation of linear and circRNA05 (578 nt) followed by PAGE analysis of collected fractions. Column and buffers as in A—IP-RP (Supplementary Information). Fractions F1 contains mostly precursor RNA; F2 contains the circular product of high purity; F3 includes linear dimers and concatemers. C Resolution of linear (lower retention time) and circular (higher retention time) RNA06 (598 nt) using IP-RP-HPLC with different ion-pairing reagents: triethylammonium acetate (TEAA), diisopropylammonium acetate (DIPAA), or n-hexylammonium acetate (HAA). A–C For purification/separation conditions please see Supplementary Information.
Translation of chemically modified circRNA
After establishing a robust methodology for preparing chem-circRNAs, we investigated the influence of chem-circRNA sequence and chemical modifications on its biological activity. Our study focused on two key aspects: (i) the biological stability and translational activity of chemically circularized mRNA and (ii) the incorporation of the N7-methylguanosine (m7G) cap structure and poly(A) tract into chemically circularized mRNAs and their functional implications. To investigate the translational properties and structure-activity relationship of chem-circRNA, we designed and synthesized a series of precursor sequences (RNA09, RNA11–RNA15), encoding a Gaussia luciferase, flanked by 5ʹ and 3′ untranslated regions (UTRs) and poly(A) sequences (Table 1).
Circularization was achieved using two strategies: chemical circularization employing the PORA method or enzymatic ligation via T4 RNA ligase II. The precursors for chemical circularization were obtained by IVT using EP5AG and EP5Cap (Fig. 1B) as initiators to yield (EP5)RNA and (EP5Cap)RNA, respectively, and were subsequently circularized via the PORA method to afford circ(EP5)RNA and circ(EP5Cap)RNA. For enzymatic circularization affording circ(p)RNA, the linear RNA precursors with a 5′ monophosphate [(p)RNA] were generated using guanosine monophosphate (GMP) as the transcription initiator (Fig. 1A). To assess the biological activity, circular RNAs and their linear RNA precursors—each bearing the corresponding 5′ end modification—were transfected into four distinct cell lines (A549, HEK 293 T, HepG2 and HeLa). Before the key experiments, we confirmed that PORA conditions do not impact the translational activity of mRNA using a mock mRNA (RNA09) treated with PORA conditions but without the oxidation step (Supplementary Fig. 11).
Next, the translational activity of chemically circularized RNA constructs was evaluated to determine whether the introduction of the EP5 chemical linker influences circRNA functionality (Fig. 6A, B). For this analysis, we employed a model mRNA (RNA12) containing the encephalomyocarditis virus (EMCV) internal ribosome entry site (IRES) sequence, which facilitates cap-independent translation initiation, followed by coding sequence only. Given that the purity of circRNA preparations was between 85% to 95%, we aimed to exclude any significant contribution of residual linear precursors to the overall translational output. The applied mRNA design minimized potential biases arising from residual linear RNA species, as the lack of 3’ UTR and poly(A) leads to their rapid degradation and low translational activity. Thus, this model system ensured that any observed translational activity was primarily derived from circRNAs rather than residual linear RNA. To ensure a fair comparison of the translational activities of linear and circular RNAs of the same sequence (Fig. 6A), we confirmed in a separate qPCR experiment that the circular and linear RNA species of the same sequence are transfected with similar efficiencies (Supplementary Fig. 12). PIE-derived circRNA16-PIE and a linear mRNA optimized for cap-dependent translation (Cap1-RNA11) served as additional references (Fig. 6B). As anticipated, the translational activity of circ(EP5)RNA12 was substantially higher than that of its linear precursor (Fig. 6B). Notably, chemically circularized circ(EP5)RNA12 showed comparable translational activity and stability to its enzymatically circularized counterpart circ(p)RNA12 (Fig. 6A, B), underscoring the biocompatibility of the internucleotide linkage formed upon chemical circularization.

A–E Translation activity and protein production profiles of linear and circular mRNAs based on two sequence designs: RNA12 (A, B) and RNA13 (C–E), both containing EMCV IRES elements upstream of the GLuc coding region. Circular RNAs were generated from linear precursors bearing either a 5′-monophosphate (p-) or ethylenediamine motif (EP5). A, C Show total protein output measured in A549, HEK293T (HEK), HeLa, and HepG2 (HEP) cells over 168 h post-transfection, normalized to canonical Cap1-RNA₁₁ (=1). B, D Show time-course of luciferase activity in A549 cells transfected with circ(p), circ(EP₅), lin(p), lin(EP₅), and control RNAs. A–D Data represent mean ± SD from three biological replicates (n = 3). A, C P values were calculated using unpaired Student’s t test. E Shows normalized protein levels obtained from RNA13 calculated as a protein level at a specific timepoint divided by the total protein collected over 168 h. The bars (left graph) represent the relative levels of the reporter protein in the cell culture medium, determined at the indicated time points and normalized to the level obtained for the reference canonical mRNA (Cap1-RNA11), means ± SEM, n = 3, * P < 0.05, *** P < 0.001, **** P < 0.0001, ordinary one-way ANOVA with Tukey’s multiple comparisons test (4 h, 96 h, 120 h, 144 h, 168 h) or Kruskal–Wallis test with Dunn’s multiple comparisons test (24 h, 48 h, 72 h). The right graph shows time-dependent protein accumulation (percentage of protein produced at all timepoints from 0 h up to the specific timepoint) calculated for each timepoint and fitted to a one-phase decay curve to determine the protein production half-life (the time after which half of the total protein level was reached). Source data are provided as a Source Data file.
