Introduction
The functional characteristics of Survivin make it a distinctive protein with diverse roles, including the regulation of cell proliferation and apoptosis. Its expression pattern is also unique; Survivin is prominently expressed during embryonic development and in fetal tissues, but is largely absent in most normal and differentiated tissues1,2,3,4. However, it is highly expressed in various human cancers5,6 (Supplementary Table 1). In cancer cells, Survivin is frequently associated with increased cell proliferation, resistance to chemotherapy, reduced apoptosis, cellular stress, and cancer recurrence4,7,8. Survivin belongs to the Inhibitor of Apoptosis Protein (IAP) family9. Apoptosis plays a crucial role in suppressing cancer progression by eliminating abnormal cells. It also helps contribute to the removal of self-reactive lymphocytes from the immune system in autoimmune diseases. In the context of a pathological immune response, Survivin prolongs the survival of self-reactive lymphocytes by inhibiting apoptotic pathways in these cells10,11. Additionally, survivin promotes metastasis and enhances the WNT/β-catenin pathway through a PI3K/AKT-dependent mechanism, facilitating self-renewal10,12. One of its primary functions is the inhibition of caspase activity. Survivin monomer forms a complex with XIAP, another member of the IAP family13,14. This interaction stabilizes XIAP and, in turn, inhibits caspase-9 activity (The interaction between XIAP BIR3 domain and caspase-9 is shown in Supplementary Fig. 1)15,16. Specifically, Survivin protects XIAP from ubiquitination and proteasomal degradation by binding to it. Another mechanism by which Survivin inhibits apoptosis involves neutralizing the Smac/DIABLO factor. Smac/DIABLO, a mitochondrial protein, initiates intrinsic apoptosis by binding to IAPs and disrupting their interaction with caspases. When Survivin binds to Smac/DIABLO, it suppresses Smac/DIABLO’s ability to interact with IAPs such as XIAP. XIAP, a critical protein in apoptosis inhibition, contains three Baculoviral IAP Repeat(BIR) domains. The third domain, BIR3, directly binds to the N-terminus of Caspase-9, thereby blocking apoptosis17. It has been demonstrated that four amino acids, at the N-terminus of the SMAC protein (AVPI), bind to the BIR3 domain, inhibiting its function and triggering apoptosis18,19,20,21(The interaction between XIAP BIR3 domain and Smac/DIABLO is shown in Supplementary Fig. 2). TheBIRC5 gene, located in the telomeric region of chromosome 17q25, encodes Survivin22. This gene consists of four exons and three introns. And through RNA splicing, it generates six protein isoforms: Survivin-WT, − 2B, − 3B, − 3α, –ΔEx3, and − 2α. Among these, WT-survivin and survivin-DEx3 regulate apoptosis, while survivin-3B and WT-survivin are involved in cell cycle regulation1,23,24,25. Survivin, composed of 142 amino acids with a molecular weight of 16.5 kDa26, consists of a 70 amino acid N-terminal BIR domain associated with apoptotic regulation, a C-terminal α-helix critical for mitotic function, and a domain facilitating dimer formation27. Found primarily in the cytoplasm and nucleus of tumor cells, survivin is also localized to the mitochondria. It is released in response to cellular stress and inhibits caspase activity in the cytoplasm. In the nucleus, Survivin associates with the kinetochore of metaphase chromosomes in dividing tumor cells.Extracellularly, it has also been identified in exosomes derived from tumor cells (100–400 nm vesicles), which are internalized by neighboring cells1. The acetylation of survivin at lysine 129 promotes homodimerization (Supplementary Fig. 3) while deacetylation results in monomer formation, which heterodimerizes with CRM1. Survivin’s nuclear export signal (amino acids 89–98), located within the homodimerization domain, allows CRM1binding, facilitating nucleocytoplasmic transport (Supplementary Fig. 4). In the cytoplasm, the non-acetylated monomeric form of Survivin inhibits caspase activation, thereby protecting tumor cells28,29. In the nucleus, monomeric Survivin is a component of the Chromosomal Passenger Complex (CPC), critical for chromosome segregation and cytokinesis during cell division. The CPC is a heterotetrameric complex that localizes to different sites during various stages of mitosis and is crucial for regulating key events in cell division, such as chromosome attachment to microtubules, proper spindle assembly, and cytokinesis30,31. Aurora B kinase is one of the CPC’s enzymatic components; the other three CPC proteins are Inner Centromere Protein (INCENP), survivin, and Borealin (Supplementary Fig. 5). Each of these proteins has specific regulatory functions, and alterations or disruptions in any of the four CPC components can lead to errors in chromosome segregation and cytokinesis, resulting in genomic instability31. Analysis of these proteins in CPC formation indicates that Aurora B kinase is directed to mitosis by the other three CPC proteins: INCENP, Survivin, and Borealin. INCENP acts as a scaffolding protein, stabilizing the complex. Borealin promotes the association of Survivin with INCENP, and together, Survivin and Borealin connect with Aurora B kinase through INCENP. INCENP binds to Aurora B kinase at its C-terminal IN-box for centromere targeting, and its N-terminal region interacts with Borealin and Survivin32,33. This illustrates that the activation of Aurora B kinase depends on various mitotic kinases, including polo-like kinase 1 (PLK1). Additionally, studies reveal that PLK1 phosphorylates survivin at Ser-2034,35. Consequently, survivin triggers the activation of Aurora B kinase, facilitating cell division. In the case of Borealin binding to Survivin, the CPC is formed34,35,36. During mitosis, the chromosomal passenger complex initially localizes at the inner centromeres during prophase and remains there until the chromosomes align at the metaphase plate. During the transition from metaphase to anaphase, the complex relocates to the newly formed microtubules between the separating chromosomes. Additionally, chromosomal passengers migrate to the cell cortex during anaphase, where the cleavage plane is established. As the cell progresses through telophase and cytokinesis, these proteins accumulate at the midbody and are ultimately discarded when the midbody is externalized, allowing the daughter cells to separate. Although CPC proteins are functionally diverse, their localization is interdependent37. Inhibition of CPC complex formation leads to polyploidy, cytokinesis failure, and mitotic arrest. This study focuses on the design and experimental evaluation of a novel peptide that competes with Borealin for binding to Survivin38, thereby inhibiting CPC complex formation. Consequently, reduced Survivin levels during the G2/M phase of the cell cycle lead to the cessation of cell division and mitotic catastrophe. On the other hand, disruption of the Survivin-CRM1 interaction triggers apoptosis and has the potential to suppress cancer cell proliferation26,39.
