Drug discovery has long faced a stubborn reality: Most disease-relevant proteins lack the deep, hydrophobic pockets required for inhibition by conventional small-molecule drugs. Beginning in the early 2000s, researchers began framing this challenge in terms of druggability—whether a given protein can bind effectively to traditional therapeutics.1 Transcriptional regulators, scaffolding proteins, and intrinsically disordered domains were therefore often considered largely undruggable.
Over the past decade, however, a wave of technological advances has begun to expand the druggable proteome. These include covalent inhibitors (such as sotorasib), chemical proteomics, and AI-enabled structural prediction tools like AlphaFold. We spoke with four leading experts to explore two additional therapeutic modalities gaining momentum in 2026: targeted protein degraders and macrocycles.
Targeted protein degradation
Targeted protein degradation (TPD) is a therapeutic strategy that aims to destroy disease-causing proteins within cells. “Traditional therapies with small-molecule drugs tend to focus on inhibiting problematic proteins. In contrast, TPD aims to completely eliminate these proteins,” explains Alessio Ciulli, PhD, professor at the University of Dundee and founder of the Centre for Targeted Protein Degradation.
More specifically, degrader drugs bring the disease-causing target protein into proximity with an E3 ligase—an enzyme that is part of the cell’s protein degradation machinery.

[Nurix Therapeutics]
“TPD works in a clever way by hijacking the cell’s own trash disposal mechanisms,” Ciulli says. “We are borrowing from nature and saying, ‘Come and help me get rid of these proteins.’”
“A major advantage of TPDs is that, unlike traditional small-molecule drugs, they bind only transiently with their intended targets and do not require a well-defined binding pocket,” notes Gwenn Hansen, PhD, CSO of Nurix Therapeutics. “This enables the elimination of previously undruggable proteins.”
PROTACs versus molecular glues
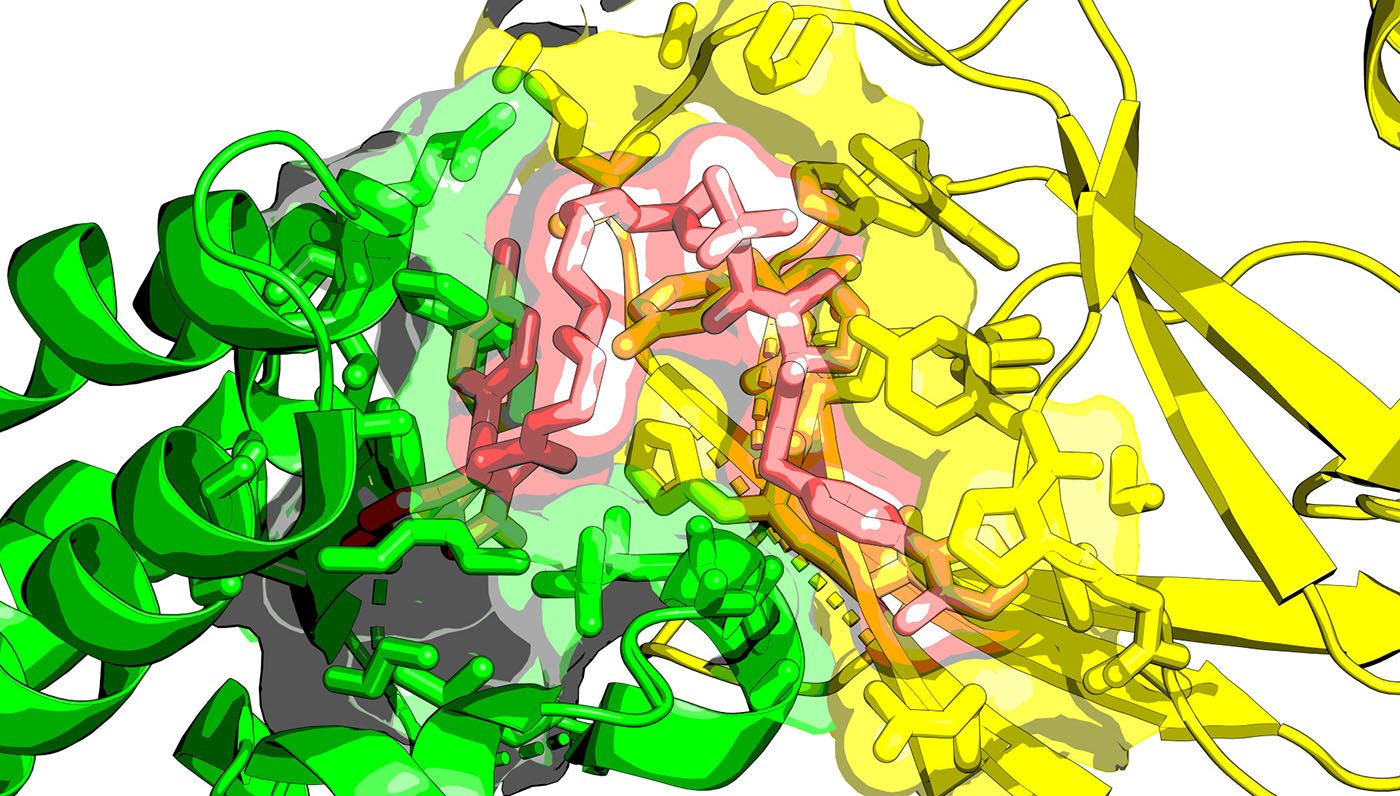
The TPD field consists of two major classes of degraders—PROTACs (PROteolysis TArgeting Chimeras) and molecular glue degraders. PROTACs are small molecules that consist of three key parts: 1) a ligand that binds the disease-causing protein, 2) a ligand that binds an E3 ligase, and 3) a chemical linker connecting the two.