Next, we examined the influence of poly(A) tract, on the durability of protein expression and the translational activity of chem-circRNA analogs. The effect of a 30-nucleotide poly(A) sequence was assessed using circRNA13 (Fig. 6C–E). As expected, the stability of linear precursors was enhanced by the poly(A) tract, leading to a transient surge in protein expression within the first 24 h post-transfection, followed by a rapid decline (Fig. 6D, E). The chemically and enzymatically circularized variants of the same sequence—circ(EP5)RNA13 and circ(p)RNA13—provided sustained protein production, showing similar durability to circRNAs lacking a poly(A) sequence [circ(p)RNA12 and circ(EP5)RNA12] (Fig. 6B, D). Notably, the circRNA13 analogs exhibited prolonged expression profiles, with estimated protein production half-lives ranging from 29 to 50 h, significantly exceeding those of their linear counterparts (8–11 h) (Fig. 6E, Supplementary Fig 13A). Comparing the results obtained for RNA12 and RNA13 we concluded that, unlike what is observed for linear mRNAs, adding a poly(A) tract downstream of the coding sequence does not increase circRNA durability. Importantly, both chemically and enzymatically circularized RNAs displayed a protein production profile similar to that of circRNA16-PIE, which incorporates a custom-designed spacer at the splicing site (Fig. 6B, D)5,6.
We next examined how adding m7G cap structure to circRNAs with IRES-EMCV will affect their translational activity. We hypothesized that enabling these two distinct translation initiation mechanisms in circRNAs could enhance protein production. To test this, we chemically circularized mRNAs containing a 5ʹ EP5Cap, followed by the EMCV IRES and GLuc reporter sequences alone (RNA12) or in combination with additional downstream elements, including a poly(A) tract (RNA13), the HBB 3ʹ UTR (RNA14), or the HBB 3ʹ UTR followed by a poly(A) tract (RNA15) (Fig. 7 and Supplementary Fig 14). A linear Cap1-RNA11 construct served as a reference.

A–H Translation activity and protein expression profiles of enzymatically circularized and chem-circRNAs derived from four RNA templates (RNA₁₂–RNA₁₅) in A549, HEK 293 T (HEK), HeLa, and HepG2 (HEP) cells. Circular RNAs were generated from linear precursors containing either a 5′ monophosphate (p-) or an EP₅Cap structure and encoded GLuc. A, C, E, G Show total protein output from transfected cells after 168 h, normalized to expression from the canonical linear Cap1-RNA₁₁ (=1); B, D, F, H Show time-course of GLuc expression in A549 cells transfected with indicated constructs. A, C, E, G Data represent mean ± SD of three biological replicates (n = 3), P values were calculated using unpaired Student’s t test. Source data are provided as a Source Data file.
Incorporating the cap structure into chemically circularized RNA [circ(EP5Cap)RNA12 and circ(EP5Cap)RNA13] increased the protein output compared to the enzymatically circularized counterparts [circ(p)RNA12 and circ(p)RNA13], with an average enhancement of 4-fold for RNA12 and 5-fold for RNA13, irrespective of the presence of a downstream poly(A) tract (Fig. 7A, C). Adding a 3’ UTR alone or 3’ UTR followed by a poly(A) tract to the mRNA sequence, amplified the difference between capped circRNAs [circ(EP5Cap)RNA14 and circ(EP5Cap)RNA15] and their enzymatically circularized counterparts [circ(p)RNA14 and circ(p)RNA15], yielding 6.5–21-fold and 6.5–17-fold increases for RNA14 and RNA15, respectively (Fig. 7E, G). However, when compared to circ(EP5Cap)RNA12 and circ(EP5Cap)RNA13, RNA14 and RNA15 showed lower overall protein production (Fig. 7A, C, E, G). This suggested that the 3’ terminal elements essential for linear RNAs (3′ UTR and poly(A)) are not beneficial for circRNA design, even if combined with the endocyclic cap. Importantly, most chem-circRNAs carrying an endocyclic cap [circ(EP5Cap)RNA12, circ(EP5Cap)RNA13, circ(EP5Cap)RNA14] showed increased durability and sustained protein production compared to their linear counterparts (Fig. 7B, D, F). However, in an RNA sequence optimized for the cap-dependent translation (i.e., containing both a 3′ UTR (HBB) and 30-nt poly(A) tract), the protein production profiles of circular RNA and linear RNAs were very similar (Fig. 7H).