Results
Computational
Docking
According to the molecular docking analysis, the shared residues of the Survivin protein that bind to anti-cancer peptides are located in the Borealin region and include the linker region of Survivin, with a high score in cluster 0.
MD analysis
Root mean square deviation analysis (RMSD)
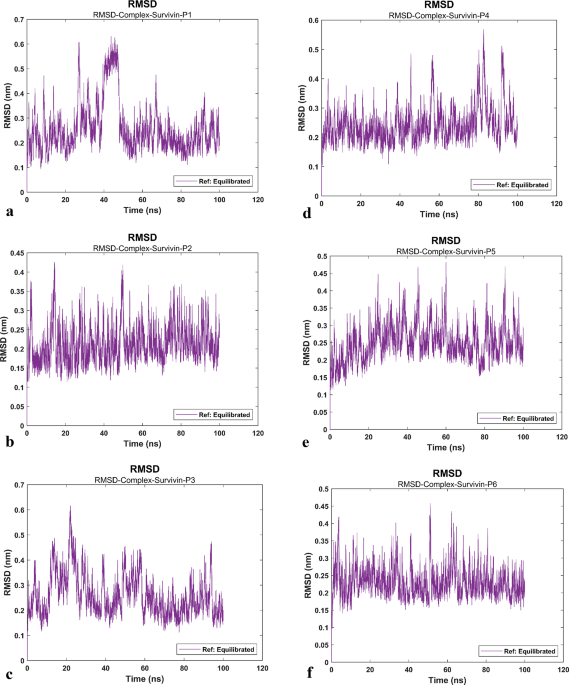
The root mean square deviation (RMSD) values of proteins and ligands were calculated against the initial structure in the protein–ligand systems and plotted to compare protein backbone stability (Fig. 1a–f)40,41. The RMSD of the protein’s backbone (Fig. 1g–l), specifically from the region where the peptide is attached (the protein linker part), which lies between the two domains of the protein, exhibited fluctuations compared to the domains of the protein.




Root Mean Square Deviation (RMSD) of Survivin protein and systems. RMSD values of the systems and the protein’s backbone are determined through molecular dynamics (MD) simulation at different time levels. The RMSD graphs indicate that all Survivin-peptide complexes exhibit fluctuation amplitudes comparable to those of the Survivin protein, confirming stable peptide interactions throughout the simulation. In each complex, the A domain of Survivin exhibits consistent fluctuations, while the B domain shows greater fluctuations, attributed to the presence of a linker. (a) RMSD values of the Survivin-P1 system (purple). (b) RMSD values of the Survivin-P2 system (purple). (c) RMSD values of the Survivin-P3 system (purple). (d) RMSD values of the Survivin-P4 system (purple). (e) RMSD values of the Survivin-P5 system (purple). (f) RMSD values of the Survivin-P6 system (purple). (g) RMSD values of the survivin protein (P1) (yellow). (h) RMSD values of the survivin protein (P2) (yellow). (i) RMSD values of the survivin protein (P3) (yellow). (j) RMSD values of the survivin protein (P4) (yellow). (k) RMSD values of the survivin protein (P5) (yellow). (l) RMSD values of the survivin protein (P6) (yellow). (m) RMSD values of domain A of Survivin protein (P1) (blue). (n) RMSD values of domain A of Survivin protein (P2) (blue). (o) RMSD values of domain A of Survivin protein (P3) (blue). (p) RMSD values of domain A of Survivin protein (P4) (blue). (q) RMSD values of domain A of Survivin protein (P5) (blue). (r) RMSD values of domain A of Survivin protein (P6) (blue). (s) RMSD values of domain B of Survivin protein (P1) (red). (t) RMSD values of domain B of Survivin protein (P2) (red). (u) RMSD values of domain B of Survivin protein (P3) (red). (v) RMSD values of domain B of Survivin protein (P4) (red). (w) RMSD values of domain B of Survivin protein (P5) (red). (x) RMSD values of domain B of Survivin protein (P6) (red).
Due to the presence of a linker in the survivin protein structure, considerable fluctuations were observed in the diagrams. There was no definitive conclusion about the stability of the protein and peptide structure. To address this, the RMSD of the protein domains (A (Fig. 1m–r), and B (Fig. 1s–x), and the peptide (Fig. 2a–f) were calculated separately. The results indicated that both the protein and peptide showed similar RMSD values, suggesting that they fluctuated together during the simulation due to the presence of the linker. The results show that the stability of the six systems was simulated and finally confirmed by the RMSD analysis. The average RMSD value demonstrates that, after 18 ns, the RMSD curves for both protein–ligand systems exhibit nearly the same conformation change pattern within an acceptable range.

Root Mean Square Deviation (RMSD) of peptides. RMSD values of the peptide’s backbone are determined through molecular dynamics (MD) simulation at different time levels, as shown in the RMSD graphs of the peptides, they exhibit similar fluctuations to their Survivin-peptide complexes,that these comparable fluctuations indicate stable binding of the anticancer peptides to the survivin protein during the simulation. (a) RMSD values of the anti-cancer peptide (P1) (black). (b) RMSD values of the anti-cancer peptide (P2) (black). (c) RMSD values of the anti-cancer peptide (P3) (black). (d) RMSD values of the anti-cancer peptide (P4) (black). (e) RMSD values of the anti-cancer peptide (P5) (black). (f) RMSD values of the anti-cancer peptide (P6) (black).
Radius of gyration (Rg)
The radius of gyration (Rg) during MD simulations diagrams shows consistent and steady conformational changes across all systems. This observation implies a stable behavior potential of interaction between each protein and ligand system41 (Fig. 3a–f).

The radius of the gyration(Rg). The Rg graph of systems remains stable during the (100 ns) simulation,as observed in the Rg graphs, the binding of the anticancer peptide to the survivin protein during the simulation does not disrupt the protein fold, demonstrating a stable and consistent fluctuation range. (a) Rg values of the Survivin-P1 system. (b) Rg values of the Survivin-P2 system. (c) Rg value of the Survivin-P3 system. (d) Rg values of the Survivin-P4 system. (e) Rg values of the Survivin-P5 system. (f) Rg values of the Survivin-P6 system.
