Introduction
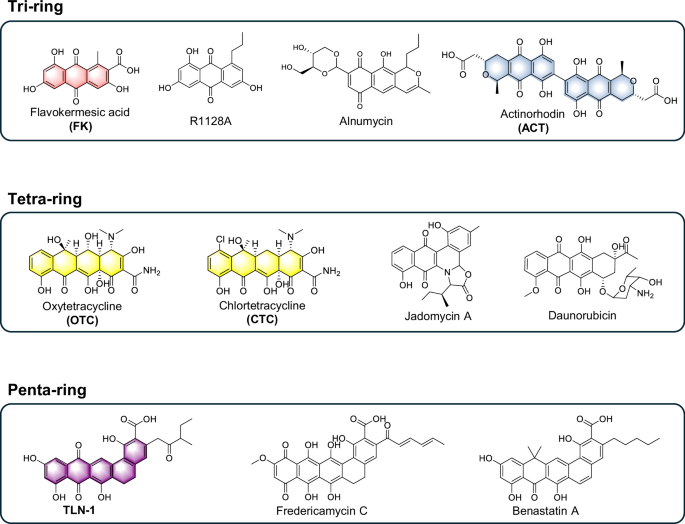
Type II polyketides (T2PKs) constitute a class of aromatic compounds distinguished by the presence of benzene rings1,2. These compounds exhibit a diverse molecular backbone, which includes three-ring naphthoquinones, four-ring tetracyclines, and angucyclines, as well as five-ring pentangular polyketides (Fig. 1). The polyconjugated structure formed by the interaction of the benzene ring with polar functional groups imparts a variety of colors to these compounds. Beyond their aesthetic appeal, these pigmented compounds fulfill essential roles as food additives, and in cosmetic formulations and pharmaceutical tablet coatings, among various other applications, exemplified by the natural red colorant carminic acid3,4. The diverse chemical groups of T2PKs also contribute to a wide array of pharmacological activities. Notable examples include daunorubicin, which demonstrates antitumor properties, and tetracyclines (TCs), which exhibit a broad spectrum of antibacterial activity5,6. To discover novel structural compounds and valuable products, the conventional strategy involves medium optimization or modification of culture conditions, such as the one-strain many-compounds (OSMAC) approach7. However, these strategies are often inefficient, challenging, and incapable of diverse T2PKs mining.

The products related to this study are highlighted in their respective colors.
Recent advancements in bioinformatics analysis and gene cloning technologies have facilitated the discovery of an increasing number of novel T2PKs8,9,10. However, a significant proportion of promising candidates remain undiscovered due to the inefficiencies associated with in situ fermentation and gene manipulation techniques8,9,10. Furthermore, to enhance the titer of valuable products, multiple rounds of metabolic engineering must be conducted, which is both time-consuming and labor-intensive.
In light of the challenges previously outlined, our objective is to formulate a universal strategy for the efficient production of T2PKs via heterologous expression. Therefore, a critical and foundational decision must be made at the outset of our research: the identification of an optimal host. Based on our prior experiences with terpene products mining in various host organisms, the efficient production of target compounds is closely associated with two key characteristics: the availability of precursors within the host and the compatibility between the product and the chassis. For instance, Bian et al. successfully synthesized 50 terpenoids within a single chassis by enhancing the supply of precursors, thereby rendering products that would otherwise be unattainable under natural conditions readily accessible11. Yuan et al. obtained 185 distinct terpenoids derived from filamentous fungi within the same species chassis, underscoring the importance of product-to-chassis compatibility12.
An optimal host can minimize both time and labor costs, thereby facilitating the efficient synthesis of the target products. Escherichia coli is recognized as the most widely utilized heterologous expression host due to its easy gene manipulation procedure and rapid growth rate. However, despite possessing some advantageous traits, previous studies indicated that the synthesis of the 6-deoxyerythronolide B (the macrocyclic core of the type I polyketides erythromycin) can be achieved using E. coli as host13, it encounters challenges regarding the soluble expression of the minimal polyketide synthase (PKS, core enzymes of T2PKs) in vivo14,15,16,17,18. To date, few reported cases have successfully addressed this issue within E. coli14,19,20. Additionally, other commonly employed eukaryotic hosts, such as Saccharomyces cerevisiae (S. cerevisiae) and Aspergillus nidulans, have also been reported to synthesize T2PKs. However, the substitution of PKS with OKS, a type III PKS, appears to be the sole approach for the synthesis of T2PKs in eukaryotic hosts, addressing only the octaketide molecular backbone, which represents a specific type of T2PKs21,22. Furthermore, the overall efficiency of the refactoring pathway is suboptimal, which necessitates an extension of the fermentation cycle, particularly in yeast23. Consequently, none of the previously mentioned organisms meet the criteria for an ideal host for T2PKs production.
It is well established that the genus Streptomyces serves as an ideal chassis for the heterologous expression of natural products owing to its extensive secondary metabolic biosynthetic gene clusters (BGC)24. It has successfully facilitated the heterologous production of diverse natural products, including ribosomal peptides, polyketides, and nonribosomal peptides25,26. Approximately 62% of the annotated T2PKs BGC available in the NCBI database originate from Streptomyces, suggesting that T2PKs may exhibit optimal chassis compatibility with it9,27. Although the genetic manipulation of the model Streptomyces chassis (S. albus, S. coelicolor, and S. lividans) is relatively simple, and they have been utilized as hosts for the synthesis of aromatic polyketide products, the efficiency of heterologous expression is unsatisfactory, typically ranging from 0.2 mg/L to 127 mg/L28,29,30. To activate or enhance the production of target products, multiple rounds of metabolic engineering must be conducted. Nevertheless, the final heterologous expression efficiency remains suboptimal31,32,33.
More recently, Duan et al. successfully knocked out the native gene cluster in high-yielding strains of tylosin, thereby facilitating the heterologous expression of a type I polyketide34. Additionally, Wang et al. employed an oxytetracycline (OTC) industrial strain S. rimosus 461 as a chassis to achieve a chlortetracycline (CTC) titer of 3.8 g/L35. These findings suggest that industrial high-yield strains exhibit enhanced potential as chassis for heterologous production of homologous natural products.
To find a versatile chassis for the efficient production of T2PKs, several streptomyces strains are compared, and the industrial CTC producer S. aureofaciens J1-022 is selected as the host. We evaluate the near-native compound OTC, characterized by a tetra-ring backbone, which is synthesized efficiently without the need for additional metabolic engineering. Subsequently, we successfully synthesized three-ring type T2PKs, specifically ACT and flavokermesic acid (FK), both of which demonstrate high production efficiency. Furthermore, we directly activate an unidentified BGC associated with penta-ring type aromatic polyketides, resulting in the discovery of a structurally distinct TLN-1 at high production levels.
Results
Selection of the optimal host for heterologous expression of T2PKs
Through a comparative analysis of the functional characteristics across distinct organism hosts, we prioritized Streptomyces hosts as the initial focus for chassis development due to their rich abundance of secondary metabolic gene clusters and the widespread distribution of T2PKs9,24. Previous studies have demonstrated that various T2PKs can be successfully synthesized in model strains of Streptomyces28,29. Therefore, two widely utilized Streptomyces model chassis S. albus J1074 and S. lividans TK24 were tested for the heterologous expression of T2PKs. Initially, the tetra-ring type T2PKs, OTC, was selected as the evaluation candidate, which also serves as an important precursor of doxycycline36,37. To obtain the OTC BGC for heterologous expression, we utilized genomic DNA from S. rimosus ATCC 10970 as the template, while ensuring BGC cloning integrity through alignment with previously validated heterologous expression work31 (Fig. 2a). The ExoCET technology was employed to construct the E. coli–Streptomyces shuttle plasmid p15A_oxy, which contains the complete OTC BGC38. The successful introduction of complete OTC BGC into these model chassis was confirmed through PCR verification and the resulting heterologous expression strains were designated as J1074_otc and TK24_otc (Supplementary Fig. 1), respectively.

a The genetic architecture of the complete OTC BGC in S. rimosus ATCC10970. b The high-performance liquid chromatography (HPLC) results of the OTC heterologous expression strains. OTC, TC, and CTC standards serve as positive controls. The J1-022 serves as control. c Comparative analysis of colony phenotypes between S. aureofaciens and S. rimosus. d Relative acetyl-CoA accumulation levels per unit cell dry weight (CDW) in three strains across distinct growth phases. e Relative malonyl-CoA accumulation levels per CDW in three strains across distinct growth phases. f Relative NADP+ levels per unit CDW in three strains across distinct growth phases. g Relative NADPH levels per unit CDW in three strains across distinct growth phases. Bar figures show mean values with error bars indicating s.d. (standard deviations, n = 3 biological replicates). Statistical significance was denoted as follows: ns (not significant), *P < 0.05, **P < 0.001 (two-tailed unpaired t-test, uncorrected for multiple comparisons). Source data for this figure can be found in the source data file.
However, neither J1074_otc nor TK24_otc demonstrated any accumulation of TCs (Fig. 2b). This finding is consistent with prior research that suggests the model Streptomyces chassis typically requires metabolic engineering strategies such as endogenous gene cluster knockout, substrate supply enhancement, or positive regulator overexpression to enable efficient activation and biosynthesis of target products31,32,39.