We further investigated the effect of incorporating the m7G cap on biological activity of circRNA using RNA sequences optimized for cap-dependent translation (i.e., without IRES; Fig. 8). To that end, we circularized capped mRNA (EP5Cap) bearing a minimal 5′ UTR (Kozak), a 3′ UTR derived from the human β-globin (HBB) gene, and a 30-nt poly(A) tract (RNA09) (Fig. 8). The translational activity of chemically circularized circ(EP5Cap)RNA09 and enzymatically circularized circ(p)RNA09 constructs was assessed against linear Cap1-RNA11 serving as a reference (Fig. 8A, C). Introducing the cap structure into circRNA09 [circ(EP5Cap)RNA09] led to a striking 13–370-fold increase in protein levels compared to its enzymatically circularized counterpart circ(p)RNA09 (Fig. 8A). Notably, the overall protein output observed for circ(EP5Cap)RNA09 was higher than observed for circRNAs containing the IRES-EMCV sequence (RNA12-15) (Figs. 7A, C, E, G and 8A). However, the overall translational activity and durability of capped circRNA09 [circ(EP5Cap)RNA09] were similar to its linear precursor [lin(EP5Cap)RNA09] (Fig. 8B). Overall, these findings further support our observation regarding the influence of mRNA sequence architecture on protein output. Specifically, as an RNA sequence becomes increasingly optimized for cap-dependent translation, the translational profile of circRNA appears to converge with that of its linear counterpart, diminishing the advantages typically associated with circularization.

A, B Show total protein output from circRNA₀₉ and linear (EP5Cap)RNA09 constructs in A549, HEK293T (HEK), HeLa, and HepG2 (HEP) cells transfected with circular RNAs derived from linear precursors bearing either a 5′ monophosphate (p-) [circ(p)RNA09] or an EP₅Cap structure [circ(EP5Cap)RNA09]. Protein expression was measured as secreted GLuc activity accumulated over 168 h and normalized to the level obtained with canonical Cap1-RNA₁₁ (=1). C Shows the time-course of GLuc expression in A549 cells transfected with circ(p)RNA₀₉, circ(EP₅Cap)RNA₀₉, respective linear precursors, and linear Cap1-RNA11. D Shows in vitro translation of circ(p)RNA₀₉, circ(EP₅Cap)RNA₀₉, and circ(p)RNA₁₅ in rabbit reticulocyte lysate (RRL) in the presence of the cap-dependent translation inhibitor (m7GpsppG), demonstrating inhibition of translation and supporting a cap-dependent translation mechanism40,41. Chem-circ RNAs containing N1-methylpseudouridine can be translationally active (E–H). Cells were transfected with circ(EP5Cap)RNA09 (Cap), circ(p)RNA15 (EMCV), Cap1-RNA11, and their respective counterparts containing N1-methylpeudouridine (+m1Ψ). E Shows the time-course of GLuc expression from circRNAs (circ(EP5Cap)RNA09 and circ(p)RNA15) and linear Cap1-RNA11 over 168 h in A549 cells. F–H Show the total protein output from four cell lines (A549, HeLa, HEK, HepG2) transfected with circ(EP5Cap)RNA09/circ(EP5Cap)RNA09 + m1Ψ (F), circ(p)RNA15/circ(p)RNA15 + m1Ψ (G), linear Cap1-RNA₁₁/Cap1-RNA₁₁ + m1Ψ (H), harvested at 168 h post-transfection. In each F–H, values are normalized to the signal from the corresponding uridine-containing mRNA. A, B, F, G, H Data represent mean ± SD of three biological replicates (n = 3), P values were calculated using unpaired Student’s t test. Source data are provided as a Source Data file.
To verify if the endocyclic cap in circRNA can engage in the cap-dependent translation mechanism, we evaluated the expression of circ(EP5Cap)RNA09, circ(p)RNA09, and circ(p)RNA15 in a cell-free system (rabbit reticulocyte lysate, RRL) in the presence of a cap-dependent translation inhibitor (m7GpsppG dinucleotide, Fig. 8D)40,41. As expected, increasing concentration of the inhibitor led to a progressive reduction in reporter protein levels from circ(EP5Cap)RNA09, whereas circ(p)RNA15, which contains an IRES sequence, remained unaffected. These findings strongly suggest that the endocyclic m7G cap structure in chem-circRNA09 is functional and engages in the cap-dependent translation mechanism.
Next, we investigated whether our chemical circularization approach is compatible with mRNA body modifications commonly used to reduce undesired immunogenicity of in vitro transcribed mRNAs. To that end, N1-methylpseudouridine (m1Ψ) is among the most critical modifications in therapeutic mRNA, as it has been shown to reduce activation of cytosolic and toll-like receptors (TLRs), thereby lowering cellular immune response and mRNA-associated toxicity42. However, the incorporation of m1Ψ and other noncanonical nucleobases can disrupt the function of RNA elements such as IRES sequences or autocatalytic introns by altering their secondary structure6. To determine whether an endocyclic cap structure could functionally substitute for the IRES element and support the translation of circRNA containing the m¹Ψ modification, we generated m¹Ψ-modified circRNAs by either chemical or enzymatic circularization. The resulting circRNAs (variants of RNA15) were transfected into four cell lines to assess their translational activity (Fig. 8E–H). As expected, IRES-dependent translation of enzymatically circularized RNA [circ(p)RNA15 (EMCV+m¹Ψ)] was abolished, likely due to m¹Ψ-induced disruption of the IRES secondary structure and consequent loss of function (Fig. 8E, G). In contrast, the chemically circularized capped RNA [circRNA09 (Cap+m¹Ψ)], which contained m¹Ψ and the HBB UTR, remained translationally active, albeit at a reduced level compared to its unmodified counterpart (Fig. 8E, F). A similar reduction in translational activity was observed upon incorporating of m¹Ψ into linear Cap1-RNA11 (Fig. 8E, H). In human monocyte-derived dendritic cells, all tested mRNAs but the ones containing virus-derived IRES sequences show negligible reactogenicity, as evidenced by the secretion of CXCL8 (Supplementary Fig. 15). Collectively, these results demonstrate that the chemical circularization method is compatible with RNA base modifications and highlight its potential for optimizing circRNAs’ biological activity in therapeutic applications.
