Introduction
Bioactive compounds play a crucial role in various biological and ecological processes, with applications extending beyond human health to agriculture, biotechnology, and environmental sustainability1. These compounds, produced by a diverse range of organisms, including higher plants and microorganisms, contribute to microbial interactions, chemical defense mechanisms, and biogeochemical cycles2.
Over 70% of the Earth’s surface is covered by the ocean. The ocean’s biodiversity is far more rich than the terrestrial environments3. Numerous bioactive compounds are produced by soil or marine organisms, including bacteria, fungi, plants, and certain animals. Recent studies have highlighted marine bacteria as a prolific source of bioactive compounds with significant potential in pharmaceutical and agricultural applications4. These bioactive compounds are being investigated as potential medications and have been employed as herbicides, insecticides, immunosuppressants, antibiotics, and anti-cancer agents5. A comprehensive analysis of the global ocean microbiome predicted approximately 64,217 biosynthetic gene clusters (BGCs) of 66 different types, underscoring the ocean’s rich reservoir of natural products6.
Bioactive compounds are encoded by biosynthetic gene clusters (BGCs)7. The updated MIBiG 4.0 database provides a comprehensive resource for understanding BGC organization and function, emphasizing their role in natural product biosynthesis through global collaborative curation8.
The most common documented microbial BGCs are polyketide synthases (PKS), non-ribosomal peptide synthetases (NRPS), ribosomally produced and post-translationally modified peptides (RiPPs), terpenoids, saccharides, and a few other hybrid chemicals7. NRPSs are large, modular enzymes that function like assembly lines to synthesize diverse natural products, many of which have medicinal properties, driving research into their pathways and chemical diversity9.
Siderophores are small-molecule iron chelators produced by a variety of microbes, which take up iron from their surroundings. Microbes use NRPS or NRPS-independent siderophore (NIS) enzymes for the biosynthesis of siderophores8. Siderophores comprise a chemically diverse family of ~ 500 molecules11. These structural variations in siderophores and their receptors have evolved due to the competition for Fe3+ uptake between bacteria or between bacteria and their hosts9.
Marine bacteria require micromolar levels of iron for their growth. However, the iron concentration of the ocean surface water is just 0.1–2 nM10. Hence, in order to avoid this iron shortage, marine bacteria have developed siderophore-mediated iron delivery systems11. In addition, marine microbial siderophores can be used as a target to block siderophore virulence factors. This leads to the development of new, broad-spectrum antibiotics, or different drug conjugates for the treatment of drug resistant bacteria11. The siderophore vibrioferrin belongs to the carboxylate class and has two α-hydroxy acid groups in it. The vibrioferrin isolated from an enteropathogenic estuarine bacterium, Vibrio parahaemolyticus, often linked to gastroenteritis caused by seafood12. Siderophores such as amphibactins (tris-hydroxamates), vibrioferrin (an α-hydroxy carboxylate), and crochelin A (a siderophore with varied chelating groups that includes a new Fe chelating moiety) with a range of Fe-binding affinities were found in γ-proteobacterium Azotobacter chroococcum13. Among these, vibrioferrin has the lowest Fe-binding affinities13. Many bacteria are able to synthesize several types of siderophore families, which is a typical strategy used by bacteria to reserve gene clusters for siderophores with both high and low Fe-binding affinities13.
The current availability of sophisticated bioinformatics techniques allows the identification of BGCs and better understanding of their pathways14,15. Genome mining using antiSMASH (antibiotics and secondary metabolite analysis shell) is a critical process in modern bioactive compounds discovery processes16.
This study aimed to evaluate the diversity and BGCs distribution in marine bacterial species belonging to the Proteobacteria, Bacteroidetes, Firmicutes, and Actinobacteria phyla. For phylogenetic diversity analysis, the rpoB gene was selected, as it is a well-established genetic marker providing reliable phylogenetic insights due to its relatively conserved nature, allowing for accurate reconstruction of evolutionary relationships among diverse bacterial strains17. Additionally, this study investigates the variation in vibrioferrin-producing NI-siderophore BGCs at both gene cluster and amino acid sequence levels across Vibrio harveyi, Vibrio alginolyticus, and Photobacterium damselae strains, including two reference genomes of vibrioferrin. This emphasis on the genetic and structural variability of vibrioferrin clusters distinguishes our study from prior broad surveys of BGC diversity.