Based on the principle of product-chassis compatibility, we consequently refined our focus on two high-yielding strains of T2PKs: S. aureofaciens, a high-yield CTC producer, and S. rimosus, recognized for its overproduction of OTC40,41. To determine the optimal chassis, we conduct a thorough comparison of the two finalists. From the standpoint of chassis development (Table 1), S. rimosus presents three significant limitations. Firstly, colony morphology assessment prioritized gene manipulation efficiency during subculturing, particularly evaluating single-colony isolation capacity and genetic tractability. While high productivity does not inherently correlate with specific morphological traits, the industrial OTC producer S. rimosus represents an exception42,43,44. Compared to S. aureofaciens, S. rimosus exhibits suboptimal colony morphology, as subcultures frequently demonstrate poor sporulation or a shriveled phenotype (Fig. 2c). This correlation suggests a certain degree of genetic instability within the organism42,43,44. Secondly, the fermentation cycle of S. rimosus is approximately twice as long as that of S. aureofaciens. The shorter fermentation cycle results in reduced contamination risk and accelerated validation of strain engineering outcomes. Finally, the genetic manipulation of high-yielding OTC strains presents considerable challenges. Based on our observations, the acquisition of the correct gene editing strain will be considerably more challenging in S. rimosus compared to S. aureofaciens, thereby complicating the gene disruption process. Furthermore, S. rimosus demonstrates a notable tolerance to apramycin, a widely utilized agent for genetic manipulation in the Streptomyces, with an effective concentration of approximately 500 μg/mL—tenfold that of S. aureofaciens35. Such elevated screening concentrations may induce a tolerance effect in the strains, potentially resulting in false-positive screening results. In conclusion, following a preliminary comparison of strain characteristic, the high-yielding strain S. aureofaciens J1-022 demonstrates enhanced potential as an ideal chassis with the potential for efficient synthesis of T2PKs.
To validate our hypothesis and elucidate the metabolic superiority of S. aureofaciens J1-022 over the conventional model Streptomyces chassis, we performed targeted metabolites analysis using high-resolution mass spectrometry (HRMS) focusing on critical T2PKs biosynthetic precursors, including acetyl-CoA, NADP+, NADPH, ATP, and malonyl-CoA. While successfully quantifying acetyl-CoA, NADP+, and NADPH, initial HRMS analysis showed limited sensitivity for malonyl-CoA detection in these chassis (Supplementary Fig. 2). Subsequent quantification using a ELISA kit measured malonyl-CoA concentrations successfully. Results revealed undetectable levels of acetyl-CoA in model Streptomyces chassis strains TK24 and J1074, while J1-022 demonstrated a discernible advantage in acetyl-CoA supply throughout both logarithmic and stationary growth phases. (Fig. 2d). Furthermore, J1-022 exhibited enhanced malonyl-CoA flux relative to TK24 and J1074. Specifically, 16% and 21% increases during log-phase growth, and 20% and 18% increases in stationary phase, respectively (Fig. 2e). Concerning reducing equivalents supply during log-phase growth, J1-022 showed significant NADP⁺ and NADPH accumulation, while model Streptomyces chassis strains exhibited undetectable levels of these cofactors. In the stationary phase, J1-022 demonstrated 72% higher NADP⁺ and a 1.4-fold greater NADPH pool compared to J1074. Meanwhile, TK24 displayed negligible reducing equivalents accumulation (Fig. 2f, g). The results revealed that industrial CTC-producing strain J1-022 exhibited significant advantages in supplying critical biosynthetic precursors and reducing equivalents.
Furthermore, to enable direct comparison with model Streptomyces chassis strains, we introduced the p15A_oxy plasmid into the S. aureofaciens J1-022 strain, generating the strain J1-022_otc (Supplementary Fig. 1). We hypothesized that model Streptomyces chassis strains might share the conserved challenge of inefficient expression of minimal PKS – a phenomenon well-documented in E. coli systems16,18. Therefore, we applied quantitative protein analysis based on ion intensities of tryptic peptides derived from OTC minimal PKS to determine relative levels in OTC heterologous expression strains. Our data revealed striking differences that the tryptic peptides corresponding to all four core proteins in OTC minimal PKS demonstrated comparable ion intensities in J1-022_otc, whereas related peptide signals remained undetectable in model Streptomyces chassis strains (Supplementary Fig. 3), which indicates that J1-022 exhibits advantages in expressing minimal T2PKSs derived from OTC BGC. HPLC analysis of the J1-022_otc fermentation broth achieving a shake-flask titer of 162.95 mg/L OTC, 2252.72 mg/L tetracycline (TC), and 5259.28 mg/L CTC (Supplementary Fig. 4), the results demonstrated successful direct activation of the OTC BGC.
Construction of the chassis strain via the deletion of the T2PKS BGC in S. aureofaciens J1-022
To enhance the efficiency of heterologous expression, it is common practice to knock out gene clusters of the same type within the host34,35. This approach effectively mitigates competition between the endogenous gene clusters and the exogenous gene clusters for synthetic precursors. The analysis conducted using antiSMASH (version 6.0.1) identified a total of 25 BGCs related to specialized metabolism in S. aureofaciens J1-022 (Supplementary Table 1)45. Among these, only two were classified as encoding T2PKs, one of these two clusters is involved in the biosynthesis of CTC, while the other is associated with spore pigment production (Supplementary Table 2)46.
Given that polyketides biosynthesis universally employs malonyl-CoA as an elongation unit, all corresponding gene clusters represent theoretically valid knockout targets. RT-PCR analysis of ketosynthase (KS) genes within polyketides BGC revealed exclusively observed transcription in the CTC-associated KS gene, whereas the sporulation pigment-related KS gene showed negligible transcription. No detectable expression was observed in other polyketides gene clusters (Supplementary Fig. 5).
Initially, our objective was to evaluate the compatibility of the OTC with the S. aureofaciens chassis. To achieve this, we knocked out the minimal PKS of the CTC BGC (Fig. 3a), resulting in the development of Chassis1.0. HPLC analysis confirmed that the TCs production in the mutant strain was abolished, as anticipated (Fig. 3b). Subsequently, we employed a similar knockout strategy on the spore pigment BGC to create Chassis1.1 (Fig. 3a, c). Although no significant differential chromatographic peaks were observed in the metabolite detection, a notable alteration in the colony phenotype occurred, shifting from dark brown to light green (Fig. 3b).

a The genetic architecture of the complete CTC BGC in S. aureofaciens J1-022. b The results of the HPLC comparing the Chassis1.0 and Chassis1.1. J1-022 serves as control; CTC standard serves as positive control. The colony phenotypes observed on the culture plate of mutants were presented. c S. aureofaciens chassis were constructed by executing iterative in-frame knockout. d Relative acetyl-CoA accumulation levels per unit CDW in J1-022 and three variant chassis across distinct growth phases. e Relative malonyl-CoA accumulation levels per CDW in J1-022 and three variant chassis across distinct growth phases. f Relative NADP+ levels per unit CDW in J1-022 and three variant chassis across distinct growth phases. g Relative NADPH levels per unit CDW in J1-022 and three variant chassis across distinct growth phases. Bar figures show mean values with error bars indicating s.d. (n = 3 biological replicates). Statistical significance was denoted as follows: ns (not significant), *P < 0.05, **P < 0.001 (two-tailed unpaired t-test, uncorrected for multiple comparisons). Source data for this figure can be found in the source data file.
Given that most T2PKs are pigmented, the lighter color phenotype may serve as a more effective indicator of product synthesis. To fully demonstrate the capacity of T2PKs synthesis in S. aureofaciens, we deleted the rest of CTC biosynthetic genes in Chassis1.1 to construct Chassis2.0 (Fig. 3c). This modified chassis was subsequently utilized to facilitate the specific synthesis of OTC.
To enable comprehensive comparison of chassis variants during endogenous gene cluster knockout, we employed HRMS for targeted monitoring of key T2PKs precursors and reducing equivalents. Compared to strains J1-022, chassis strains exhibited significant elevation in intracellular acetyl-CoA and malonyl-CoA pools (Fig. 3d, e). Overall NADP+ and NADPH levels in chassis strains decreased during logarithmic growth but showed marked accumulation advantages during stationary phase in Chassis2.0 (Fig. 3f, g). We also systematically analyzed the accumulation levels of ATP and S-adenosylmethionine (SAM) in chassis strains, which play crucial roles in multiple biochemical and T2PKs post-modification processes. Following endogenous T2PKs gene clusters inactivated, ATP concentrations exhibited a significant increase, while SAM, another critical cofactor in OTC post-modification process, showed no statistically significant elevation (Supplementary Fig. 6).
Collectively, Chassis2.0 demonstrates enhanced precursor supply capacity and reduced cross-talk potential with heterologous T2PKs BGCs. These benefits enable focused production of target compounds in Chassis2.0, establishing this chassis as the preferred platform strain for subsequent studies.
Tetra-ring type T2PKs were efficiently synthesized in Chassis2.0
To assess the capacity for T2PKs synthesis in S. aureofaciens strains, the plasmids p15A_oxy was introduced into Chassis1.1 and Chassis2.0, respectively, resulting in the creation of the OTC heterologous expression strains Chassis1.1_otc and Chassis2.0_otc. To verify the integrity of the transferred OTC BGC, we designed three pairs of primers for PCR verification (Supplementary Fig. 1). Notably, the synthesis of OTC was activated in both chassis without additional chassis and metabolic engineering (Fig. 4a).

a HPLC results of metabolite accumulation in different OTC heterologous expression chassis. OTC, TC and CTC standards serve as positive controls. b Quantification of the OTC titer in different OTC heterologous expression chassis. c The depiction of the downstream biosynthetic pathways for OTC and CTC in Chassis2.0_otc. d Comparison of minimal PKS expression levels per unit CDW between J1-22_otc and the variant chassis heterologously expressing OTC BGC across distinct growth phases. Bar figures show mean values with error bars indicating s.d. (n = 3 biological replicates). Statistical significance was denoted as follows: ns (not significant), *P < 0.05, **P < 0.001, ****P < 0.0001 (two-tailed unpaired t-test, uncorrected for multiple comparisons). Source data for this figure can be found in the source data file.