Finally, we have verified if chem-circRNAs obtained by the PORA method are compatible with translation in vivo. To that end, we prepared two chem-circRNAs, RNA18 and RNA19, encoding human erythropoietin. These RNAs were formulated into LNPs (Supplementary Fig. 16) and injected into C57BL/6 mice, followed by blood collection at 6, 24, and 48 h and determination of hEPO levels by ELISA (Fig. 9). We observed a significantly higher overall protein output for chem-circ RNAs, compared to their linear counterparts, confirming both the biocompatibility of the internucleotide linkage formed during circularization and the advantage of chem-circRNAs over their linear counterparts. However, the overall protein output and translation durability were relatively low, emphasizing the need for further optimization of the chem-circRNA sequence.

A C57BL/6 female mice were intravenously inoculated with 1 µg of SM-102 -based LNP-formulated RNA. Blood was collected at the indicated time points to measure hEPO concentration with ELISA. Experimental setup. Created in BioRender. Chmieliński, S. (2025) https://BioRender.com/txhelkn. B hEPO concentration in serum over time, means ± SD, n = 5. Green stars: P < 0.01 for circ(EP5Cap)RNA18 vs each of the linear RNA group; teal stars: P < 0.01 for circ(EP5Cap)RNA19 vs each of the linear RNA group; ns—not significant (circ(EP5Cap)RNA18 vs circ(EP5Cap)RNA19), 2 way ANOVA with Tukey’s multiple comparisons test. Source data are provided as a Source Data file.
Discussion
In this work, we report the first method for the chemical circularization of in vitro transcribed (IVT) mRNA. Exogenously delivered circRNA represents a promising drug modality due to its extended lifespan in the cytosol. Since circRNAs lack free ends, they are not susceptible to exonuclease-mediated degradation, which is the main pathway for the bulk removal of cytoplasmic RNA2,43. To date, the most well-characterized methods for circularizing linear mRNA involve enzymatic ligation (RNA or DNA ligases) and ribozymatic methods based on self-splicing introns (Fig. 1)5,8,9,10,11,12,13,14,15. Chemical methods have been limited to short RNA sequences applicable to rolling circle amplification (RCA) of short peptides44. Our group has been involved for many years in developing chemical modifications of RNA ends. In this work, we leveraged this experience to develop a method of joining modified 5′ and 3′ ends of in vitro transcribed RNA, i.e. to chemically circularize full-length mRNAs. We were inspired by recent observations, made by us and others26,34 that even in long RNA macromolecules, the 5′ and 3′-ends are usually sufficiently close to each other to enable fluorescence energy transfer (<10 nm), which should mean they are also close enough to efficiently undergo an intramolecular chemical reaction. As a result, our study describes a convenient method for obtaining chemically circularized RNAs (chem-circRNAs) based on a direct and selective chemical reaction called PORA (Periodate Oxidation and Reductive Amination). The starting material for this reaction is RNAs obtained by a standard in vitro transcription in the presence of transcription primers carrying an ethylenediamine motif. PORA is performed on the RNA product in a one-pot two-step reaction relying on simple reagents, taking less than 2 h and providing conversions in the range of 35–63%.
Notably, our methodology enables incorporating chemical modifications into circRNAs previously incompatible with this modality. For example, the translational activity of circRNAs reported in the literature depends on IRES sequences, highly structured regions within the 5′ UTR, that recruit ribosomes. Although m1Ψ can be incorporated into these sequences, it disrupts their secondary structure, abolishing translational activity. In contrast, our circRNA design, which features an endocyclic cap structure, supports cap-dependent translation and, hence, can be modified with m1Ψ without dramatically compromising the translational activity of the circRNA.
Chemical circularization represents an attractive alternative to existing methods for mRNA circularization, particularly for full-length coding mRNAs. In comparison to ribozyme-based approaches utilizing permuted group I catalytic introns, chemical circularization produces a simplified impurity profile (Fig. 5 vs. Supplementary Fig. 5) and does not require magnesium ions during the reaction, which may contribute to higher RNA homogeneity.