Methods
Bacterial strain selection and genome retrieval
To investigate the potential production of bioactive compounds, a total of 21 marine bacterial species (199 strains) were selected for this study. Selection was based on previous isolation and molecular identification of marine bacteria from marine sediment in the Oman Sea. The following bacteria were included for the study: Shewanella corallii, Shewanella submarina, Shewanella waksmanii, Photobacterium damselae, Photobacterium leiognathi, Microbulbifer agarilyticus, Alteromonas macleodii, Alteromonas mediterranea, Halomonas organivorans, Ruegeria atlantica, Ruegeria arenilitoris, Pseudoalteromonas donghaensis, Vibrio campbellii, Vibrio harveyi, Vibrio alginolyticus, Tenacibaculum singaporense, Tenacibaculum mesophilum, Tenacibaculum aiptasiae, Virgibacillus halodenitrificans, Bacillus licheniformis, and Micrococcus luteus.
Genome sequences of 199 marine bacterial strains, representing 21 species and 17 genera, were retrieved from the NCBI database. Complete genomes were used when available, while high-quality contig-level assemblies were included for species without complete genomes (see Supplementary Table S1 for accession numbers). The scientific names of the strains, accession numbers, genome assembly, and genome information, including the number of genes, genome size, and number of protein coding genes, were obtained as well (Supplementary Table S1).
Biosynthetic gene cluster prediction and analysis
To identify and compare biosynthetic gene clusters (BGCs) among the selected marine bacterial genomes, the antiSMASH 7.0 bacterial version14 was used to screen each strain for the presence of BGCs. Analysis was performed using antiSMASH 7.0 (bacterial version) with default detection settings, enabling KnownClusterBlast, ClusterBlast, SubClusterBlast, and Pfam domain annotation. Following the antiSMASH analysis, the results were systematically compiled into an Excel spreadsheet, where the total number of BGCs and their classifications were recorded for each genome. This dataset was then used to compare BGC abundance and diversity across all 199 strains (Supplementary Table S2).
Phylogenetic analysis based on rpoB Gene
Microbial phylogenetic analysis based on the rpoB gene was conducted using MEGA11 (Molecular Evolutionary Genetics Analysis version 11). The rpoB gene sequences (192) were retrieved from NCBI nucleotide and aligned using the ClustalW multiple alignment tool in BioEdit software. The aligned sequences were exported to MEGA11 to construct a maximum likelihood phylogeny with 1000 bootstrap replicates and other parameters kept as default.
The phylogenetic tree was exported as a Newick format to the Interactive Tree of Life [iToL] to visualize and annotate with all BGCs to explore the evolutionary diversity of the selected strains and their potential for producing natural bioactive compounds.
Genomic analysis of NI-Siderophore BGCs
NI-siderophore BGCs region GenBank files of all strains that have NI-siderophore BGC predicted for vibrioferrin compound (58 strains) belonging to Vibrio harveyi, Vibrio alginolyticus, and Photobacterium damselae and NI-siderophore BGC regions of the two reference genomes of vibrioferrin: Vibrio alginolyticus and Vibrio parahaemolyticus were downloaded from antiSMASH as BioEdit files. Then, all nucleotide sequences were copied to Geneious Prime software and translated to amino acid sequences and then aligned using Clustal Omega with the default setting.
All NI-siderophore BGCs predicted by antiSMASH were assigned to vibrioferrin. No cryptic NI-siderophore clusters were identified in this dataset (Supplementary Table S4). Thus, the downstream comparative analysis was based exclusively on vibrioferrin BGCs.
Further alignment was done to the NI-siderophore BGC regions of the two reference genomes of vibrioferrin along with additional alignment for the core genes. All alignments were annotated using Geneious Prime.