GROMACS protein–ligand interaction energy analysis
To recalculate the system energy from the existing simulation path, we called mdrun with the rerun option. The total interaction energy was calculated and plotted for six protein-peptide systems. The average short-range Coulombic interaction energy for P1, P2, P3, P4, P5, and P6 peptides, respectively, are (− 215.865 kJ mol−1, − 232.263 kJ mol−1, − 229.382 kJ mol−1, − 216.896 kJ mol−1, − 157.223 kJ mol−1, − 212.905 kJ mol−1). The short-range Lennard–Jones energy for P1, P2, P3, P4, P5, and P6 peptides, respectively, is (− 195.9 kJ mol−1, − 200.542 kJ mol−1, − 189.717 kJ mol−1, − 198.668 kJ mol−1, − 169.833 kJ mol−1, − 202.029 kJ mol−1). The average calculation results of both the Coulombic interaction energy and the short-range Lennard–Jones energy for Survivin-P1, Survivin-P2, Sutrvivin-P3, Survivin-P4, Survivin-P5, and Survivin-P6 respectively are (− 411.764, − 432.805, − 419.099, − 415.564, − 327.56, − 414.934) (Fig. 4a–f).

The GROMACS Interaction energy (IE). The Average of Coulombic IE and Lennard–Jones IE were calculated with the help of protein–ligand analysis by GROMACS software version 2023. (a) IE values of the Survivin-P1 system. (b) IE values of the Survivin-P2 system. (c) IE value of the Survivin-P3 system. (d) IE values of the Survivin-P4 system. (e) IE values of the Survivin-P5 system. (f) IE values of the Survivin-P6 system.
gmx_MMPBSA analysis
The free energy for all simulations was calculated using GROMACS files. Input files were generated by selecting Poisson-Boltzmann (PB) and Generalized Born (GB) calculations. Subsequently, Van der Waals, Electrostatic, Desolvation, and Total interaction energies were computed for each system23. The calculated interaction affinities of the protein-peptide systems are summarized in Table 1.
Experimental
MTT assay
Cell viability in human breast cancer cells was assessed using the MTT assay. As illustrated in the diagrams (Fig. 5a–e) and Table 2, MCF-7 cells were treated with the anti-cancer peptide (P3) at ten different concentrations and incubated for 24 and 48 h. The antiproliferative effects of the anti-cancer peptide(P3) were measured using an ELISA reader. The results showed a significant reduction in cell viability and growth compared to the control groups.

MTT assay. Assessment of the impact of serial dilutions of peptide on MCF-7 cells’ proliferation and viability. (a) The data shown in the diagram represent the means of independent experiments conducted at 24 h (gray column) and 48 h (black column), with error bars indicating the standard deviation. (b) MTT line graph at 24 h. (c) MTT line graph at 48 h. (d) MTT line graph showing cell viability across different concentrations. (e) MTT bar graph comparing viability at 24 and 48 h. The IC50 values for both 24 and 48 h are marked with stars on the graphs. Each concentration was analyzed in triplicate. The data are expressed as the mean ± standard deviation (s.d.) of data from triplicate wells.*P < 0.05.
Flow cytometry apoptosis
Based on the results of the MTT analysis, the IC50 concentrations were determined to be 40 μM at 24 h and 25 μM at 48 h. Therefore, these two concentrations of the medicinal peptide were used for the tests.
To ascertain whether the peptide-induced growth inhibition in MCF-7 cells corresponded with the induction of apoptotic cell death, the apoptosis index was measured using the Annexin V-FITC/PI co-staining assay. According to this method, MCF-7 cells were examined by flow cytometry 24 h (40 µM) and 48 h (25 µM) post-treatment with the anti-cancer peptide (P3). Untreated MCF-7 cells served as the negative control. All data were analyzed using the dedicated software, FlowJo V10.10. The data for 24 h (Fig. 6a–c) and 48 h (Fig. 6d–f) are presented in diagrams. indicate that the apoptotic fraction of MCF-7 cells, as measured by flow cytometry, exceeded 80% at a concentration of 25 μM after 48 h.

Apoptosis flow cytometry. Flow cytometry observations and measurements at 24 h for the treated group indicated a small percentage of apoptotic cells. (a) Control cells after 24 h of incubation. (b) treated cells after 24 h at 40 µM incubation. At 48 h, a significant increase in apoptosis was observed compared to the control group. (c) Control cells after 48 h of incubation. (d) treated cells after 48 h at 25 µM incubation. Q1:Necrosis, Q2: Late Apoptosis, Q3: Early Apoptosis, Q4: Live Cells (e) A line graph illustrating the apoptosis assessment of the treated groups compared to controls after 24 h of incubation. (f) A line graph illustrating the apoptosis assessment of the treated groups compared to controls after 48 h of incubation. The data are expressed as the mean ± standard deviation (s.d.) of data from triplicate wells. *P < 0.05, **P < 0.001, and ***P < 0.0001.
Cell cycle analysis
In this study, the DNA content of MCF-7 cells was measured during the cell cycle to gather information about cell cycle progression. Bioinformatic analysis revealed that the peptide induces cell cycle arrest from the G2 to M phase. Therefore, the peptide’s effect on halting the MCF-7 cell cycle was investigated.
The curves illustrate the distribution of DNA content across the cell cycle phases in MCF-7 cells following 24-h (Fig. 7a–c) and 48-h (Fig. 7d–f) treatments. In control samples (untreated MCF-7 cells), 35.08% of cells remained in the G0/G1 phase at 24 h and 39.53% at 48 h, 45.27% in the S phase at 24 h and 41.82% at 48 h, and 18.83% in the G2/M phase at 24 h and 21.17% at 48 h. Treatment of MCF-7 cells with the anti-cancer peptide (P3) at a concentration of 40 µM for 24 h and 25 µM for 48 h increased the proportion of cells in the G0/G1 phase, suggesting enhanced apoptosis. Additionally, a significant arrest of cells was observed in the G2/M phase.

Cell cycle flow cytometry, Curves estimate the percentages of MCF-7 cell populations in different cell cycle phases. (a) Control cells after 24 h of incubation. (b) treated cells after 24 h at 40 µM incubation. (c) Control cells after 48 h of incubation. (d) treated cells after 48 h at 25 µM incubation. (e) A line graph illustrating the cell cycle assessment of the treated groups compared to controls after 24 h of incubation. (f) A line graph illustrating the cell cycle assessment of the treated groups compared to controls after 48 h of incubation. The data are expressed as the mean ± standard deviation (s.d.) of data from triplicate wells. *P < 0.05, **P < 0.001, and ***P < 0.0001.