In Chassis1.1_otc, the minimal PKS of OTC probably engaged in cross-talk with the remaining post-modification genes of CTC cluster as anticipated, resulting in the co-production of various TCs (Fig. 4b, c). The resulting titer of OTC reached 436 mg/L, which is comparable to the levels observed in heterologous expression strains of OTC following multiple rounds of metabolic engineering (Fig. 4b)31. This was accompanied by the accumulation of 2092.89 mg/L TC and 4696.78 mg/L CTC.
In Chassis2.0_otc, the endogenous CTC BGC was completely deleted, resulting in OTC becoming the primary product, with a titer of 8531.4 mg/L, to the best of our knowledge, which is the highest titer of heterologous expression reported to date, being 120 times greater than that of the host strain ATCC10970 from which the gene cluster was derived40, and a 370% increase relative to commercial OTC production strain M4018, even surpasses the production strains after metabolic engineering (Fig. 4b)47. The productivity achieved a level at 70.8 mg/L/h. This productivity is the highest documented to date, exceeding that of the industrial OTC strain referenced in the literature35,47.
Furthermore, we performed quantitative protein analysis across successful OTC heterologous expression strains based on ion intensities of tryptic peptides derived from OTC minimal PKS. The data demonstrated enhanced expression efficiency of the OTC minimal PKS in Chassis2.0, directly correlating with its superior OTC biosynthetic capacity (Fig. 4d).
The superiority of heterologous expression of the T2PKs biosynthetic pathway in Chassis 2.0
In the elucidation of the T2PKs biosynthesis pathway, S. aureofaciens Chassis2.0 also exhibits distinct advantages over other hosts. The downstream biosynthetic gene OxyS is limited to hydroxylating at the C6 position when utilizing anhydrotetracycline (ATC) as a precursor in S. cerevisiae (Fig. 5)48, a finding that contradicts previous in vitro and in vivo results.

In vitro experiments have demonstrated that OxyS can sequentially complete the dihydroxylation process at the C6 and C5 positions. This result was corroborated in vivo in this study, wherein the Chassis2.0_otc strain not only achieved high-efficiency OTC synthesis but also reduced monohydroxylated intermediates to yield TC (Figs. 4b, 5). This finding further substantiates the notion that the adaptability of the chassis is instrumental not only in achieving a high titer of the target product but also in providing a more authentic in vivo environment for the heterologous reconstituted biosynthesis pathway, thereby restoring the natural process of product synthesis.
Moreover, Chassis2.0 demonstrates a more robust capacity for product synthesis compared to other model Streptomyces, with side product TC accumulating at concentrations reaching 348.28 mg/L (Fig. 4b). The high biosynthetic capacity of the Chassis2.0 facilitates the rapid identification of intermediate product structures and simplifies the elucidation of the biosynthesis process.
Versatile applications in the synthesis of tri-ring T2PKs within Chassis2.0
Following the efficient production of TCs, we proceeded to conduct a thorough evaluation of the T2PKs synthesis capacity of the S. aureofaciens chassis. The tri-ring type T2PKs, which exhibit an aesthetically pleasing color, were selected as the subsequent test candidate. Initially, we selected the BGC of ACT, a naphthoquinone-type compound that is well-characterized as a pigment. We constructed the plasmid p15A-act via ExoCET technology. This plasmid harbors the complete native ACT BGC, which was successfully heterologously expressed49 (Fig. 6a). This plasmid was subsequently introduced into Chassis2.0 via conjugation. The resulting exconjugants, designated Chassis2.0_ACT, were confirmed through PCR verification utilizing three pairs of primers (Supplementary Fig. 7). Consequently, the plate culture of Chassis2.0_ACT exhibited a darker gray-blue coloration compared to Chassis2.0 on ISP2 (Fig. 6b). Following the extraction of the fermentation culture, the crude extract demonstrated a significant difference between the samples treated with acid and those treated with base, displaying a red color in an acidic environment and a blue color in an alkaline environment (Fig. 6c). These results were in strong agreement with prior research and indicated a successful activation of the ACT BGC50. To visually demonstrate the activation capability of Chassis2.0 for actinorhodin biosynthesis, we conducted parallel fermentations of Chassis2.0_ACT and S. coelicolor A3(2). HRMS analysis of the fermentation products detected both actinorhodin and its derivative γ-actinorhodin (Supplementary Figs. 8, 9)50. The biosynthesis levels of both lactone γ-actinorhodin and actinorhodin were significantly higher than those observed in the native producer (Fig. 6d).

a The genetic architecture of the complete ACT BGC in S. coelicolor A3 (2). b Different coloration of phenotype on agar plates. c The coloration of crude extract in different pH environments. d The HRMS-based per-unit CDW quantification result of ACT and its derivatives in Chassis2.0_ACT versus S. coelicolor A3(2). e The color comparison of crude extract and the Q-TOF LC/MS analysis of primary accumulated products in Chassis2.0_FK. f The quantification result of FK in Chassis2.0_FK. The Chassis2.0 served as the blank control check. Bar figures show mean values with error bars indicating s.d. (n = 3 biological replicates). Statistical significance was denoted as follows: ns (not significant), *P < 0.05, **P < 0.001 (two-tailed unpaired t-test, uncorrected for multiple comparisons). Source data for this figure can be found in the source data file.
The initial successful activation of ACT prompted us to investigate another naphthoquinone-type compound, FK, which serves as a significant intermediate in the synthesis of the natural red colorant carminic acid. It has been synthesized in various host organisms, facilitating a more comprehensive comparison of the Chassis2.0 with other hosts21,23,51,52. We selected the minimal PKS of ACT, along with the cyclases ZhuI and ZhuJ derived from Streptomyces sp. R1128, which results in an artificial pathway for the biosynthesis of FK53. The native operon architecture of the ACT-derived minimal PKS was preserved to ensure the efficient expression14. This structural fidelity is critical for efficient reconstruction of T2PKs, particularly regarding minimal PKS units assembly14,19. Cyclases ZhuI and ZhuJ were synthesized de novo due to the unavailability of Streptomyces sp. R1128. To ensure that the genes across the three-strain system (two gene donor strains, S. coelicolor A3(2), Streptomyces sp. R1128, and one chassis strain, S. aureofaciens) are efficiently expressed, we implemented promoter optimization strategies. stnYp54, a recently characterized constitutive promoter in Streptomyces, drives the minimal PKS. kasOp*55, a strong constitutive promoter, drives the acyl carrier protein (ACP). ermEp*56, a widely used constitutive promoter, drives ZhuI/J expression to direct metabolic flux of unsaturated polyketide intermediates into downstream cyclization pathways (Supplementary Fig. 10). The related biosynthetic genes were integrated into the plasmid pSET152, resulting in the construct designated pSET152-fk. This construct was subsequently introduced into Chassis2.0, designated Chassis2.0_FK.
The crude fermentation extract from Chassis2.0_FK exhibited a dark red coloration compared to Chassis2.0 (Fig. 6e). Furthermore, several significant differential chromatographic peaks were observed in the sample from Chassis2.0_FK, indicating the successful activation of the introduced biosynthetic pathway. The primary product in Chassis2.0_FK, with a corresponding molecular ion [M − H]− = 313.0358 at a retention time of 64.5 min, was further validated based on a qualitative strategy established in prior research, which confirmed the retention times of compounds in the carminic acid biosynthetic pathway via trace FK derivatives contained within carminic acid standards52. We confirmed the retention time (64.5 min) of putative FK using EIC profiles, HRMS/MS fragmentation patterns, and nuclear magnetic resonance (NMR) analysis (Supplementary Fig. 11–13). To maximize quantitative accuracy, FK was re-purified and subjected to full-wavelength UV scanning, with absorption peak area proportions exceeding 99%, thereby meeting the purity benchmarks required for analytical standards (Supplementary Fig. 14 and Supplementary Table 3). The accumulation of FK in shake-flask culture reached 566.4 mg/L, representing the highest titer reported to date (Fig. 6f)51. To comprehensively demonstrate the efficiency of Chassis2.0 and the FK biosynthetic pathway, we also analyzed other shunt products derived from the pathway. Notably, the results indicated that the accumulation levels of the shunt products were significantly lower than those of FK (Supplementary Fig. 11b, 15). Moreover, a chromatographic peak at 62.01 min in the EIC exhibited the same exact mass and HRMS/MS signatures consistent with FK, which could represent a probable FK isomer (Supplementary Fig. 11b). These results demonstrate the feasibility of employing Chassis2.0 and its potential for the efficient production of T2PKs, whether in the case of a native BGC or an artificial pathway.
The potential of penta-ring type T2PKs discovery within Chassis2.0
Following the successful synthesis of known T2PKs products featuring three-ring and four-ring backbones, our focus now shifts to the final category: T2PKs with a five-ring backbone, designated as pentangular-type T2PKs. This represents the last component necessary to demonstrate the synthetic diversity of T2PKs products within the S. aureofaciens chassis. The synthesis of final products from pentangular-type polyketides theoretically necessitates a greater quantity of malonyl-CoA precursors compared to tri-ring and tetra-ring type T2PKs. Consequently, the BGC associated with pentangular-type polyketides typically requires activations through metabolic engineering, which impedes the rate of discovery for such products57,58,59,60,61. In our previous work involving the gentamicin-producing strain M. echinospora J1-020, we identified an uncharacterized BGC, which possesses three cyclases and two C19-ketoreductases, and it is typically associated with the biosynthesis of pentangular-type polyketides (Supplementary Table 4)62. In the final section of this study, we aimed to illustrate the penta-ring type T2PKs mining capabilities of Chassis2.0 by cloning and conducting the heterologous expression of this BGC, which is designated as TLN (Fig. 7a).