BGC clustering and network analysis
Clustering of the annotated NI-siderophore BGCs was performed using BiG-SCAPE (Biosynthetic Gene Similarity Clustering and Prospecting Engine) version 2.0 to group them into Gene Cluster Families (GCFs) based on domain sequence similarity18. The analysis was conducted across multiple similarity cutoffs with final clustering results interpreted at 10% and 30% cutoffs. BiG-SCAPE similarity networks were generated using distance cutoffs of 30% and 10%. The 30% threshold follows prior benchmarking, where it has been applied to define broad gene cluster families21. In addition, we also applied a more stringent 10% cutoff to resolve fine-scale families within GCFs, which has been adopted in subsequent comparative BGC studies to capture subtle sequence-level diversity.
The resulting networks were visualized using Cytoscape version 3.10.3. Each GCF was further analyzed to assess core and accessory gene conservation, providing insights into diversity in vibrioferrin biosynthesis.
Results
Biosynthetic gene cluster (BGC) diversity in marine bacteria
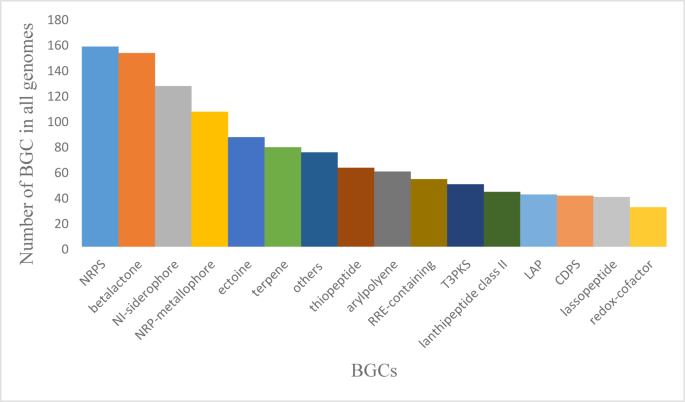
A total of 29 different regions potentially encoding BGCs for the production of bioactive compounds were predicted by antiSMASH within the 199 bacterial genomes, which gave insight into the wide range of bioactive compounds (Supplementary Table S2). Fifteen regions, including arylpolyene, betalactone, CDPS (RNA-dependent cyclodipeptide synthases), ectoine, LAP (Linear azol(in)e-containing peptides), lanthipeptide class II, lassopeptide, NI-siderophore, NRPS (Non-rRibosomal Peptide Synthetase), NRP-metallophore (Non-Ribosomal Peptide metallophores), redox-cofactor, RRE-containing (RRE-element containing cluster), T3PKS, terpene, and thiopeptide, were common in most species. Fourteen regions were found in a few species or only one, namely hglE-KS (Heterocyst glycolipid synthase-like PKS), hserlactone, lanthipeptide class I, lanthipeptide class II, NAPAA (Non-Alpha Poly-Amino Acids like e-Polylysin), NRPS-like (NRPS-like fragment), oligosaccharide, phosphonate, PUFA (Polyunsaturated Fatty Acid), ranthipeptide, resorcinol, RiPP-like (Other unspecified ribosomally synthesized and post-translationally modified peptide product (RiPP)), T1PKS (Type I PKS (Polyketide synthase)) and thioamitides, these were the rare BGCs. The total number of BGCs per genome ranged between 0 and 11 (5.5 ± 3.45) in the 199 bacterial genomes investigated. Bacillus licheniformis, contained the highest number of BGCs, three strains contained 11 BGCs (Bacillus licheniformis NWMCC0046, Bacillus licheniformis SCDB 34, and Bacillus licheniformis RCM100141), and the others (36 strains) contained 10 BGCs. No BGCs were detected in the two genomes of Shewanella waksmanii ATCC BAA-643 and Vibrio alginolyticus K05K4 (Supplementary Table S1). The highest number of NRPS copies was 3, found in 4 genomes: Vibrio campbellii LJC011, Vibrio campbellii DS40M4, Bacillus licheniformis NWMCC0046, and Bacillus licheniformis RCM100141 (Supplementary Table S2). Some BGCs were rare and found only in one genome, such as lanthipeptide class I, lanthipeptide class III, oligosaccharide, phosphonate, ranthipeptide, and thioamitides. Other rare BGCs are found in only two genomes, such as resorcinol, PUFA, and hglE-KS. Although present in only a few genomes (≤ 5), these loci may encode unusual metabolites and represent potential sources of novelty for natural product discovery. Some other rare BGCs were found only in 4 or 5 genomes, such as T1PKS and NRPS-like, respectively (supplementary Table S2). The most dominant BGCs across species were NRPS (157), betalactone (152), NI-siderophore (127), and NRP-metallophore (106) (Fig. 1). However, because a genome can comprise numerous copies of the same BGC, as in NRPS and NRP-metallophore, they existed only in 91 and 84 genomes, respectively. While betalactone and NI-siderophore, which are found in most genomes, were present in 157 and 127 genomes, respectively. The genus-level distribution of BGC classes is detailed in Supplementary Table S2, which provides the underlying counts for Fig. 1. This integration highlights both common classes such as NRPS and betalactones and the presence of rare clusters with potential novelty. To complement Fig. 1, genus-level distributions of the top BGC classes are shown as a heatmap (Supplementary Figure S6), which integrates the summary counts provided in Supplementary Tables S1–S2.