“PROTACs are like matchmakers,” notes Ciulli. “They follow a plug-and-play strategy in the sense that you can mix and match any target protein with any E3 ligase. It is like two hooks and a way to connect the hooks.”
“It is almost like your PROTACs build bridges on demand,” he continues. “You say, ‘Hey, I want to get from protein A to protein B. Let’s build the bridge that allows us to do that.’ Because you know exactly what A and B are.”
Although dozens of PROTACs are advancing through clinical trials as of early 2026, none of them have yet received full FDA approval.
Molecular glues are another type of TPD. Instead of generating entirely new protein–protein interactions, as with PROTACs, molecular glues require a pre-existing surface complementarity between the two proteins. More specifically, they enhance the binding of the target protein to the E3 ligase, in a mechanism called gluing. Thalidomide, lenalidomide, and pomalidomide are clinically approved molecular glue degraders.
Ciulli relates how the precise molecular mechanism of thalidomide wasn’t established until the 21st century, when researchers identified its target, the E3 ligase cereblon. Tragically, the drug led to thousands of congenital abnormalities in children whose mothers had taken it during early pregnancy in the 1950s and 1960s.
Like PROTACs, molecular glues are small molecules (they lack a linker and only feature one of the two binders). “Compared with PROTACs, glues are much more minimalistic,” notes Ciulli. “They don’t have this plug-and-play of PROTACs. The proteins already have some intimacy, but it is not quite there. And then, BOOM, molecular glues make it happen.”
Which is more promising for the future, PROTACs or molecular glues? “They are both important. They are absolutely both the future,” argues Ciulli. “They both need to be deployed conceptually and practically if we really want to drug some of these cancers.”
“To some extent, PROTACs and molecular glues are really a continuum,” adds Ciulli. “It is not like we are talking about apples and pears. It is more like oranges and mandarins. The mechanism is exactly the same. We are degrading the protein in the end.”
Hansen notes that Nurix is primarily focused on PROTACs, which she describes as “the most tractable modality for targeted protein degradation.” However, the company is also developing molecular glues as well as a newer TPD modality called degrader antibody conjugates (DACs), which couple PROTACs to
antibodies.
TPD for new targets
“Nurix is currently emphasizing previously undruggable targets that sit at the heart of disease biology—including a lot of signaling proteins and transcriptional regulators,” notes Hansen.
Nurix’s high-throughput approach leverages a combination of proprietary DNA-encoded libraries, synthetic chemistry, and machine learning to rapidly identify and optimize their TPDs. The company’s current pipeline consists of several oncology and immunology targets.
Likewise, Ciulli stresses that his lab is “working on really tough targets that we previously wouldn’t dare to touch.” More specifically, he is focused on cancers driven by specific genetic alterations, such as mutations, translocations, and genetic fusions. “These events generate proteins that are very, very hard to target,” he says. Such proteins tend to have multiple structures that are hard to predict with technologies like AlphaFold. In other cases, these proteins are recycled very, very quickly.
Ciulli notes the success of PROTACs in targeting KRAS,2 which is often highly mutated in cancer. Small-molecule drugs typically target only very specific KRAS mutations that affect around three percent of patients.
“In contrast, we published a PROTAC degrader that we were able to show removes 13 out of 17 of all of the known oncogenic drivers of cancer. With PROTACs, a single molecule can degrade all of the KRAS mutations.”
Finally, Hansen highlights the ability of TPDs to degrade disease proteins that have developed mutations or acquired resistance following treatment. “By eliminating a protein entirely rather than transiently inhibiting its function, degraders can achieve deeper and more durable control or even elimination of disease,” she notes.
“Looking forward, I expect the field to move away from proving that degradation works and toward proving where it wins,” she predicts. “The companies that win will be the ones that treat degradation as a drug-discovery discipline, not an alternate and occasional modality.”
Introducing macrocycles
David J. Earp, PhD, president and CEO of Circle Pharma, explains how the company is developing a class of compounds called macrocycles to treat cancer. “Macrocycles offer the potential to drug targets that have eluded other drug modalities,” he notes.