Live/dead staining
Using fluorescence microscopy, the viability of42cells was assessed by labeling and quantifying both live and dead cell populations. Cells exhibiting intact membranes displayed green fluorescence, signifying viability, whereas cells with compromised membranes or those that were non-viable emitted orange and red fluorescence, respectively. Treatment with the anti-cancer peptide (P3) agent significantly increased the proportion of non-viable cells compared to the control group (Fig. 8a–e). These findings suggest that the P3 promotes apoptosis in MCF-7 cells.

Live/Dead Cell Double Staining, viable and dead cells were evaluated using acridine orange/propidium iodide dual staining in MCF-7 cells. Each concentration was analyzed in triplicate. (a) Control cells after 24 h of incubation. (b) Control cells after 48 h of incubation. Not-viable cells were noticeable at 24 and 48 h in the anti-cancer peptide(P3) treated groups. (c) Treated cells after 24 h at 40 µM incubation. (d) Treated cells after 48 h at 25 µM incubation. Scale bar, 20 µm. Magnification, × 200. Green and red/orange fluorescence shows viable and non-viable cells, respectively. (e) A line graph illustrating the Live/Dead assessment (AO/PI Double Staining) of the studied groups. The data are expressed as the mean ± standard deviation (s.d.) of data from triplicate wells. *P < 0.05, **P < 0.001, and ***P < 0.0001.
Discussion
Protein–protein interactions (PPIs) are fundamental to various biological processes and provide essential insights into protein function43,44,45, assembly, and communication. Disruption of Survivin’s interactions with other proteins, especially given its overexpression in cancer cells, is considered a promising approach for cancer diagnosis and treatment46,47. Peptides have emerged as a notable class of drugs with diverse applications, spanning antitumor, antidiabetic, antimicrobial, and other properties42,48,49,50. The therapeutic potential of peptides represents a rapidly advancing field of research47, offering several advantages, notably fewer side effects compared to small molecules51,52. Here, we designed and identified anticancer peptides using LigPlot + version 2.2, NCC-Finder, YASARA version 22.5.22, ChimeraX version 1.7, GROMACS version 2023, and gmx_MMPBSA version 1.6.2. Previous experimental studies have demonstrated that peptide function is strongly correlated with the stability of secondary structures, such as α-helix or β-strand content, and with protein binding affinity. We utilized the structural stability of the Borealin protein as a foundation for peptide design.. Eventually, six candidate peptides were designed using NCC-Finder, LigPlot + version 2.2, and YASARA version 22.5.22. After the molecular dynamics simulation, interaction energy calculation, and RMSD analysis of all five designed protein-peptide systems compared to the Native Survivi-P1 system, it was evident that the mutating positions 71 to valine and 69 to glutamine yielded enhanced variants. Their performance surpassed that of the native (unmutated) P1 peptide. that of the Native peptide (P1) (no mutant). Transfection experiments, exposing MCF-7 cells to the peptide P3 for 4 h, indicate that the cell-penetrating sequence at the peptide’s N-terminal enhances internalization38,53,54,55,56. Based on the results, peptide (P3) effectively competes with Borealin for Survivin binding, inhibiting CPC formation and suppressing cancer cell division. P3 blocks Survivin’s intraction with CRM1,therby interfering with its nuclear-cytoplasmic transport. This reduces XIAP protection, weakness anti apoptotic defenses in the cytosol and ultimately leads to apoptosis. Future studies should explore Survivin’s role in stabilizing other IAPs. Blocking Survivin’s nuclear export signal (NES) impacts the localization and function of CPC components26. Consistent results across MD simulations, gmx_MMPBSA analysis, and experimental data support the clinical potential of these peptides in cancer treatment. In MTT analysis, IC5057 values were achieved at 40 µM and 25 μM concentrations over 24 and 48 h, respectively. Flow cytometry revealed 86.6% apoptosis at 25 µM after 48 h. Cell cycle arrest in the G2/M phase was observed, demonstrating inhibition of uncontrolled proliferation. Inducing apoptosis inhibits tumor progression, metastasis, and angiogenesis. These findings suggest that peptide-based drugs may improve patient outcomes, as chemical therapies often cause nonspecific toxicity, drug resistance, and damage to healthy tissues. In contrast, peptides show lower toxicity, reduced resistance, and longer-term therapeutic viability. Although, in this study we employed advanced bioinformatics tools , including simulations and docking, to design, screen, and optimizepeptides prior to laboratory validations58, Comprehensive evaluation of off-target toxicity and long-term biocompatibility remains essential. Peptides, while generally less stable and prone to rapid degradation, penetrate tissue more efficiently than small molecules48,59. Still, effective delivery systems remain a challenge. Nanoliposomes are promising vehicles for targeted delivery due to their ability to encapsulate both hydrophilic and hydrophobic drugs, protect peptides from enzymatic breakdown, and prolong systemic circulation. To further enhance the systemic delivery and stability of anticancer peptides, future studies should explore advanced encapsulation strategies such as liposomal nanocarriers. Nanoliposomes present a versatile and biocompatible platform for targeted peptide delivery, offering protection against enzymatic degradation and enabling sustained drug release. Moreover, surface modifications, such as PEGylation or conjugation with tumor-specific ligands, could optimize biodistribution, minimize off-target effects, and enhance therapeutic efficacy60,61,62. Integrating these nanocarrier technologies with peptide-based treatments may significantly improve pharmacokinetics and clinical performance. As such, the development of liposome-based delivery systems should be considered a promising direction in the advancement of peptide therapeutics for cancer. Peptide P3 can also be conjugated with cargo molecules or chemotherapy agents via linkers, creating multifunctional therapeutic compounds59,63. This approaches enhances tereatment precision and minimizes the side effects of traditional chemotheraphy. Animal studies and biophysical assays are essential for validating in vivo efficacy. Our designed peptide stands out as a versatile and potent cancer therapeutic, capable of disrupting critical cellular pathways. Specifically, the peptide inhibits the CPC complex, halting cell division, while concurrently targeting XIAP–Survivin interactions to trigger apoptosis. By blocking Survivin’s nuclear role, the peptide combines the strengths of Sur-X derivatives with mitotic disruption, indicating superior performance17. Moreover, freeing the Smac/DIABLO binding site boosts the effectiveness of Smac mimetics64. Structurally, the peptide exceeds small molecules like YM155 in cell permeability and tissue penetration65, allowing effective activity at lower doses and reducing off-target toxicity and resistance48. In conclusion, the Borealin-derived anticancer peptide P3 exemplifies a novel class of rationally engineered biotherapeutics that disrupt critical molecular interactions within cancer cells. Its dual functionality, hindering mitotic progression and promoting apoptosis, positions it as a compelling candidate for next-generation cancer treatments, especially in cases resistant to conventional chemotherapies.