Using simple 35 nt RNA models (RNA01 and RNA02), we demonstrated that circularization yields are predominantly dependent on the local secondary structure of RNA (end-to-end distance) rather than its length (Fig. 2). We further showed that circularization is equally efficient for RNA of various lengths and sequences, up to almost 4000 nt long. A reduction in circularization efficiency was observed for longer RNAs and those with unstructured 3′ ends (incorporating 3′ terminal polyA tracts), probably because of higher conformational flexibility. This issue was addressed by secondary structure control with oligonucleotide DNA splints, achieving circularization efficiencies ranging from 35% to 60%, even for complex macromolecular RNA constructs (Table 1). We also developed methods that enable efficient separation of chem-circRNAs from their unreacted linear precursors and provided molecular tools to verify the structure and homogeneity of chem-circRNAs. Our results demonstrate that chem-circRNAs undergo translation in living cells, similarly, to enzymatically obtained circRNAs, and remain translationally active in a mouse model. Moreover, the duration of expression in cells for most chem-circRNA analogs was extended compared to their linear precursors (Fig. 6E). We also investigated the impact of elements required for cap-dependent translation, such as the m7G cap and the poly(A) tract, on the durability of protein production and the translational activity of our chem-circRNA analogs. Adding m7G cap increased the translational activity of chem-circRNAs, although the effect was sequence dependent. We demonstrated that the endocyclic cap in chem-circRNA maintains its functionality, as evidenced by enhanced protein production compared to unmodified circRNAs (Fig. 7) and susceptibility to inhibition by compounds targeting the cap-binding translation factor, eIF4E (Fig. 8D).
We investigated the effect of introducing an EP5Cap structure to RNAs containing an internal ribosome entry site (IRES) in the 5′ untranslated region (UTR). In our experiments, we employed the well-characterized EMCV IRES, which has been widely used in both linear and circular RNAs. However, the design of our circular RNAs could potentially be improved by exploring other IRES sequences with higher activity, such as those derived from HCV or HRV-B3 45,46.
Overall, our data reveal that chemical circularization is compatible with RNA base-modifications and that the endocyclic cap moiety could serve as an alternative to IRES for promoting circRNA translation (Fig. 8E–H). However, further extensive optimization of chem-circRNA sequences and chemical structures is crucial to fully realize the potential of this methodology. Nonetheless, to our best knowledge, this work describes several unprecedented achievements in RNA circularization: (i) the chemical circularization of mRNA encoding full-length protein, (ii) the chemical circularization of IVT RNA, (iii) the development of circular RNAs that undergo cap-dependent translation, and (iv) the circularization of m1Ψ-modified RNA. While this work was under review, two other manuscripts independently showing the creation of circular RNAs equipped with m7G and their superior translational activity were published, highlighting that chemical modifications and topology alterations are the future directions for the development of circRNA field47,48. Unlike our work, these methods were based on the enzymatic ligation approach. Compared to enzymatic ligation and autocatalytic circularization, our approach allows for site-selective introduction of RNA chain modifications simply by adding an analog of a substrate for RNA polymerase during the IVT reaction. This gives users the freedom of choice of chemical modifications that could help expand the repertoire of known and tested circRNA modifications. In light of that, we believe this work opens an unexplored avenue for capped circular RNA and potentially a new generation of RNA therapeutics. Still, more research on sequence optimization and other aspects of chem-circRNA design is needed to fully embrace the potential of this technology.
Methods
Software and tools used
The MFE secondary structure model of the mRNA sequences was predicted with RNAfold web server, with energy parameters: RNA parameters (Turner model, 2004). The three-dimensional RNA models were created with the RNA Composer web server tool. The circular mRNA content was determined by densitometric analysis of the raw PAGE images using ImageJ or CLIQS (Core Laboratory Image Quantification Software).
Preparation of linear 5′ end modified and unmodified mRNA as circularization precursors
The synthesis of the respective IVT initiators and intermediates is detailed in the Supplementary Information.
Unmodified and modified linear mRNA precursors were synthesized by in vitro transcription from a linearized plasmid DNA template using T7 RNA Polymerase, HC (Thermo) followed by treatment with DNase I (Thermo). mRNA was purified using a silica-based column followed by RP-HPLC. After HPLC purification, the recovered RNA represents 75–90% of the initial crude mRNA.
Chemical circularization
An aqueous solution of the precursor RNA (25–50 µg in 490 µl, ~140 µM) was diluted with buffer (70 µl, 800 mM KH2PO4, 200 mM NaCl, pH 7.0) and incubated at 65 °C for 5 min, followed by cooling to r.t. After 10-15 min at r.t., a fresh solution of sodium periodate (70 µl, 10 mM) was added, and the reaction mixture was immediately placed in the dark and incubated at 25 °C for 30 min. A fresh solution of sodium cyanoborohydride (70 µl, 200 mM) was added to the final volume of 700 µL, and the incubation was continued for 90 min. The crude circRNA product was purified using a silica-based column (Monarch RNA cleanup kit, 50 µg, NEB) followed by RP-HPLC. After HPLC purification, the recovered RNA represented 50–70% of the initial crude mRNA. For additional purification and removal of linear RNA, the product was either treated with RNase R as described below (see Section RNase R digestion) or isolated via electroelution (for details see Supplementary Information).
Chemical circularization with complementary splint
The reaction was carried out following the procedure outlined above, with the modification that a complementary oligonucleotide (~11 µl, 10 µM, 1.5 eq, see Supplementary Table 2 for sequence) was employed.
RNase R digestion
A solution of RNA (43 µl, 10 µg) was incubated at 65 °C for 5 min and rapidly cooled in the ice bath. After 5 min at 0-4 °C, RNase R buffer ×10 (5 µl, 200 mM Tris-HCl, 1 M KCl, 1 mM MgCl2, pH 7.5, ABM), RiboLock RNase inhibitor (1.25 µl, 40 U/µl, Thermo), and RNase R (1 µl, 10 U/µl, ABM) were added. After 30 min of incubation at 37 °C the products of digestion were analyzed with PAGE directly or isolated from the reaction mixture using Monarch RNA cleanup kit (10 µg, NEB).