a The genetic architecture of the unknown pentangular polyketide BGC in M. echinospora J1-020. b The color comparison of crude extract and the LC-UV/Q-TOF MS analysis of primary accumulated products in Chassis2.0_FK. c The product ion profiles of the activated products are presented. The red arrow indicates the precursor ion. d The quantification result of activated products in Chassis2.0_TLN. e The appearance of the purified products for compounds 1–3 is presented. The structure of TLN-1 (1) has been confirmed through NMR analysis and HRMS/MS analysis. Bar figures show mean values with error bars indicating s.d. (n = 3 biological replicates). Statistical significance was denoted as follows: ns (not significant), **P < 0.001 (two-tailed unpaired t-test, uncorrected for multiple comparisons). Source data for this figure can be found in the source data file.
To maximize cloned cluster integrity, the TLN BGC was defined based on pentangular T2PKs biosynthetic logic and structural hallmarks, which include minimal PKS, cyclases, and commonly flanking transporter/regulatory genes2. We constructed the plasmid p15A-tln using the genomic DNA of M. echinospora J1-020 as a template. This plasmid was subsequently transferred to Chassis2.0, and the resulting heterologous expression strains were designated as Chassis2.0_TLN. The integrity of the transferred gene clusters was verified using four primers (Supplementary Fig. 16). Notably, the crude extract of Chassis2.0_TLN exhibited a dark purple coloration in comparison to Chassis2.0, indicating the direct activation of the introduced gene cluster (Fig. 7b, Supplementary Fig. 17). The metabolic analysis of Chassis2.0_TLN revealed some different peaks exhibiting specific UV absorption maxima, which is consistent with the characteristics of pentangular T2PKs (Supplementary Fig. 18a). The chromatographic peak corresponding to a molecular ion [M − H]− = 515.1341 at a retention time of 21.0 min, a molecular ion [M − H]− = 513.1341 at a retention time of 33.3 min, and a molecular ion [M − H]− = 500.1480 at a retention time of 44.7 min were proposed as a series of related products compound 1, 2 and 3 (Supplementary Fig. 18b). The exact masses of the products exceed 500 Da, suggesting that a total of twelve or thirteen malonyl-CoA units participated in the biosynthesis, which is typical for the extender number in pentangular-type T2PKs. LC-HRMS/MS analysis of these peaks revealed a consistent pattern of product ion fragments, which further indicated these products may belong to a series of derivatives (Fig. 7c). Subsequently, we isolated compounds 1–3 and quantified them at respective maximum UV absorption for purity identification (Supplementary Figs. 19–21 and Supplementary Tables 5–7). Surprisingly, the further titer quantification results indicated a comparable production efficiency for these compounds in Chassis2.0. The titers reached 483.46 mg/L for TLN-1, 751.45 mg/L for compound 2, and 866.56 mg/L for compound 3 (Fig. 7d). Notably, structural elucidation of analogous pentangular T2PKs in prior reported heterologous systems required multi-liter scale fermentation (typically 4-20 L)29,57,58,59,60,61, whereas the Chassis2.0 achieves sufficient titers for structure characterization in standard shake-flask cultures (<0.5 L), demonstrating its exceptional capacity to accelerate natural product discovery and biosynthetic pathway elucidation. Furthermore, the structure of compound 1, designated as TLN-1, predicted a molecular formula C29H24O9 by HRMS result, is new in nature and features a rare 2-methylbutyryl-CoA starter unit (Fig. 7e, Supplementary Figs. 22, 23, Supplementary Table 8). Unfortunately, compounds 2 and 3 exhibit instability in liquid, which has hindered the elucidation of their structures. Overall, these results demonstrate the potential of Chassis2.0 for the efficient discovery of novel T2PKs.
Discussion
Heterologous expression has been widely acknowledged as a valuable approach for the investigation of T2PKs63. Despite the numerous hosts reported to successfully facilitate the heterologous synthesis of these compounds, challenges remain, including the poor product-chassis compatibility and suboptimal expression efficiency. In this study, we identified the high-yielding strain S. aureofaciens J1-022 as the optimal chassis by adhering to the principles of product-chassis compatibility and evaluating host characteristics, the difficulty of genetic manipulation, and the efficiency of heterologous synthesis of T2PKs. We proceeded to delete the endogenous T2PKs BGC in S. aureofaciens J1-022 and selected OTC, a product compatible with the chassis, as our first test subject. The resulting accumulation of OTC reached 8531.4 mg/L. Notably, this titer surpassed native OTC production strain after metabolic engineering, establishing the highest known level of heterologous expression for OTC to date. The productivity reached 70.8 mg/L/h, which is the highest documented in literature to date.
Two critical observations emerged during heterologous expression of the OTC BGC. First, Chassis2.0 demonstrates enhanced polyketide precursor supply compared to the strain J1-022, resulting in superior overall TCs production levels in Chassis2.0_otc versus J1-022_otc. However, despite comparable precursor accumulation to Chassis1.1, Chassis2.0_otc maintains higher TCs production levels than Chassis1.1_otc. We hypothesize this phenomenon arises from synergistic advantages in Chassis2.0, including the levels of ATP, NADP+, NADPH, and SAM, coupled with its enhanced expression efficiency of the OTC minimal PKS. Second, chassis strains with inactivated T2PKs BGC exhibited systemic advantages in precursor supply compared to the strain J1-022. However, J1-022 maintained higher NADP+ and NADPH levels during the logarithmic phase. We speculate that this phenomenon may arise from unknown regulatory mechanisms within the metabolic network. Collectively, knockout of the endogenous T2PKs BGC resulted in enhanced NADP+ and NADPH accumulation during the stationary phase.
Furthermore, to assess the versatility of Chassis 2.0, the tri-ring type products ACT and FK were selected as candidates for testing. Initially, the native BGC of ACT, derived from S. coelicolor A3(2), was successfully activated without the need for additional metabolic engineering. This success motivated us to conduct further evaluations. Consequently, FK, which serves as a significant intermediate in the biosynthesis of carminic acid, was chosen for assessment due to its successful applications across various host organisms. This selection facilitates a comprehensive comparative analysis between Chassis2.0 and other chassis systems. Given that the native pathway for FK has not been identified in native insect species, we have opted to employ a completely artificial biosynthetic pathway for the synthesis of this compound. The original study suggested that the shunt product could represent a substantial proportion of the metabolites, a phenomenon observed in artificial biosynthetic pathways due to the inherent inefficiencies related to enzyme identification and catalysis53. In this work, we achieved heterologous biosynthesis of FK using a minimal PKS capable of condensing eight ketone building blocks, coupled with cyclases derived from R1128 biosynthetic pathway. Unlike previous studies, the minimal PKS in this study employs an ACT-native ACP instead of the R1128-derived counterpart53. Furthermore, pathway optimization utilized constitutively active promoters commonly adopted in Streptomyces systems to regulate cyclases expression. This strategy was expected to direct metabolic flux toward the downstream cyclization process while minimizing spontaneous ring formation. Surprisingly, Chassis2.0 not only activates the pathway but also achieves the highest titer recorded in shake-flask culture to date51. It is crucial to note that the biosynthetic pathway for FK in this work differs from prior reports. Consequently, the achieved titer may be attributed to synergistic interactions between the inherent advantages of Chassis2.0 and pathway optimization.
To conduct a thorough evaluation of the synthesis efficiency of various T2PKs, we selected an uncharacterized BGC associated with a pentangular polyketide for assessment. In previous research, the synthesis of pentangular polyketides either necessitated additional gene editing for activation or required large-scale fermentation to ensure sufficient accumulation for subsequent analysis, resulting in increased time and labor costs57,59,60. The BGC associated with pentangular polyketides derived from M. echinospora J1-020 was directly activated in Chassis2.0, yielding associated products that exhibited a high production level. This finding underscores the robustness of the S. aureofaciens Chassis 2.0 in the heterologous expression of T2PKs.
It is noteworthy that the mining of T2PKs will exhibit greater efficiency in Chassis2.0 in comparison to other hosts. In other hosts, the expression of the T2PKs core units is often unsuccessful, necessitating the substitution of alternative polyketide synthases to address this challenge; however, the final efficiency remains suboptimal21,22,23. Furthermore, the heterologous expression of T2PKs BGC may not accurately reflect the natural processes48. In the S. aureofaciens Chassis2.0, the efficiency of T2PKs synthesis is significantly enhanced, as evidenced by the OTC, the intermediate FK, and the penta-ring products. This enhanced efficiency can aid in the structures identification of the intermediates and accelerate the elucidation of biosynthetic pathways.
Despite the successful deletion of the minimal PKS associated with spore pigment, which led to the creation of a phenotype with diminished pigmentation, the post-modification genes remained present in Chassis2.0. Prior research has indicated that spore pigment gene clusters are typically expressed during the initial stages of spore hyphae germination, whereas specialized metabolism occurs in the intermediate and later stages of Streptomyces fermentation. Cross-talk between spore pigment biosynthetic genes and secondary metabolite gene clusters remains challenging unless coordinated transcriptional activation is achieved through inducible promoter systems64. Additionally, stringent orthogonality between sporulation-associated KS/ACP domains and their antibiotic-producing counterparts imposes intrinsic barriers to such promiscuity, though the mechanistic underpinnings of this functional segregation remain unresolved65. Consequently, the residual post-modified genes are theoretically unlikely to exert a significant impact on the heterologous expression of T2PKs. Given that T2PKs are predominantly colored, chassis strains exhibiting lighter pigmentation phenotypes may be advantageous for evaluating the efficiency of heterologous gene cluster synthesis during plate culture.
In this work, we develop a plug-and-play S. aureofaciens Chassis2.0. Diverse T2PKs are synthesized in Chassis2.0, thereby establishing a foundation for future large-scale production. Furthermore, Chassis2.0 facilitates the direct activation and efficient synthesis of various T2PKs molecules, positioning it as a promising platform for the mining and overproduction of T2PKs.