Materials and methods
Computational
Designing, modeling, and MD simulation of peptides
The initial step in designing an inhibitory peptide for the Survivin protein involves identifying the residues responsible for the interaction between Survivin and Borealin within the CPC complex, as well as those involved in the Survivin linker. To determine the exact binding sites of Survivin in the CPC complex with Borealin and its dimeric form, the 3D structures of the CPC complex (PDB ID: 2QFA) and the Survivin dimer (PDB ID: 1E31) were utilized as templates. The NCCFinder and LigPlot + version 2.266 software (available at https://www.ebi.ac.uk/thornton-srv/software/LigPlus/) were employed to pinpoint these binding sites. Based on this analysis, an 11-residue fragment of the Borealin protein (residues 65–75), which interacts with Survivin, was identified as a potential anticancer peptide and named P1.
Peptide mutation
The FoldX plugin, version 4 (available at https://foldxsuite.crg.eu/pluginInstall) of the YASARA software version 22.5.22 (available at https://www.yasara.org/) was employed to introduce effective mutations in peptides67. In the initial step, the C chain and fragments 1–65 and 75 to the end of the B chain were removed from the CPC complex using YASARA software version 22.5.22. Subsequently, the N69Q, L71V, and Y73F mutations were introduced at residues 69, 71, and 73, respectively, using the YASARA FoldX plugin. Specifically, asparagine 69 was mutated to glutamine (N69Q), leucine 71 to valine (L71V), leucine 71 to isoleucine (L71I), and tyrosine 73 to phenylalanine (Y73F), and a double mutation of N69Q_Y73F. The mutations were named P2, P3, P4, P5, and P6, respectively (Table 3).
Molecular docking
The ClusPro server protocol established the connection between Survivin and peptides68,69,70. The results, ranging from zero to three cluster scores, were examined. The common residues involved in this connection were identified through LigPlot + software version 2.2.
Molecular dynamics simulations
The six protein-peptide systems underwent Molecular Dynamics Simulations using GROMACS, version 2023 (available at https://www.gromacs.org/), with the CHARMM36 force field71,72. The systems were inserted into a cubic simulation water box using a simple point charge water model, and the water thickness in the solvation box was set to 10 Å. Counter ions (Cl and Na) were added by replacing water molecules at the most positive and negative electrical potentials to achieve a neutral simulation cell. Each electro-neutralized system underwent steepest-descent energy minimization to remove wrong contacts or clashes, followed by a two-step equilibration through NVT and NPT ensembles. All simulations were equilibrated at 300 K and 1 bar, using 100 ps in the NVT ensemble and 100 ps in the NPT ensemble, respectively41. Electrostatic interactions were calculated using the particle-mesh Ewald method. After the two-step equilibration, each system underwent a production MD run for 100 ns. The parameters and procedures of the MD simulation were adopted from earlier studies73. The trajectory files of each system were analyzed using the built-in modules of GROMACS. The dynamic stability of each system, including the backbone root mean squared deviation (RMSD) and radius of gyration (Rg), was analyzed through 2D plots. Protein-peptide interactions were further analyzed using gmx_MMPBSA version 1.6.2 (available at https://valdes-tresanco-ms.github.io/gmx_MMPBSA/dev/)74 and GROMACS Protein–Ligand Interactions and visualized using Ligplot + version 2.2 and YASARA version 22.5.22.
Experimental
Cell lines and culture
The MCF7 cell line, a human breast cancer cell line, was obtained from Tehran University in Tehran, Iran. MCF7 cells were cultured in DMEM (Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS) (Gibco), 100 U/mL penicillin, and 100 μg/mL streptomycin(Sigma-Aldrich) in a humidified atmosphere with 5% CO2 at 37 °C75. MCF-7 cells at passage 3 (80–90% confluence) were seeded in 96-well cell culture plates one day before transfection60.
Cell viability assay
gmx_MMPBSA analysis consistently showed that P3 exhibited stronger binding affinity than P2. Based on these comprehensive computational assessments, we prioritized P3 for synthesis and subsequent experimental validation. The anti-cancer peptide (P3) was synthesized by Shine-Gene Molecular Biotech, Inc. (Shanghai, China) with a purity of over 98%, as confirmed by HPLC, and a molecular weight of 1912 g/mol as determined by mass spectrometry. The peptide sequence includes three arginines at the N-terminus, serving as a cell-penetrating peptide (CPP) to enhance cellular uptake76,77, (Supplementary Note 1), (Supplementary Fig. 6), (Supplementary Fig. 7). To dissolve the peptide powder, a few drops of 25% acetic acid and sterile distilled water were used to achieve a concentration of 100 μM. MCF7 cells (2 × 104 cells/mL) were seeded individually in 96-well plates and incubated for 24 h until they adhered to the plate surface. The cells were incubated in a serum-free medium for 6 h. They were then treated with the anti-cancer peptide (P3) at ten concentrations: 5 μM, 10 μM, 15 μM, 20 μM, 25 μM, 30 μM, 35 μM, 40 μM, 45 μM, and 50 μM and incubated for 4 h in serum-free DMEM for transfection experiments60. Following transfection, the medium was replaced with DMEM containing 10% FBS, and the cells were incubated for an additional 24 and 48 h. The anti-proliferative effect of the anti-cancer peptide was evaluated using the MTT assay at both 24 and 48 h. Briefly, 20 µL of MTT solution (5 mg/mL) (Sigma-Aldrich) was added to each well, and the cells were incubated in the dark at 37 °C for 4 h. The formazan product was then dissolved in 100 µL of 10% sodium dodecyl sulfate (Sigma-Aldrich) containing 15 mM hydrochloric acid. Color intensity was measured using a microplate reader at a test and reference wavelength of 570 nm78.
Apoptosis flowcytometry
A suspension of MCF7 cells (5 × 105 cells/mL) was seeded into four wells of a 6-well culture plate to assess the apoptotic effect of the designed anti-cancer peptide (P3) on cancer cells. Two wells were treated with different concentrations of the anti-cancer peptide (P3) (40 µM and 25 µM), while two wells were left untreated as negative controls. The cells were incubated at 37 °C in a 5% CO2 incubator for 24 and 48 h, respectively. Following incubation, the cells were detached, centrifuged at 1500 rpm for 5 min, and washed twice with phosphate-buffered saline (PBS) (Sigma-Aldrich). The cells were then resuspended in 1X binding buffer, stained with 5 µL of Annexin V-FITC (Sigma-Aldrich) and 10 µL of propidium iodide (Sigma-Aldrich)79, and fluorescence was measured using flow cytometry80. Flow cytometric analysis was conducted using FlowJo V.10.10 software(Supplementary Method 1)81.