RNase H nicking analysis
A solution of circular RNA (2.60 µl, 500 ng) in water (17.8 µl) was heated at 70 °C for 5 min in the presence of a DNA probe (0.85 µl, 6 eq., 10 µM, 5′CCTTCAGCTCGATGCGGTTCA 3′) and gradually cooled to 37 °C for 20 min. Next, RNase H buffer ×10 (2.5 µl, 50 mM Tris-HCl, 75 mM KCl, 3 mM MgCl2, 10 mM DTT, pH 8.3), and RNase H (1.25 µl, 1 U/µl, Thermo) were added to final volume of 25 µl. After 15 min of incubation at 37 °C the products of digestion were directly analyzed with PAGE.
Translation inhibition in RRL system
Flexi Rabbit Reticulocyte Lysate (4 μL, Promega) was diluted with 4 μL solution containing amino acid mixture (100 μM), KOAc (500 mM), and MgCl2 (2.5 mM), and incubated for 1 h at 30 °C. Next, 1 μL of RNA (100 ng/µl) mixed with 1 μL of m7GpsppG (0.32–200 μM) was added to 8 μL of the lysate mixture and incubated for 1 h at 30 °C. Next, the reaction was immediately placed into a microplate reader (Synergy H1, BioTek), 50 μL of h-coelenterazine in 1× PBS was added and the luminescence was measured at 25 °C. The assay was performed in duplicate to calculate the mean and standard error (SEM).
Cell culture and transfection (A549, HEK 293 T, HeLa, Hep G2)
One day before transfection, 8 × 103 cells were seeded per well of a 96-well plate and incubated under standard conditions (37 °C, 5% CO2, saturating humidity). Transfection was performed with Lipofectamine Messenger MAX (Invitrogen, Waltham, MA, USA), according to the manufacturer’s instructions. Each RNA variant (65 ng) was transfected in triplicate. At appropriate time points, 20 µL of medium from each well was withdrawn to test the amount of produced protein, and the remaining medium was completely replaced with a fresh one. The amount of luciferase released into the medium was analyzed using the GLuc GLOW Assay (NanoLight Technologies, Norman, OK, USA) reagents, according to the manufacturer’s instructions using an EnVision plate reader (Perkin Elmer, Waltham, MA, USA)
In vivo experiment
Mice
The experiments were carried out in 10–12-week-old ( ~ 25 g) female C57BL6/cmdb under the protocols approved by the II Local Ethical Committee for Experiments on Animals in Warsaw, Poland (WAW2/111/2024). All experiments were conducted in accordance with the Directive of the European Parliament and Council No. 2010/63/EU on the protection of animals used for scientific purposes. Mice were obtained from the Breeding Facility of the Medical University of Bialystok, Bialystok, Poland. All animals were maintained in a specific pathogen-free (SPF) environment in the individually ventilated cages (IVC) under the conditions of a 12-h day/night cycle, 55% humidity and +22 °C air temperature with unrestricted access to food and drinking water.
Formulation
The respective linear mRNAs and their circular counterparts were generated according to the general procedure described in the Supplementary Information, with the exception that circRNAs were enriched using RNase R digestion.
The mRNA in 100 mM citrate buffer (pH 4) and lipid mixture (25 mM in ethanol) containing 50 mol% SM-102, 10 mol% DSPC, 38.5 mol% cholesterol, and 1.5 mol% DMG-PEG 2000 were combined to form lipid nanoparticles using the NanoAssemblr Spark (Precision Nano-systems). The obtained mRNA-LNPs were diluted in dPBS, and the buffer was exchanged by concentrating the solution through 50 kDa spin filters (MilliporeSigma, UFC505024) to remove ethanol. Concentrations and encapsulation efficiency of mRNA were determined using the Quant-it Ribo-Green RNA Assay Kit (ThermoFisher, R11490). The mean particle size and particle size distribution of the LNPs were analyzed by DLS using a Zetasizer Ultra (Malvern Instruments Ltd., Worcestershire, UK).
In vivo human erythropoietin mRNA expression studies
All mice were randomly assigned to experimental groups (n = 5 C57BL/6 mice per group). Each mouse received intravenously 100 µL of LNP-encapsulated human erythropoietin (hEPO) mRNA (1 µg per mouse). Blood samples were collected via cheek pouch from submandibular veins at 6, 24, 48, and 72 h after mRNA-LNPs injection. The blood was allowed to clot at room temperature for 30 min, and the serum was separated by centrifugation at 10,000×g for 10 min. Human EPO levels in serum were quantified using an ELISA kit (Invitrogen, Human EPO ELISA Kit, # BMS2035-2) according to the manufacturer’s instructions.
Statistical analysis
Statistical analysis was performed using GraphPad Prism version 9. Figures were generated using the same software and statistical significance was determined using appropriate tests. For normal data distribution evaluation the Shapiro-Wilk test was used. Details of all remaining statistical tests used are provided in the figures’ captions.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the findings of this study are available within the paper and its Supplementary Information. Complete DNA sequences, detailed descriptions of chemical syntheses, characterization of chemical compounds, experimental procedures involving in vitro transcription, RNA circularization and preparation, nucleolytic assays, FRET probes, cell culture, and cell-free experiments are provided in the Supplementary Information. Source data are provided with this paper.