Methods
Strains, culture medium, and cultivation conditions
The strains used in this study are listed in Supplementary Table 9. The strain S. aureofaciens J1 − 022, the CTC high-yielding industrial strain generously provided by Jinhe Biotechnology Co., Ltd.41, was used as a heterologous expression chassis. The strain S. rimosus ATCC10970 was used as sample to isolate genomic template for cloning OTC BGC. The strain S. coelicolor A3 (2) was used as sample to isolate genomic template for cloning ACT BGC. The strain Micromonospora echinospora J1-020 was used as sample to isolate genomic template for cloning unidentified pentangular T2PKs BGC. S. aureofaciens J1 − 022, S. albus J1074, S. lividans TK24, and their derivative strains were cultured at 30 °C about 4–5 d on solid ISP2 medium (per liter containing 4 g yeast extract, 10 g maltose extract, 4 g glucose, 2 g CaCO3, and 18 g agar powder, pH=7.0) for sporulation and conjugation. S. rimosus ATCC10970, S. coelicolor A3 (2) and M. echinospora J1-020 were cultured at 30 °C about 4–5 d on solid SFM medium (per liter containing 20 g soybean flour, 20 g mannitol, 20 g agar powder, pH=7.2) for sporulation. The liquid medium TSB (per liter containing 30 g tryptone soya broth) was used to activate spores and isolate genomic DNA, incubating at 30 °C about 12–24 h. The fermentation medium (per liter containing 80.0 g corn starch, 40.0 g soya flour, 1.0 g yeast extract, 14.0 g tryptone, 8.0 g corn milk, 7.0 g CaCO3, 3.5 g (NH4)2SO4, 2.5 g NaCl, 0.25 g MgSO4 and 15 mL soya bean oil), which is consistent with previous research41, was used to fermentation, incubating at 30 °C about 5 d. All Streptomyces strains were cultured in 500 mL Erlenmeyer flasks with 40 mL liquid medium at 220 rpm and analyzed at the end of the fermentation unless otherwise specified. Escherichia coli strains were cultivated at 37 °C in Luria−Bertani (LB) liquid medium or on LB agar plate.
Construction of plasmids
The plasmids and primers used for strain construction are listed in Supplementary Data 1 and 2, respectively. Polymerase chain reaction (PCR) was performed using Phanta Max Super-Fidelity DNA polymerase (Vazyme Biotech, Nanjing, China). The primers for PCR amplification were synthesized by Sangon Biotech (Shanghai, China). As the strain hosting the target cyclases were inaccessible, the relevant genes were synthesized de novo by GeneCreate (Wuhan, China) with codon optimization performed through their proprietary platform. Codon-optimized genes, including cyclases ZhuI (GenBank accession number: Q9F6D3) and ZhuJ (GenBank accession number: Q9F6D2) from streptomyces sp. R1128 were codon-optimized for S. aureofaciens and synthesized (Supplementary Data 3). Restriction endonucleases were purchased from Vazyme Biotech (Nanjing, China). PCR products or endonuclease digestion products were purified using a gel extraction kit (Aowei Biotech, GuangZhou, China). All plasmids derived from the pJTU1278 vector were used to gene knockout by the homologous double exchange mechanism. All plasmids derived from the p15A-cm-ccdB or pSET152 vector were subjected to ExoCET or Hieff Clone Multi-One Step Cloning Kit (Yeasen, Shanghai, China) for cloning BGC, which was based on a previous study38 and was described in the p15A_oxy construction.
The derivatives of vector pJTU1278 were utilized for the gene deletion via homologous double exchange. The insertion of homologous arms were amplified using the S. aureofaciens genomic DNA as template, of which relevant primers were listed in Supplementary Data 1 under the subheading of “plasmid construction for deletion of minimal PKS” and “plasmid construction for deletion of CTC post-modification genes”, respectively. The vector pJTU1278 was independently digested using XbaI and HindIII, purified, and ligated with the PCR products according to the Gibson assembly cloning Kit to construct the plasmids pJTU1278-KOSP, pJTU1278-KOCTC, pJTU1278-KOON, pJTU1278-KOMH, pJTU1278-KOGD, and pJTU1278-KOPQ (Supplementary Figs. 24, 25). The promoter ermEp* was amplified from plasmid PN5 using primers ermEp-F/R, while cyclase genes ZhuI and ZhuJ were amplified from synthetic gene templates via primer pairs ZhuI-F/R and ZhuI-F/R, respectively; these fragments were ligated into the first DNA fragment via three-fragment overlap extension PCR (OE-PCR). The promoter stnYp was amplified from pSET152-stnYp–indC with primers stnYp-F/R, and PKS genes were amplified from S. coelicolor genomic DNA using primers actKS-F/R and actCLF-F/R, followed by three-fragment OE-PCR to assemble the second DNA fragment. The promoter kasOp*, amplified from plasmid PN1 with kasOp-F/R and ACP, amplified from S. coelicolor genome using actACP-F/R, were ligated via two-fragment OE-PCR as the third DNA fragment. Vector backbone pSET152 was linearized via XbaI/BamHI digestion as the fourth DNA fragment. The final plasmid pSET152-fk was constructed via four-fragment Gibson assembly. (Supplementary Fig. 10).
p15A_oxy was constructed for on the p15A-cm-ccdB plasmid backbone with the insertion of OTC BGC via ExoCET DNA assembly method. In the first step, the S. rimosus M4018 was cultured in TSB liquid broth for about 12 h. The mycelium was harvested and then the resulting pellet was extracted using a Bacteria DNA Kit (TIANGEN Biotech, Beijing, China). The purified genomic DNA (about 20 μg) was digested using EcoRV for 8 h and then the resulting reaction product was extracted using DNA Extraction Phenol Reagent (Solarbio Biotech, Beijing, China). The resulting digested genomic DNA was prepared for next step. Next, the p15A-cm-ccdB vector was amplified using the primer pair oxy-DHA-F/oxy-UHA-R. The resulting DNA fragments were flanked by 80-bp homologous arm of OTC BGC. Finally, the linear p15A-cm fragment and digested genomic DNA were ligated via T4 polymerase (NEB, cat. no. M0203, USA) reaction in vitro (according to the manufacturer’s instructions) and then transformed into the E. coli GB05-dir through electroporation. The recombinants were screened using NcoI restriction analysis to ensure the integrity of OTC BGC (Supplementary Fig. 26a). All manipulations related to the extraction of genomic DNA must be conducted with utmost care to avoid physical shear forces that could compromise the integrity of the genome. The construction procedure of p15A-act and p15A-tln were similar to the p15A_oxy. In the case of p15A-act, the purified genomic DNA of S. coelicolor A3 (2) was digested using NdeI and AclI, the p15A-cm-ccdB vector was amplified using the primer pair ACT-UHA-F/ACT-DHA-R and the recombinants were screened using BamHI (Supplementary Fig. 26b). In the case of p15A-tln, the purified genomic DNA of M. echinospora J1-020 was digested using EcoRV and HindIII, the p15A-cm-ccdB vector was amplified using the primer pair TLN-UHA-F/TLN-DHA-R and the recombinants were screened using BamHI (Supplementary Fig. 26c).
Conjugation
All plasmids were transformed into Streptomyces via triparental conjugation (Supplementary Table 10). The plasmids were transformed into E. coli DH10B via electroporation and the transformants will serve as donors. The plasmid donors and E. coli ET12567/pUB307 were cultured overnight, inoculated at a 1:10 dilution in fresh LB medium, and cultured until the OD 600 reached 0.4-0.6. These cells were pelleted via centrifugation at 1500 g for 5 min, washed twice in LB broth, and finally suspended in a smaller volume of LB broth. Streptomyces spores were washed twice in TES (2-(2-[hydroxy-1,1-bis(hydroxymethyl) ethyl] amino) ethane sulfonic acid) buffer (0.05 M, pH = 8.0), following which heat shock (50 °C, 10 min) and germination (37 °C, 2 h) steps were performed to activate spores. These recipient and donor cells were mixed via a dilution plating method, which involved TES buffer mixed at a 1:1 ratio, and on to IPS2 agar plate supplemented with 20 mM MgCl2. The plate was incubated at 30 °C for 12–14 h and then overlaid with 1 mL of sterile water containing trimethoprim (50 μg/mL) and apramycin (50 μg/mL). Approximately incubation at 30 °C for 4–5 d, resultant exconjugants were picked, following which they were examined via PCR to confirm the genotype using the primers in Supplementary Data 2. All related chemicals were purchased from Sangon Biotech Co., Ltd (Shanghai, China). The standard of carminic acid was purchased from Titan Technology Co., Ltd (Shanghai, China).
Extraction and isolation of T2PKs products
For the extraction of TCs of all Streptomyces strains, spores were incubated at 30 °C in seed medium for 1 d and transferred with a volume of 10% to fermentation medium for another 5 d for fermentation. After incubation, the broth was adjusted to pH = 1.5-2.0 with oxalic acid to release the tetracyclines products from cells. The lysate mixture was centrifuged at full speed for 20 min. The supernatant was filtered through a 0.22 μm syringe filter and then analyzed. For the extraction of the pigment T2PKs. The fermentation broth was adjusted to pH = 3.5 adding formic acid and extracted with an equal volume of ethyl acetate. The organic phase was evaporated under reduced pressure. The residue was dissolved in methanol (containing 1% formic acid). The concentrated liquid samples was filtered through a 0.22 μm syringe filter and then analyzed.