Cell cycle analysis
Flow cytometry was the key technique used to evaluate the DNA content during the cell cycle steps82. In brief, 5 × 105 cells/mL were treated with the anti-cancer peptide (P3) at concentrations of 40 µM and 25 µM. After 24 h (40 µM) and 48 h (25 µM), the cell pellets were washed and resuspended in 2 mL of 1% paraformaldehyde in PBS and incubated for 15 min at 4 °C. Then, the cells were centrifuged, and 1 mL of cold Perm Buffer III solution was added. The cells were incubated for 30 min at 4 °C and washed twice in PBS. Next, 500 µL of propidium iodide staining buffer (containing 50 µg/mL PI and 10 µg/mL RNase in PBS) was added and incubated for 1 h at room temperature in the dark. After staining the DNA with PI, the samples were evaluated using a flow cytometer(BD FACSCalibur, BD Biosciences, USA). Flow cytometric analysis was performed using FlowJo V.10.10 software.
Live/dead cell double staining
For the Staining, MCF-7 cells (80–90% confluence) (5 × 105 cells/mL) were seeded in 24-well plates on glass slides at 37 °C, with one plate incubated for 24 h and the other for 48 h. After incubation, the cells in the control group and the treated MCF-7 cells (24 h, 40 µM, and 48 h, 25 µM) were collected and rinsed twice with PBS. The glass slides were stained with 10 µL of the prepared AO (100 µg/mL) (Sigma-Aldrich) and PI (100 µg/mL) (Sigma-Aldrich) for 15 min in the dark at 37 °C83, followed by washing with PBS three times. The stained cells were then examined under a fluorescence microscope84,85 with an excitation wavelength of 488 nm and a magnification of × 200.
Data availability
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.
References
-
Garg, H., Suri, P., Gupta, J. C., Talwar, G. P. & Dubey, S. Survivin: A unique target for tumor therapy. Cancer Cell Int. 16, 49 (2016).
-
Jaiswal, P. K., Goel, A. & Mittal, R. D. Survivin: A molecular biomarker in cancer. Indian J. Med. Res. 141(4), 389–397 (2015).
-
Cai, X. et al. Survivin regulates the expression of VEGF-C in lymphatic metastasis of breast cancer. Diagn. Pathol. 7, 52 (2012).
-
Fang, X. L. et al. Overview of role of survivin in cancer: Expression, regulation, functions, and its potential as a therapeutic target. J. Drug Target. 32(3), 223–240 (2024).
-
Kim, P. J., Plescia, J., Clevers, H., Fearon, E. R. & Altieri, D. C. Survivin and molecular pathogenesis of colorectal cancer. Lancet 362(9379), 205–209 (2003).
-
Blum, B., Bar-Nur, O., Golan-Lev, T. & Benvenisty, N. The anti-apoptotic gene survivin contributes to teratoma formation by human embryonic stem cells. Nat. Biotechnol. 27(3), 281–287 (2009).
-
Siddharth, S., Das, S., Nayak, A. & Kundu, C. N. SURVIVIN as a marker for quiescent-breast cancer stem cells-An intermediate, adherent, pre-requisite phase of breast cancer metastasis. Clin. Exp. Metastasis. 33(7), 661–675 (2016).
-
Wang, Q. & Greene, M. I. Survivin as a therapeutic target for the treatment of human cancer. Cancers (Basel). 16(9), 1705 (2024).
-
Berthelet, J. & Dubrez, L. Regulation of apoptosis by inhibitors of apoptosis (IAPs). Cells 2(1), 163–187 (2013).
-
Zafari, P., Rafiei, A., Esmaeili, S. A., Moonesi, M. & Taghadosi, M. Survivin a pivotal antiapoptotic protein in rheumatoid arthritis. J. Cell Physiol. 234(12), 21575–21587 (2019).
-
Shomali, N. et al. Survivin: A novel therapeutic target that correlates with survival of autoreactive T lymphocytes obtained from patients with ankylosing spondylitis. Gene 844, 146829 (2022).
-
Fernández, J. G. et al. Survivin expression promotes VEGF-induced tumor angiogenesis via PI3K/Akt enhanced β-catenin/Tcf-Lef dependent transcription. Mol. Cancer 13(1), 209 (2014).
-
Verdecia, M. A. et al. Structure of the human anti-apoptotic protein survivin reveals a dimeric arrangement. Nat. Struct. Biol. 7(7), 602–608 (2000).
-
Singh, R., Letai, A. & Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 20(3), 175–193 (2019).
-
Barzegar Behrooz, A., Talaie, Z., Jusheghani, F., Łos, M. J., Klonisch, T. & Ghavami, S. Wnt and PI3K/Akt/mTOR survival pathways as therapeutic targets in glioblastoma. Int. J. Mol. Sci. 23(3), (2022).
-
Bu, H., Liu, D., Cui, J., Cai, K. & Shen, F. Wnt/β-catenin signaling pathway is involved in induction of apoptosis by oridonin in colon cancer COLO205 cells. Transl. Cancer Res. 8(5), 1782–1794 (2019).
-
Fang, W. et al. Sur-X, a novel peptide, kills colorectal cancer cells by targeting survivin-XIAP complex. J. Exp. Clin. Cancer Res. 39(1), 82 (2020).
-
de Souza, T. L. et al. Conformational selection, dynamic restriction and the hydrophobic effect coupled to stabilization of the BIR3 domain of the human X-linked inhibitor of apoptosis protein by the tetrapeptide AVPI. Biophys. Chem. 152(1–3), 99–108 (2010).
-
Cheung, C. H. A., Chang, Y. C., Lin, T. Y., Cheng, S. M. & Leung, E. Anti-apoptotic proteins in the autophagic world: An update on functions of XIAP, Survivin, and BRUCE. J. Biomed. Sci. 27(1), 31 (2020).
-
Sun, H. et al. Design of small-molecule peptidic and nonpeptidic Smac mimetics. Acc. Chem. Res. 41(10), 1264–1277 (2008).
-
Carneiro, B. A. & El-Deiry, W. S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 17(7), 395–417 (2020).
-
Wheatley, S. P. & Altieri, D. C. Survivin at a glance. J. Cell Sci. 132(7), jcs223826 (2019).