Macrocycles are large, ring-shaped molecules that provide structural support and allow for effective binding to biological targets. Cyclosporine and erythromycin are two examples of macrocycle drugs derived from nature. Over the past decade, there has also been a strong interest in developing fully synthetic macrocycles, and these efforts have generated early clinical success.

“Peptidic macrocycles occupy a compelling middle ground between biologics and conventional small molecules,” he notes. Unlike biologics, macrocycles are small enough to enter cells and can be orally available when properly engineered. On the other hand, macrocycles are larger than small-molecule drugs. In larger targets, this allows for multiple contacts across a broader area.
Like Circle, Curve Therapeutics is also developing macrocycle peptides. Ali Tavassoli, PhD, CSO of Curve and professor of chemical biology at the University of Southampton, explains that the company is focused on small cyclic peptides—called microcycles—composed of only six amino acids in a head-to-tail ring. They are generated in mammalian cells during a post-translational process called intein (internal protein segment) splicing.
Unlike larger cyclic peptides from other technologies, the key binding regions of microcycles (pharmacophores) work through just three or four amino acids. “Therefore, they are ideally positioned for further peptide engineering and modification with non-peptide components. They also have the potential for scaffold hopping into small molecules,” says Tavassoli.
Curve is currently producing and screening libraries of around a billion microcycles directly inside mammalian cells, selecting hits based on function inside a disease-relevant environment. Curve’s function-based approach is unique, as other companies emphasize binding as the primary readout.
“Binding in a test tube doesn’t guarantee function in a cell,” emphasizes Tavassoli. “We screen for biological function inside live mammalian cells, where targets exist in native conformations with disease-relevant modifications. They ask, ‘What binds?’ while we ask, ‘What functions?’” Curve’s intracellular screening process involves genetically encoded cyclic peptide libraries and fluorescence detection.

Success with macrocycles
Curve’s lead program is a first-in-class dual HIF-1 and HIF-2 inhibitor that disrupts proteins that enable cancer to thrive in low-oxygen tumor environments. The company is also pursuing FOXA1 inhibitors for hormone-refractory breast cancer, for which no inhibitors exist despite significant R&D interest in the target.
Meanwhile, Earp highlights two programs in Circle’s macrocycle pipeline that directly target cyclins, master regulators of cell division that are frequently dysregulated in cancer. “Historically, cyclins were considered undruggable, as prior efforts using traditional small molecules failed to effectively target these highly dynamic intracellular protein–protein interfaces.”
Circle’s lead program, CID-078 (an oral macrocycle cyclin A/B RxL inhibitor), is currently in late Phase I clinical development for solid tumors.3 “We are progressing very efficiently through dose escalation and have excellent clinician engagement,” notes Anne E. Borgman, MD, chief medical officer. Clinical data will be available in 2026.
The company also anticipates filing an IND for a second program (CID-165, a cyclin D1 RxL inhibitor) with potential applications in ER-positive breast cancer and lymphomas. Both programs represent first-and-only-in-class approaches, says Earp.
Beyond cyclins, Circle hopes to apply its macrocycle platform to other high-value oncology targets that remain inaccessible to conventional small-molecule drugs—including transcriptional regulators and scaffolding proteins. The company aims to access intracellular targets and achieve oral bioavailability.
Tavassoli and Earp agree that intracellular delivery remains a major hurdle for cyclic peptides. Fortunately, AI-driven design, new cyclization chemistries, and non-natural amino acids are transforming what is possible, notes Tavassoli. He predicts that multiple drugs from cyclic peptide platforms will be approved within the next decade to target proteins.
“As these tools mature, we expect macrocycles to play an increasingly central role in expanding the druggable proteome, particularly in oncology and other areas driven by challenging intracellular protein–protein interactions,” adds Earp.
References
- Zhang Z, Shokat KM. Introduction: drugging the undruggable. Chem Rev. 2025;125(14):6433-6434. doi: 10.1021/acs.chemrev.5c00246.
- Popow J, Farnaby W, Gollner A, et al. Targeting cancer with small-molecule pan-KRAS degraders. Science. 2024;385(6715):1338-1347. doi: 10.1126/science.adm8684.
- Singh S, Gleason CE, Fang M, et al. Targeting G1-S-checkpoint-compromised cancers with cyclin A/B RxL inhibitors. Nature. 2025;646(8085):734-745. doi: 10.1038/s41586-025-09433-w.
- Gadd MS, Testa A, Lucas X, et al. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat Chem Biol. 2017;13(5):514-521. doi: 10.1038/nchembio.2329.
- Zengerle M, Chan KH, Ciulli A. Selective small molecule induced degradation of the BET bromodomain protein BRD4. ACS Chem Biol. 2015;10(8):1770-7. doi: 10.1021/acschembio.5b00216.
The post Expanding the Druggable Proteome with Tech Advances appeared first on GEN – Genetic Engineering and Biotechnology News.