References
-
Rohner, E., Yang, R., Foo, K. S., Goedel, A. & Chien, K. R. Unlocking the promise of mRNA therapeutics. Nat. Biotechnol. 40, 1586–1600 (2022).
-
Garneau, N. L., Wilusz, J. & Wilusz, C. J. The highways and byways of mRNA decay. Nat. Rev. Mol. Cell Biol. 8, 113–126 (2007).
-
Suzuki, H. et al. Characterization of RNase R-digested cellular RNA source that consists of lariat and circular RNAs from pre-mRNA splicing. Nucleic Acids Res. 34, e63 (2006).
-
Suzuki, H. & Tsukahara, T. A View of Pre-mRNA Splicing from RNase R Resistant RNAs. Int. J. Mol. Sci. 15, 9331–9342 (2014).
-
Wesselhoeft, R. A., Kowalski, P. S. & Anderson, D. G. Engineering circular RNA for potent and stable translation in eukaryotic cells. Nat. Commun. https://doi.org/10.1038/s41467-018-05096-6 (2018).
-
Wesselhoeft, R. A. et al. RNA circularization diminishes immunogenicity and can extend translation duration in vivo. Mol. Cell 74, 508–520.e504 (2019).
-
Enuka, Y. et al. Circular RNAs are long-lived and display only minimal early alterations in response to a growth factor. Nucleic Acids Res. 44, 1370–1383 (2016).
-
Chen, X. & Lu, Y. Circular RNA: biosynthesis in vitro. Front. Bioeng. Biotechnol. 9, 787881 (2021).
-
Dolinnaya, N. G. et al. Oligonucleotide circularization by template-directed chemical ligation. Nucleic Acids Res. 21, 5403–5407 (1993).
-
Fedorova, O. A., Gottikh, M. B., Oretskaya, T. S. & Shabarova, Z. A. Cyanogen bromide-induced chemical ligation: mechanism and optimization of the reaction conditions. Nucleosides Nucleotides 15, 1137–1147 (1996).
-
Nakamoto, K. et al. Chemically synthesized circular RNAs with phosphoramidate linkages enable rolling circle translation. Chem. Commun. 56, 6217–6220 (2020).
-
Rigden, J. E. & Rezaian, M. A. In vitro synthesis of an infectious viroid: analysis of the infectivity of monomeric linear CEV. Virology 186, 201–206 (1992).
-
Chen, C.-Y. & Sarnow, P. Initiation of protein synthesis by the eukaryotic translational apparatus on circular RNAs. Science 268, 415–417 (1995).
-
Qu, L. et al. Circular RNA vaccines against SARS-CoV-2 and emerging variants. Cell 185, 1728–1744.e1716 (2022).
-
Kaufmann, G., Klein, T. & Littauer, U. Z. T4 RNA ligase: substrate chain length requirements. FEBS Lett. 46, 271–275 (1974).
-
Litke, J. L. & Jaffrey, S. R. Highly efficient expression of circular RNA aptamers in cells using autocatalytic transcripts. Nat. Biotechnol. 37, 667–675 (2019).
-
Kumar, A. et al. Extensive in Vitro and in Vivo Protein Translation Via in Situ Circularized RNAs (Cold Spring Harbor Laboratory, 2022).
-
Ford, E. & Ares, M. Synthesis of circular RNA in bacteria and yeast using RNA cyclase ribozymes derived from a group I intron of phage T4. Proc. Natl Acad. Sci. USA 91, 3117–3121 (1994).
-
Puttaraju, M. & Been, M. Group I permuted intron-exon (PIE) sequences self-splice to produce circular exons. Nucleic Acids Res. 20, 5357–5364 (1992).
-
Sinha, T., Panigrahi, C., Das, D. & Chandra Panda, A. Circular RNA translation, a path to hidden proteome. WIREs RNA https://doi.org/10.1002/wrna.1685 (2022).
-
Lei, M., Zheng, G., Ning, Q., Zheng, J. & Dong, D. Translation and functional roles of circular RNAs in human cancer. Mol. Cancer https://doi.org/10.1186/s12943-020-1135-7 (2020).
-
Ramanathan, A., Robb, G. B. & Chan, S.-H. mRNA capping: biological functions and applications. Nucleic Acids Res. 44, 7511–7526 (2016).
-
Yang, Y. & Wang, Z. IRES-mediated cap-independent translation, a path leading to hidden proteome. J. Mol. Cell Biol. 11, 911–919 (2019).
-
Koch, A., Aguilera, L., Morisaki, T., Munsky, B. & Stasevich, T. J. Quantifying the dynamics of IRES and cap translation with single-molecule resolution in live cells. Nat. Struct. Mol. Biol. 27, 1095–1104 (2020).
-
Kim, B., Seol, J., Kim, Y. K. & Lee, J.-B. Single-molecule visualization of mRNA circularization during translation. Exp. Mol. Med. https://doi.org/10.1038/s12276-023-00933-1 (2023).