-
Dallaglio, K., Marconi, A. & Pincelli, C. Survivin: A dual player in healthy and diseased skin. J. Invest. Dermatol. 132(1), 18–27 (2012).
-
Wang, Q. & Greene, M. I. Survivin as a therapeutic target for the treatment of human cancer. Cancers 16(9), 1705 (2024).
-
Espinosa, M. et al. Survivin isoform Delta Ex3 regulates tumor spheroid formation. Cancer Lett. 318(1), 61–67 (2012).
-
Meiners, A. et al. Specific inhibition of the Survivin–CRM1 interaction by peptide-modified molecular tweezers. Nat. Commun. 12(1), 1505 (2021).
-
Kondapuram, S. K., Ramachandran, H. K., Arya, H. & Coumar, M. S. Targeting survivin for cancer therapy: Strategies, small molecule inhibitors and vaccine based therapeutics in development. Life Sci. 335, 122260 (2023).
-
Wang, H. et al. Acetylation directs survivin nuclear localization to repress STAT3 oncogenic activity. J. Biol. Chem. 285(46), 36129–36137 (2010).
-
Pavlyukov, M. S. et al. Survivin monomer plays an essential role in apoptosis regulation. J. Biol. Chem. 286(26), 23296–23307 (2011).
-
Hadders, M. A. & Lens, S. M. A. Changing places: Chromosomal Passenger Complex relocation in early anaphase. Trends Cell Biol. 32(2), 165–176 (2022).
-
Carmena, M., Wheelock, M., Funabiki, H. & Earnshaw, W. C. The chromosomal passenger complex (CPC): From easy rider to the godfather of mitosis. Nat. Rev. Mol. Cell Biol. 13(12), 789–803 (2012).
-
Jeyaprakash, A. A. et al. Structure of a Survivin-Borealin-INCENP core complex reveals how chromosomal passengers travel together. Cell 131(2), 271–285 (2007).
-
Xu, Z. et al. INCENP-aurora B interactions modulate kinase activity and chromosome passenger complex localization. J. Cell Biol. 187(5), 637–653 (2009).
-
Garlapati, C. et al. PLK1 and AURKB phosphorylate survivin differentially to affect proliferation in racially distinct triple-negative breast cancer. Cell Death Dis. 14(1), 12 (2023).
-
Chu, Y. et al. Aurora B kinase activation requires survivin priming phosphorylation by PLK1. J. Mol. Cell Biol. 3(4), 260–267 (2011).
-
Colnaghi, R. & Wheatley, S. P. Liaisons between survivin and Plk1 during cell division and cell death. J. Biol. Chem. 285(29), 22592–22604 (2010).
-
Wheatley, S. P. & McNeish. I. A. Survivin: A protein with dual roles in mitosis and apoptosis. international review of cytology. Vo. 247, pp. 35–88, (Academic Press; 2005).
-
Nozaki, I. et al. Borealin-derived peptides as survivin-targeting cancer imaging and therapeutic agents. Bioconjug. Chem. 33(11), 2149–2160 (2022).
-
Li, C. M. et al. Novel peptide therapeutic approaches for cancer treatment. Cells 10(11), 2908 (2021).
-
Aier, I., Varadwaj, P. K. & Raj, U. Structural insights into conformational stability of both wild-type and mutant EZH2 receptor. Sci. Rep. 6, 34984 (2016).
-
Ghavamipour, F. et al. Development of highly potent anti-angiogenic VEGF8–109 heterodimer by directed blocking of its VEGFR-2 binding site. FEBS J. 281, 4479–4494 (2014).
-
Yin, H. et al. Design, synthesis and anticancer evaluation of novel oncolytic peptide-chlorambucil conjugates. Bioorg. Chem. 138, 106674 (2023).
-
Bendel, A. M. et al. The genetic architecture of protein interaction affinity and specificity. Nat. Commun. 15(1), 8868 (2024).
-
Zeng, M. et al. Protein–protein interaction site prediction through combining local and global features with deep neural networks. Bioinformatics 36(4), 1114–1120 (2020).
-
Scott, D. E., Bayly, A. R., Abell, C. & Skidmore, J. Small molecules, big targets: Drug discovery faces the protein-protein interaction challenge. Nat. Rev. Drug. Discov. 15(8), 533–550 (2016).
-
Mita, A. C., Mita, M. M., Nawrocki, S. T. & Giles, F. J. Survivin: Key regulator of mitosis and apoptosis and novel target for cancer therapeutics. Clin. Cancer Res. 14(16), 5000–5005 (2008).
-
Vallet, C. et al. Functional disruption of the cancer-relevant interaction between survivin and histone H3 with a guanidiniocarbonyl pyrrole ligand. Angew Chem. Int. Ed. Engl. 59(14), 5567–5571 (2020).
-
Wang, L. et al. Therapeutic peptides: Current applications and future directions. Signal Transduct. Target. Ther. 7(1), 48 (2022).
-
Fu, X.-Y. et al. Three rounds of stability-guided optimization and systematical evaluation of oncolytic peptide LTX-315. J. Med. Chem. 67(5), 3885–3908 (2024).
-
Yin, H. et al. The hybrid oncolytic peptide NTP-385 potently inhibits adherent cancer cells by targeting the nucleus. Acta Pharmacol. Sin. 44, 201 (2023).
-
Rossino, G. et al. Peptides as therapeutic agents: Challenges and opportunities in the green transition era. Molecules 28(20), 7165 (2023).
-
Mougel, A. et al. Synergistic effect of combining sunitinib with a peptide-based vaccine in cancer treatment after microenvironment remodeling. Oncoimmunology 11(1), 2110218 (2022).
-
Schmidt, N., Mishra, A., Lai, G. H. & Wong, G. C. Arginine-rich cell-penetrating peptides. FEBS Lett. 584(9), 1806–1813 (2010).
-
Lunde, P. K. et al. Polyarginine cell-penetrating peptides bind and inhibit SERCA2. Cells 12(19), 2358 (2023).
-
Madani, F., Lindberg, S., Langel, U., Futaki, S. & Gräslund, A. Mechanisms of cellular uptake of cell-penetrating peptides. J. Biophys. 2011, 414729 (2011).
-
Majidi, A., Nikkhah, M., Sadeghian, F. & Hosseinkhani, S. Development of novel recombinant biomimetic chimeric MPG-based peptide as nanocarriers for gene delivery: Imitation of a real cargo. Eur. J. Pharm. Biopharm. 107, 191–204 (2016).