For the isolation of FK, a total of 100 mL of fermentation culture was extracted and concentrated. The extract was dissolved in methanol (containing 1% formic acid) and filtered. Then, the isolation procedure was performed on Shimadzu LC-20AP preparative HPLC system. Filtrate was injected into a XBridegeTM Prep C18 reversed chromatographic column (5 μm, 250 mm × 10 mm, Waters, USA) at 25 °C. The HPLC parameters were as follows: at a constant flow rate of 3 mL/min with 70% buffer A (ultrapure water) and 30% buffer B (acetonitrile with 0.1% formic acid) for 50 min; all peaks were monitored at 280 nm. The fraction collector was activated when the ultraviolet (UV) absorbance unit exceeded 100 mAU during the time interval of 43 to 45 min. The collected fraction liquid was evaporated under reduced pressure and 4.8 mg FK were obtained. The final powder was dissolved in methanol-d and confirmed through NMR analysis. For the isolation of compounds 1 to 3, the principal conditions were identical to the FK, a total of 200 mL fermentation culture was extracted and concentrated. The fraction collector was activated when the UV absorbance unit exceeded 100 mAU during the time intervals of 16.5 to 18.5 min, 25 to 27 min, and 34.5 to 38 min, respectively. 24.5 mg compound 1, 29.6 mg compound 2, and 27.8 mg compound 3 were obtained, respectively. Compound 1 was dissolved in dimethyl sulfoxide-d6. The structures of these compounds were determined using 600 MHz 1H NMR, 13C NMR, 1H-1H COSY, HSQC, and HMBC analyses (Supplementary Figs. 27–31).
Targeted protein analysis by LC-HRMS/MS
Protein extraction was performed by adding lysis buffer (1% SDC/100 mM Tris-HCl, pH 8.5) to the samples, followed by sonication and centrifugation at 12,000 × g for 5 min to collect the supernatant. Protein concentration in the supernatant was quantified using the BCA assay. Equal amounts of protein were adjusted to identical volumes using 1% SDC/100 mM Tris-HCl (pH 8.5). Reduction and alkylation were achieved by adding TCEP and CAA, followed by incubation at 60 °C for 30 min. The SDC concentration was diluted below 0.5% by adding an equal volume sof ddH2O, and trypsin was added at a 1:50 (w/w) enzyme-to-protein ratio for overnight digestion at 37 °C with agitation. The enzymatic reaction was terminated with TFA the next day, followed by centrifugation at 12,000 × g to collect the supernatant. Desalting was performed using in-house prepared SDB columns.
For the LC-HRMS/MS analysis, samples were analyzed using an Ultimate 3000 RSLC nano system (Thermo scientific, USA) coupled to a tims TOF Pro mass spectrometer (Bruker, Germany). Peptides were loaded via an autosampler onto a C18 trap column (75 µm × 2 cm, 3 µm particle size, 100 Å pore size; Thermo scientific, USA) and separated on an analytical column (75 µm × 15 cm, 1.7 µm particle size, 100 Å pore size; Ion Opticks, Australia) with mobile phase A (0.1% formic acid) and B (0.1% formic acid in acetonitrile) under a gradient elution. Data acquisition was performed in diaPASEF mode with a capillary voltage of 1500 V. MS1 and MS2 scans covered a range of 100–1700 m/z, and ion mobility was set to 0.6–1.6 Vs/cm². Accumulation and ramp times were fixed at 50 ms. Collision energy linearly decreased from 59 eV (1/K0 = 1.6 Vs/cm²) to 20 eV (1/K0 = 0.6 Vs/cm²) based on ion mobility. Acquisition windows were defined using timsControl software according to m/z-ion mobility distributions.
For the targeted protein analysis, raw HRMS/MS data were analyzed in library-free mode using DIA-NN (v1.9.2) and based on a customized protein sequence databases (four entries, OxyA, OxyB, OxyC, and OxyD)66. Default parameters were applied with key settings: precursor prediction enabled; trypsin/P digestion (≤ 1 missed cleavage); fixed carbamidomethyl (C) modification; variable oxidation (M) and protein N-terminal acetylation; MS1/MS2 mass tolerances of 15 ppm; match-between-runs (MBR) and heuristic protein inference enabled; FDR ≤ 1%. Protein quantification utilized MaxLFQ normalization. Quantitative analysis at the protein level was performed using the output from DIA-NN. The output ion intensities of tryptic peptides derived from OTC minimal PKS were normalized to CDW for cross-strain comparability.
Targeted metabolites analysis and sample preparation
The sample pretreatment process was performed using a tissue homogenizer JXFSTPRP-48L (Jingxin, Shanghai, China). strain samples were homogenized with 300 μL of methanol/acetonitrile/water (2:2:1, v/v/v) containing 2 μg/mL phenylalanine and 2–3 steel grinding beads at 4 °C for 2 min, followed by centrifugation at 10,000 × g for 10 min at 4 °C to collect the supernatant into a clean tube. The residual pellet was re-extracted with 200 μL of the same extraction solvent, vortexed for 30 s, and centrifuged under identical conditions; the supernatant was combined with the first extract (total ~500 μL). An appropriate volume of chloroform and ultrapure water was added to the pooled extract, vortexed for 30 s, and centrifuged at 10,000×g for 20 min at 4 °C. The upper aqueous layer was collected, dried under vacuum, and reconstituted in 100 μL of methanol/water (3:7, v/v). After centrifugation, the supernatant was subjected to analysis.
Chromatographic separation was performed on a UPLC system (Thermo scientific, USA) coupled to a Q Exactive Plus mass spectrometer (Thermo scientific, USA) equipped with a SeQuant ZIC-HILIC column (100 mm × 2.1 mm, 3.5 µm; Merck, Germany) maintained at 45 °C. The mobile phase consisted of water containing 10 mM ammonium formate as phase A and 90% acetonitrile containing 10 mM ammonium formate as phase B, with a linear gradient from 10% to 60% A. The flow rate was set to 0.4 mL/min, and the injection volume was 1 μL. Mass spectrometry analysis utilized a heated electrospray ionization source in both positive and negative ionization modes. Full-scan spectra (67–1000 m/z) were acquired in data-dependent acquisition mode with one full scan (resolution: 70,000; automatic gain control (AGC): 1 × 106; ion injection time (IT): 100 ms) followed by five MS/MS scans (resolution: 17,500; AGC: 5 × 105; IT: 50 ms) using normalized collision energies of 15, 30, and 45 eV with nitrogen as the collision gas. The spray voltage was set to 3.2 kV in positive mode and 3.0 kV in negative mode, with a capillary temperature of 320 °C and an S-lens RF level of 50 V. The targeted metabolites was confirmed via both the retention time and the mass Metabolites identification was performed via retention time alignment with authentic standards and mass accuracy (<3 ppm). Integrated EIC areas were normalized CDW for cross-strain comparability.
LC/Q-TOF MS and HPLC analysis of T2PKs products
For the quantification analysis, the procedure was performed on Agilent 1260 Infinity II HPLC system and Agilent 1290 Infinity II-6546 LC/Q-TOF MS system. Filtrate was injected into a TC-C18 reversed chromatographic column (5 μm, 250 mm × 4.6 mm, Agilent, USA) at 25 °C. For the quantification analysis of TCs, the HPLC parameters were as follows: at a constant flow rate of 1.0 mL/min with 80% solvent A (contained 20 mM oxalic acid and 20 mM triethylamine, pH = 2.0) and 20% solvent B (contained authentic acetonitrile) for 30 min; All TCs were monitored at 280 nm. Using commercially available authentic standards, a five-point calibration curve was established by plotting UV absorption peak areas against corresponding concentrations (R² >0.99). TCs samples prepared following the protocol detailed in the preceding subsection (with three independent biological replicates per experimental group) were analyzed under identical HPLC conditions. Target compound concentrations were determined by substituting the UV peak areas at matching retention times (± 0.1 min) into the calibration curve equation. For the quantitative analysis of ACT, fermentation broth was extracted thrice with ethyl acetate containing 1% (v/v) formic acid. After phase separation by centrifugation (6500 × g, 10 min), the organic phases were concentrated via rotary evaporation under vacuum and reconstituted in 1 mL HPLC-grade methanol. Following previously reported work, characteristic molecular ions were identified by HRMS with a mass accuracy <1 ppm, and MS/MS spectra were aligned to those of the original producer S. coelicolor A3(2) (Supplementary Figs. 8, 9). Relative quantitation of ACT and its derivatives between original producer and heterologous expression strains was performed based on characteristic extracted molecular ion peak areas. For the quantitative analysis of FK, since the absence of commercially available reference standards, we isolated FK from Chassis2.0_FK fermentation broth as a quantitative standard. Structural validation was performed through NMR analysis, HRMS to confirm characteristic molecular weight differences, and MS/MS spectral comparisons with published data, thereby verifying the reliability of the FK standard (Supplementary Fig. 11–15). Purity assessment of the isolated FK was conducted via HPLC-DAD. The solvents A (ultrapure water with 0.1% formic acid) and B (authentic acetonitrile) were used with the following gradient: 0–25 min, 35–80% B; 25–25.2 min, 80–100% B; 25–30 min, 100% B; 30–30.2 min, 100–35% B; 35–38 min, 35% B; at a constant flow rate of 1.0 mL/min; UV full-wavelength scanning demonstrated that the absorption peak of FK at its retention time significantly exceeded background signals, with a peak area percentage of 99.835% at the maximum absorption wavelength (Supplementary Fig. 14). A five-point calibration curve (R2 > 0.999) was established using this standard. Fermentation samples (with three independent biological replicates per experimental group) were processed identically to ACT controls, with quantification achieved by correlating UV peak areas to the calibration curve. Regarding the detailed parameter settings for HPLC analysis of the FK and ACT, the solvents A (ultrapure water with 0.1% formic acid) and B (acetonitrile with 0.1% formic acid) were used with the following gradient: 0–20 min, 10–23% B; 20–50 min, 23–32% B; 50–60 min, 32–50% B; 60–70 min, 50–100% B; 70–75 min, 100% B; 75–76 min, 100–10% B; 76–80 min, 10% B; at a constant flow rate of 0.8 mL/min; All products were monitored at 280 nm. For the quantitative analysis of TLN-1 and its derivatives. since the absence of commercially available reference standards, we isolated TLN-1 and its derivatives from Chassis2.0_TLN fermentation broth as a quantitative standard. Owing to the relatively stable chemical properties of TLN-1, its chemical structure were successfully resolved via NMR analysis. Although sufficient solid powders of compound 2 and compound 3 were obtained, their instability in deuterated solvents precluded structural characterization by NMR. However, by comparing their MS/MS pattern with that of TLN-1, we confirmed that they belong to derivatives of TLN-1 (Fig. 7c). HPLC-DAD analysis revealed that the UV absorption peak areas of these pentangular T2PKs at their maximum absorption wavelengths significantly exceeded the baseline, with TLN-1 accounting for 97.675%, compound 2 for 96.341%, and compound 3 for 95.009% (Supplementary Figs. 19–21). The elution program was identical to that described for the FK method. Based on their respective purities, we established five-point concentration gradient calibration curves, achieving correlation coefficients of 0.9998 for TLN-1, 0.9997 for compound 2, and 0.9941 for compound 3. Using the same fermentation broth processing method as for FK, we prepared samples from the Chassis2.0_TLN and quantified their concentrations by substituting UV absorption peak areas into the corresponding calibration curves. Regarding the detailed parameter settings for HPLC analysis of the TLN-1 and its derivatives, the solvents A (ultrapure water with 0.1% formic acid) and B (acetonitrile with 0.1% formic acid) were used with the following gradient: 0-35 min, 60-80% B; 30-31 min, 80-100% B; 31-40 min, 100% B; 40-41 min, 100-60% B; 41-50 min, 60% B; all peaks were monitored at 280 nm and analyzed at full UV wavelength scanning.