-
Mamot, A. et al. Ethylenediamine derivatives efficiently react with oxidized RNA 3′ ends providing access to mono and dually labelled RNA probes for enzymatic assays and in vivo translation. Nucleic Acids Res. 50, e3 (2022).
-
Fan, X., Yang, Y., Chen, C. & Wang, Z. Pervasive translation of circular RNAs driven by short IRES-like elements. Nat. Commun. https://doi.org/10.1038/s41467-022-31327-y (2022).
-
Chen, C.-K. et al. Structured elements drive extensive circular RNA translation. Mol. Cell 81, 4300–4318.e4313 (2021).
-
Mullgan, M. J. et al. Phase I/II study of COVID-19 RNA vaccine BNT162b1 in adults. Nature 586, 589–593 (2020).
-
Coleman, T. M., Wang, G. & Huang, F. Superior 5′ homogeneity of RNA from ATP-initiated transcription under the T7 ϕ2.5 promoter. Nucleic Acids Res. 32, e14 (2004).
-
Lorenz, R. et al. ViennaRNA Package 2.0. Algorithms Mol. Biol. https://doi.org/10.1186/1748-7188-6-26 (2011).
-
Sarzynska, J., Popenda, M., Antczak, M. & Szachniuk, M. RNA tertiary structure prediction using RNAComposer in CASP15. Proteins 91, 1790–1799 (2023).
-
Popenda, M. et al. Automated 3D structure composition for large RNAs. Nucleic Acids Res. https://doi.org/10.1093/nar/gks339 (2012).
-
Lai, W. et al. mRNAs and lncRNAs intrinsically form secondary structures with short end-to-end distances. Nat. Commun. https://doi.org/10.1038/s41467-018-06792-z (2018).
-
Karikó, K., Muramatsu, H., Ludwig, J. & Weissman, D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. https://doi.org/10.1093/nar/gkr695 (2011).
-
Abe, B., Wesselhoeft, R., Chen, R., Anderson, D. & Chang, H. Circular RNA migration in agarose gel electrophoresis. Mol. Cell 82, 1768–1777 (2022).
-
Starke, S. et al. Exon circularization requires canonical splice signals. Cell Rep. 10, 103–111 (2015).
-
Dodbele, S., Mutlu, N. & Wilusz, J. Best practices to ensure robust investigation of circular RNAs: pitfalls and tips. EMBO Rep. https://doi.org/10.15252/embr.202052072 (2021).
-
Masuda, N., Ohnishi, T., Kawamoto, S., Monden, M. & Okubo, K. Analysis of chemical modification of RNA from formalin-fixed samples and optimization of molecular biology applications for such samples. Nucleic Acids Res. 27, 4436–4443 (1999).
-
Perzanowska, O., Smietanski, M., Jemielity, J. & Kowalska, J. Chemically modified poly(A) analogs targeting PABP: structure activity relationship and translation inhibitory properties. Chemistry https://doi.org/10.1002/chem.202201115 (2022).
-
Kowalska, J. et al. Synthesis and characterization of mRNA cap analogs containing phosphorothioate substitutions that bind tightly to eIF4E and are resistant to the decapping pyrophosphatase DcpS. RNA 14, 1119–1131 (2008).
-
Nance, K. D. & Meier, J. L. Modifications in an emergency: the role of N1-methylpseudouridine in COVID-19 vaccines. ACS Cent. Sci. 7, 748–756 (2021).
-
Chen, L. & Yang, L. Regulation of circRNA biogenesis. RNA Biol. 12, 381–388 (2015).
-
Abe, N. et al. Rolling circle translation of circular RNA in living human cells. Sci. Rep. https://doi.org/10.1038/srep16435 (2015).
-
Chen, R. et al. Engineering circular RNA for enhanced protein production. Nat. Biotechnol. 41, 262–272 (2023).
-
Unti, M. J. & Jaffrey, S. R. Highly efficient cellular expression of circular mRNA enables prolonged protein expression. Cell Chem. Biol. 31, 163–176 (2024).
-
Fukuchi, K. et al. Internal cap-initiated translation for efficient protein production from circular mRNA. Nat. Biotechnol. https://doi.org/10.1038/s41587-025-02561-8 (2025).
-
Chen, H. et al. Chemical and topological design of multicapped mRNA and capped circular RNA to augment translation. Nat. Biotechnol. https://doi.org/10.1038/s41587-024-02393-y (2024).
Acknowledgements
This project was supported by Virtual Research Institute Łukasiewicz Research Network—PORT Polish Center for Technology Development project “Horizon for Excellence in messenger RNA applications in immunOncology”[HERO] financed by the Polish Science Fund.
Ethics declarations
Competing interests
J.K., J.J., A.M., M.W.K., M.W., A.A.R., K.F., K.C., D.N., and J.G. are co-inventors of patent application related to chemical circularization of RNA (EP23461592.0A). The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wasinska-Kalwa, M., Mamot, A., Czubak, K. et al. Chemical circularization of in vitro transcribed RNA for exploring circular mRNA design. Nat Commun 16, 6455 (2025). https://doi.org/10.1038/s41467-025-61775-1
-
Received:
-
Accepted:
-
Published:
-
DOI: https://doi.org/10.1038/s41467-025-61775-1