-
He, Y. et al. The changing 50% inhibitory concentration (IC50) of cisplatin: A pilot study on the artifacts of the MTT assay and the precise measurement of density-dependent chemoresistance in ovarian cancer. Oncotarget 7(43), 70803–70821 (2016).
-
Zahraee, H., Arab, S. S., Khoshbin, Z., Taghdisi, S. M. & Abnous, K. Molecular dynamics simulation as a promising approach for computational study of liquid crystal-based aptasensors. J. Biomol. Struct. Dyn., 2024:1–13.
-
Vadevoo, S. M. P. et al. Peptides as multifunctional players in cancer therapy. Exp. Mol. Med. 55(6), 1099–1109 (2023).
-
Samadikhah, H. R., Majidi, A., Nikkhah, M. & Hosseinkhani, S. Preparation, characterization, and efficient transfection of cationic liposomes and nanomagnetic cationic liposomes. Int. J. Nanomed. 6, 2275–2283 (2011).
-
Pande, S. Liposomes for drug delivery: Review of vesicular composition, factors affecting drug release and drug loading in liposomes. Artificial Cells Nanomed. Biotechnol. 51(1), 428–440 (2023).
-
Cui, J., Wen, Z., Zhang, W. & Wu, W. Recent advances in oral peptide or protein-based drug liposomes. Pharmaceuticals 15(9), 1072 (2022).
-
Naeem, A., Noureen, N., Al-Naemi, S. K., Al-Emadi, J. A. & Khan, M. J. Computational design of anti-cancer peptides tailored to target specific tumor markers. BMC Chem. 18(1), 39 (2024).
-
Bai, L., Smith, D. C. & Wang, S. Small-molecule SMAC mimetics as new cancer therapeutics. Pharmacol. Ther. 144(1), 82–95 (2014).
-
Nakahara, T. et al. YM155, a novel small-molecule survivin suppressant, induces regression of established human hormone-refractory prostate tumor xenografts. Cancer Res. 67(17), 8014–8021 (2007).
-
Laskowski, R. A. & Swindells, M. B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 51(10), 2778–2786 (2011).
-
Buß, O., Rudat, J. & Ochsenreither, K. FoldX as protein engineering tool: Better than random based approaches?. Comput. Struct. Biotechnol. J. 16, 25–33 (2018).
-
Desta, I. T. et al. The ClusPro AbEMap web server for the prediction of antibody epitopes. Nat. Protoc. 18(6), 1814–1840 (2023).
-
Alekseenko, A., Ignatov, M., Jones, G., Sabitova, M. & Kozakov, D. Protein-protein and protein-peptide docking with ClusPro server. Methods Mol. Biol. 2165, 157–174 (2020).
-
Porter, K. A., Desta, I., Kozakov, D. & Vajda, S. What method to use for protein-protein docking?. Curr. Opin. Struct. Biol. 55, 1–7 (2019).
-
Huang, J. et al. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 14(1), 71–73 (2017).
-
Kumar, V. & Yaduvanshi, S. Protein-protein interaction studies using molecular dynamics simulation. Methods Mol. Biol. 2652, 269–283 (2023).
-
Hollingsworth, S. A. & Dror, R. O. Molecular dynamics simulation for all. Neuron 99(6), 1129–1143 (2018).
-
Valdés-Tresanco, M., Valdés-Tresanco, M., Valiente, P. & Moreno, F. E. gmx_MMPBSA: A new tool to perform end-state free energy calculations with GROMACS. J. Chem. Theory Comput. 17(10), 6281–6291 (2021).
-
Muthusamy, T. et al. Serine restriction alters sphingolipid diversity to constrain tumour growth. Nature 586(7831), 790–795 (2020).
-
Saw, P. E. & Song, E. W. Phage display screening of therapeutic peptide for cancer targeting and therapy. Protein Cell. 10(11), 787–807 (2019).
-
Zhou, M. et al. The role of cell-penetrating peptides in potential anti-cancer therapy. Clin. Transl. Med. 12(5), e822 (2022).
-
Razak, N. A. et al. Cytotoxicity of eupatorin in MCF-7 and MDA-MB-231 human breast cancer cells via cell cycle arrest, anti-angiogenesis and induction of apoptosis. Sci. Rep. 9(1), 1514 (2019).
-
Fard, S. S., Jeddi-Tehrani, M., Akhondi, M. M., Hashemi, M. & Ardekani, A. M. Flow cytometric analysis of 4-HPR-induced apoptosis and cell cycle arrest in acute myelocytic leukemia cell line (NB-4). Avicenna J. Med. Biotechnol. 2(1), 53–61 (2010).
-
Abdullah, H., Ismail, I., Suppian, R. & Zakaria, N. M. Natural gallic acid and methyl gallate induces apoptosis in hela cells through regulation of intrinsic and extrinsic protein expression. Int. J. Mol. Sci. 24(10), 8495 (2023).
-
Qian, J. et al. Development of therapeutic monoclonal antibodies against DKK1 peptide-HLA-A2 complex to treat human cancers. J. Immunother. Cancer 12(1), e008145 (2024).
-
Pozarowski, P. & Darzynkiewicz, Z. Analysis of cell cycle by flow cytometry. Methods Mol. Biol. 281, 301–311 (2004).
-
Liu, H., Dai, L., Wang, M., Feng, F. & Xiao, Y. Tunicamycin induces hepatic stellate cell apoptosis through calpain-2/Ca2+-dependent endoplasmic reticulum stress pathway. Front. Cell Dev. Biol. 9, 684857 (2021).
-
Chatnarin, S. & Thirabunyanon, M. Potential bioactivities via anticancer, antioxidant, and immunomodulatory properties of cultured mycelial enriched β-D-glucan polysaccharides from a novel fungus Ophiocordyceps sinensis OS8. Front. Immunol. 14, 1150287 (2023).
-
Khan, H. et al. Structure based docking and biological evaluation towards exploring potential anti-cancerous and apoptotic activity of 6-Gingerol against human prostate carcinoma cells. BMC Complement. Med. Therapies 24(1), 8 (2024).
Acknowledgements
The authors are grateful for the support of the Central Tehran Branch, Islamic Azad University, Tehran, Iran, for this work.
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ahmadi, S.F., Arab, S.S. & Samadikhah, H. Computational and experimental discovery of peptide inhibitors targeting survivin for therapeutic potential in cancer. Sci Rep 15, 30599 (2025). https://doi.org/10.1038/s41598-025-13110-3
-
Received:
-
Accepted:
-
Published:
-
DOI: https://doi.org/10.1038/s41598-025-13110-3