Regarding the detailed parameter settings for mass spectrometry, the Mass Spectrum was operated with a 12.0 L/min drying gas flow rate at 350 °C, under 35 psi nebulizer pressure and negative ion mode. Fragments between 200 and 1000 m/z were detected. Chromatographic column model specifications. Auto-MS/MS mode was selected for the product ion analysis, the collision energy was set as 30 eV. Flow procedures are consistent with HPLC analysis assays except for that buffer A were all changed to 0.1% formic acid water solution. All chemical standards were purchased from Sangon Biotech Co., Ltd (Shanghai, China). All the HPLC and Mass Spectrum results were analyzed using the Agilent Mass Hunter software.
Measurements of malonyl-CoA
Cell suspensions of the strains cultured in 50 mL of fermentation medium for 16 h (during the logarithmic growth phase) were removed by centrifuging at 4000 × g for 10 min. The cells were then re-suspended in 4 mL of ddH2O, and the cell wall was destroyed by ultrasonication, after which the suspensions were centrifuged at 4000 × g for 10 min. The concentration of malonyl-CoA was measured using a microbial malonyl-CoA ELISA kit (CAT. 20154-A, Fankew, Shanghai Kexing Trading Co., Ltd)67,68. This kit employs a sandwich ELISA method to quantify malonyl-CoA levels in samples. Purified anti-malonyl-CoA antibodies were coated onto microplate wells to prepare solid-phase antibodies. Subsequently, samples were added to the antibody-coated wells, followed by incubation with horseradish peroxidase (HRP)-conjugated anti-malonyl-CoA antibodies to form antibody-antigen-enzyme-labeled antibody complexes. For generating standard curves, the secondary antibody was serially diluted at ratios of 1:1, 1:4, 1:8, 1:16, and 1:32. For test samples, a uniform 1:5 dilution ratio was applied. After rigorous washing, substrate 3,3′,5,5′-Tetramethylbenzidine (TMB) was added for chromogenic reaction. TMB is catalyzed by HRP to produce a blue coloration, which converts to yellow under acidic conditions. The color intensity directly correlates with malonyl-CoA concentration in samples. Absorbance (OD value) was measured at 450 nm using a microplate reader, with malonyl-CoA concentrations calculated against a standard curve and normalized to CDW for cross-strain comparability. The detection protocol was manually performed according to the manufacturer’s guidelines.
Statistical and reproducibility
At least three independent replicates were performed for each culture. All the data were expressed as mean ± standard deviation (SD) and were analyzed via unpaired two-tailed Student’s t-test, conducted using SPSS Statistics. The gene clusters are visualized using Chiplot69. The quantification assay were performed by utilizing GraphPad Prism 8.0 software (GraphPad Inc., LaJolla, San Diego, CA, USA). The P value was utilized to assess the statistical significance of the data (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Associated gene cluster information were collected online from NCBI, the complete oxytetracycline gene cluster sequence is available with the GenBank accession number DQ143963.1., the complete chlortetracycline gene cluster sequence is available with the GenBank accession number HM627755, the complete actinorhodin gene cluster sequence is available with the GenBank accession number AL645882.2, at the Loci of 5513809 – 5535091, the complete pentangular polyketides gene cluster sequence is available with the GenBank accession number FJ915123.1. The associated data of targeted protein analysis is deposited in the iProX database under accession number IPX0012493001. The associated data for targeted metabolites analysis have been deposited in the figshare https://doi.org/10.6084/m9.figshare.29453375.v170 and https://doi.org/10.6084/m9.figshare.29453060.v171. All other data supporting the findings of this study are available within the article, source data, and supplementary information. Source data are provided with this paper.
References
-
Zhang, Z. et al. New insights into bacterial type II polyketide biosynthesis. F1000Res 6, 172 (2017).
-
Hertweck, C. et al. Type II polyketide synthases: gaining a deeper insight into enzymatic teamwork. Nat. Prod. Rep. 24, 162–190 (2007).
-
Meloan, S. N. et al. On the structure of carminic acid and carmine. Histochemie 27, 87–95 (1971).
-
Ferreyra-Suarez, D. et al. Extraction pathways and purification strategies towards carminic acid as natural-based food colorant: A comprehensive review. Adv. Colloid Interface Sci. 323, 103052 (2024).
-
Aubel-Sadron, G. & Londos-Gagliardi, D. Daunorubicin and doxorubicin, anthracycline antibiotics, a physicochemical and biological review. Biochimie 66, 333–352 (1984).
-
Ramachanderan, R. & Schaefer, B. Tetracycline antibiotics. ChemTexts 7, 18 (2021).
-
Bode, H. B. et al. A. Big effects from small changes: possible ways to explore nature’s chemical diversity. Chembiochem 3, 619–627 (2002).
-
Zhao, L. Y. et al. Hybrid type I and II polyketide synthases yield distinct aromatic polyketides. J. Am. Chem. Soc. 146, 29462–29468 (2024).
-
Chen, S. et al. Investigation of the molecular landscape of bacterial aromatic polyketides by global analysis of type II polyketide synthases. Angew. Chem. Int. Ed. Engl. 61, e202202286 (2022).
-
Li, L. Y. et al. Resistance and phylogeny guided discovery reveals structural novelty of tetracycline antibiotics. Chem. Sci. 13, 12892–12898 (2022).
-
Bian, G. et al. Releasing the potential power of terpene synthases by a robust precursor supply platform. Metab. Eng. 42, 1–8 (2017).
-
Yuan, Y. et al. Efficient exploration of terpenoid biosynthetic gene clusters in filamentous fungi. Nat. Catal. 5, 277–287 (2022).
-
Pfeifer, B. A. et al. Biosynthesis of complex polyketides in a metabolically engineered strain of E. coli. Science 291, 1790–1792 (2001).
-
Cummings, M. et al. Assembling a plug-and-play production line for combinatorial biosynthesis of aromatic polyketides in Escherichia coli. PLoS. Biol. 17, e3000347 (2019).
-
JX Zawada, R. & Khosla, C. Heterologous expression, purification, reconstitution and kinetic analysis of an extended type II polyketide synthase. Chem. Biol. 6, 607–615 (1999).
-
Zhang, W. et al. Engineered biosynthesis of bacterial aromatic polyketides in Escherichia coli. Proc. Natl Acad. Sci. Usa. 105, 20683–20688 (2008).
-
Li, K. et al. Strategic acyl carrier protein engineering enables functional type II polyketide synthase reconstitution in vitro. ACS Chem. Biol. 20, 197–207 (2025).
-
Klein, J. G. et al. Widening the bottleneck: heterologous expression, purification, and characterization of the Ktedonobacter racemifer minimal type II polyketide synthase in Escherichia coli. Bioorg. Med. Chem. 28, 115686 (2020).
-
Liu, X. et al. Heterologous biosynthesis of type II polyketide products using E. coli. ACS Chem. Biol. 15, 1177–1183 (2020).
-
Gu, B. et al. Strategic engineering for overproduction of oviedomycin, a type II polyketide, in Escherichia coli. Metab. Eng. 90, 154–164 (2025).
-
Frandsen, R. J. N. et al. Heterologous production of the widely used natural food colorant carminic acid in Aspergillus nidulans. Sci. Rep. 8, 12853 (2018).
-
Jakočiūnas, T. et al. Programmable polyketide biosynthesis platform for production of aromatic compounds in yeast. Synth. Syst. Biotechnol. 5, 11–18 (2020).
-
Zhang, Q. et al. De novo biosynthesis of carminic acid in Saccharomyces cerevisiae. Metab. Eng. 76, 50–62 (2023).
-
Liu, R. et al. Streptomyces species: ideal chassis for natural product discovery and overproduction. Metab. Eng. 50, 74–84 (2018).
-
Yuan, Y. et al. Self-resistance-gene-guided, high-throughput automated genome mining of bioactive natural products from Streptomyces. Cell Syst. 16, 101237 (2025).
-
Libis, V. et al. Multiplexed mobilization and expression of biosynthetic gene clusters. Nat. Commun. 13, 5256 (2022).
-
Hillenmeyer, M. E. et al. Evolution of chemical diversity by coordinated gene swaps in type II polyketide gene clusters. Proc. Natl Acad. Sci. USA. 112, 13952–13957 (2015).
-
Wang, P. et al. Heterologous expression and manipulation of three tetracycline biosynthetic pathways. Angew. Chem. Int. Ed. Engl. 51, 11136–11140 (2012).
-
Zhu, X. et al. Biosynthesis of diverse type II polyketide core structures in Streptomyces coelicolor M1152. ACS Synth. Biol. 10, 243–251 (2021).
-
Yan, X. et al. Cloning and heterologous expression of three type II PKS gene clusters from Streptomyces bottropensis. ChemBioChem 13, 224–230 (2012).
-
Yin, S. et al. Heterologous expression of oxytetracycline biosynthetic gene cluster in Streptomyces venezuelae WVR2006 to improve production level and to alter fermentation process. Appl. Microbiol. Biotechnol. 100, 10563–10572 (2016).
-
Liu, W. et al. Heterologous reconstitution of oxytetracycline producing strain in Streptomyces albus Del14. Acta Micro Sin. 62, 3858–3870 (2022).
-
Gu, B. et al. Heterologous overproduction of oviedomycin by refactoring biosynthetic gene cluster and metabolic engineering of host strain Streptomyces coelicolor. Microb. Cell. Fact. 22, 212 (2023).
-
Duan, Y. et al. Exploration of Streptomyces fradiae J1–021 as a potential host for the heterologous production of spinosad. J. Agric. Food Chem. 72, 8983–8992 (2024).
-
Wang, X. et al. Heterologous production of chlortetracycline in an industrial grade Streptomyces rimosus host. Appl. Microbiol. Biotechnol. 103, 6645–6655 (2019).
-
Finlay, A. C. et al. Terramycin, a new antibiotic. Science 111, 85–85 (1950).
-
Boothe, J. H. et al. 6-deoxytetracyclines. I. chemical modification by electrophilic substitution. J. Am. Chem. Soc. 82, 1253–1254 (1960).
-
Wang, H. et al. ExoCET: exonuclease in vitro assembly combined with RecET recombination for highly efficient direct DNA cloning from complex genomes. Nucleic Acids Res. 46, e28 (2018).
-
Novakova, R. et al. Increased heterologous production of the antitumoral polyketide mithramycin A by engineered Streptomyces lividans TK24 strains. Appl. Microbiol. Biotechnol. 102, 857–869 (2018).
-
Pšeničnik, A. et al. Oxytetracycline hyper-production through targeted genome reduction of Streptomyces rimosus. mSystems 9, e00250–24 (2024).
-
Zhu, T. et al. Deciphering and engineering of the final step halogenase for improved chlortetracycline biosynthesis in industrial Streptomyces aureofaciens. Metab. Eng. 19, 69–78 (2013).
-
Kuhl, M. et al. Microparticles enhance the formation of seven major classes of natural products in native and metabolically engineered actinobacteria through accelerated morphological development. Biotechnol. Bioeng. 118, 3076–3093 (2021).
-
Petković, H. et al. Genetics of Streptomyces rimosus, the oxytetracycline producer. Microbiol. Mol. Biol. Rev. 70, 704–728 (2006).
-
Slemc, L. et al. Molecular biology methods in Streptomyces rimosus, a producer of oxytetracycline. Methods Mol. Biol. 2296, 303–330 (2021).
-
Blin, K. et al. AntiSMASH 6.0: improving cluster detection and comparison capabilities. Nucleic Acids Res. 49, W29–W35 (2021).
-
Novakova, R. et al. Characterization of the polyketide spore pigment cluster whiESa in Streptomyces aureofaciens CCM3239. Arch. Microbiol. 182, 388–395 (2004).
-
Yin, S. et al. Identification of a cluster-situated activator of oxytetracycline biosynthesis and manipulation of its expression for improved oxytetracycline production in Streptomyces rimosus. Microb. Cell. Fact. 14, 46 (2015).
-
Herbst, E. et al. Heterologous catalysis of the final steps of tetracycline biosynthesis by Saccharomyces cerevisiae. ACS Chem. Biol. 16, 1425–1434 (2021).
-
Xie, F. et al. Autologous DNA mobilization and multiplication expedite natural products discovery from bacteria. Science 386, eabq7333 (2024).
-
Bystrykh, L. V. et al. Production of actinorhodin-related ‘blue pigments’ by Streptomyces coelicolor A3(2). J. Bacteriol. 178, 2238–2244 (1996).
-
Yang, D. et al. Production of carminic acid by metabolically engineered Escherichia coli. J. Am. Chem. Soc. 143, 5364–5377 (2021).
-
Massicard, J.-M. et al. Modular cloning tools for Streptomyces spp. and application to the de novo biosynthesis of flavokermesic acid. ACS Synth. Biol. 13, 3354–3365 (2024).
-
Tang, Y. et al. Engineered biosynthesis of regioselectively modified aromatic polyketides using bimodular polyketide synthases. PLoS Biol. 2, E31 (2004).
-
Guo, W. et al. Identification and characterization of a strong constitutive promoter stnYp for activating biosynthetic genes and producing natural products in Streptomycetes. Microb. Cell. Fact. 22, 127 (2023).
-
Wang, W. et al. An engineered strong promoter for Streptomycetes. Appl. Environ. Microbiol. 79, 4484–4492 (2013).
-
Bibb, M. J. et al. Cloning and analysis of the promoter region of the erythromycin resistance gene (ermE) of Streptomyces erythraeus. Gene 38, 215–226 (1985).
-
Liu, X.-F. et al. Activation and characterization of lanthomicins A–C by promoter engineering in Streptomyces chattanoogensis L10. Front. Microbiol. 13, (2022).
-
Ogasawara, Y. et al. Expanding our understanding of sequence-function relationships of type II polyketide biosynthetic gene clusters: bioinformatics-guided identification of frankiamicin A from Frankia sp. EAN1pec. PLoS One 10, e0121505 (2015).
-
Tian, J. et al. Discovery of pentangular polyphenols hexaricins A-C from marine Streptosporangium sp. CGMCC 4.7309 by genome mining. Appl. Microbiol. Biotechnol. 100, 4189–4199 (2016).
-
Zhang, M. M. et al. CRISPR–Cas9 strategy for activation of silent Streptomyces biosynthetic gene clusters. Nat. Chem. Biol. 13, 607–609 (2017).
-
Hofeditz, T. et al. Lysoquinone-TH1, a new polyphenolic tridecaketide produced by expressing the lysolipin minimal PKS II in Streptomyces albus. Antibiotics 7, 53 (2018).
-
Wu, L. et al. Efficient synthesis of gentamicin and its related products in industrial chassis cells. J. Syn. Bio. 3, 1277–1291 (2022).
-
Malpartida, F. & Hopwood, D. A. Molecular cloning of the whole biosynthetic pathway of a Streptomyces antibiotic and its expression in a heterologous host. Nature 309, 462–464 (1984).
-
Yu, T.-W. & Hopwood, D. A. Ectopic expression of the Streptomyces coelicolor whiE genes for polyketide spore pigment synthesis and their interaction with the act genes for actinorhodin biosynthesis. Microbiology 141, 2779–2791 (1995).
-
Lee, T. S., Khosla, C. & Tang, Y. Orthogonal protein interactions in spore pigment producing and antibiotic producing polyketide synthases. J. Antibiot. 58, 663–666 (2005).
-
Demichev, V. et al. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 17, 41–44 (2020).
-
Li, J. et al. A non-carbonylative route for the efficient synthesis of central metabolite malonyl-CoA and its derived products. Nat. Catal. 7, 361–374 (2024).
-
Tang, J. et al. Bacterioferritin: a key iron storage modulator that affects strain growth and butenyl-spinosyn biosynthesis in Saccharopolyspora pogona. Microb. Cell Fact. 20, 157 (2021).
-
Xie, J. et al. Tree Visualization by one table (tvBOT): a web application for visualizing, modifying and annotating phylogenetic trees. Nucleic Acids Res. 51, W587–W592 (2023).
-
Zou, Y., Liu, T., Liu, R. Source_Data_posMS_NCOMMS-25-07339B. Data sets. figshare. https://doi.org/10.6084/m9.figshare.29453375.v1 (2025).
-
Zou, Y., Liu, T., Liu, R. Source_Data_negMS_NCOMMS-25-07339B. Data sets. figshare. https://doi.org/10.6084/m9.figshare.29453060.v1 (2025).
-
Pickens, L. B. & Tang, Y. Oxytetracycline biosynthesis. J. Biol. Chem. 285, 27509–27515 (2010).
-
Wang, P. et al. Uncovering the enzymes that catalyze the final steps in oxytetracycline biosynthesis. J. Am. Chem. Soc. 135, 7138–7141 (2013).
Acknowledgements
This work was supported by Shanghai Municipal Science and Technology Major Project, the National Key Research and Development Program of China (2021 YFC2101000 to R.L.), the National Natural Science Foundation of China (U23A20527 to T. L., 32100053 to R.L., and 82273826 to F.Z.), the National Key Research and Development Program of China (2022YFC2804300 to F.Z.), Young Elite Scientists Sponsorship Program by China Association for Science and Technology (YESS20210068 to R.L.), and the Hohhot Jinhe Technology Co., Ltd.
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Kira Weissman who co-reviewed with Delphine Noel and Koji Ichinose who co-reviewed with Makoto Hashimoto for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zou, Y., Wang, R., Liu, Z. et al. Development of a versatile chassis for the efficient production of diverse type II polyketides. Nat Commun 16, 7321 (2025). https://doi.org/10.1038/s41467-025-62659-0
-
Received:
-
Accepted:
-
Published:
-
DOI: https://doi.org/10.1038/s41467-025-62659-0