Frequency distribution of dominant BGC classes across 199 marine bacterial genomes. Data source: antiSMASH 7.0 outputs compiled into Microsoft Excel spreadsheets (see Methods, Supplementary S1–S2). BGCs detected in only 1–5 genomes were considered rare and are grouped as “Other” (details in Supplementary Table S2).
Phylogenetic distribution of marine bacteria and BGCs
Totally 192 marine bacterial strains were used for phylogenetic analysis for the rpoB gene in this study. The phylogenetic tree showed two major groups, with group 1 (38 strains), only comprising strains belonging to Bacillus licheniformis. Group 2 (154 species, green labeled) comprises strains belonging to species of the other genera: Shewanella, Photobacterium, Microbulbifer, Alteromonas, Halomonas, Ruegeria, Pseudoalteromonas, Vibrio, Tenacibaculum, Virgibacillus, and Micrococcus (Fig. 2). Based on the rpoB gene, species in group 2 are very closely related, the bootstrap value was 73%. Despite the taxonomic diversity, these strains are grouped together based on their high similarity in the rpoB gene, suggesting a conserved evolutionary history for this gene across these genera. The phylogenetic tree, which includes the strain names is provided in supplementary Figure S3.

Maximum-likelihood phylogeny of 192 marine bacterial genomes based on the rpoB gene, annotated with 15 BGC classes. Rare BGCs (detected in only 1–5 genomes) were grouped as “Other.” Sequences were aligned with ClustalW in BioEdit, and the tree was constructed in MEGA11 using the maximum-likelihood method with 1,000 bootstrap replicates. Bootstrap support values ≥ 70 are indicated by filled circles. The phylogeny was visualized and annotated in iTOL. The complete tree with strain names and genera is provided in Supplementary Figure S3.
Comparative analysis of BGCs variation among genera and strains
To investigate the variation in BGCs content between different genera and strains, we focus on sections from group 2. The first section comprises Ruegeria, Halomonas, Microbulbifer, and Micrococcus (Fig. 3A) and the second section comprises BGCs variation among Tenacibaculum species (Fig. 3B).

Biosynthetic gene cluster (BGC) contents of selected clades from the rpoB phylogeny (Fig. 2). (A) Strains from four genera (Ruegeria, Halomonas, Microbulbifer, and Micrococcus) showing distinct BGC repertoires. (B) Strains of Tenacibaculum species highlighting intra-genus diversity in BGC composition. Each row represents a genome, and colored blocks correspond to antiSMASH-predicted BGC classes. This figure illustrates how BGC diversity correlates with phylogenetic relationships among marine bacterial strains.
Dominance and functional predictions of betalactone and NI-Siderophore BGCs
Among the 15 major classes of BGCs in the 199 genomes, betalactone BGC was found in most genomes (around 79%) of the selected bacterial strains. Betalactone BGC was predicted for different compounds in some strains like Ruegeria arenilitoris HKCCA0515, Shewanella submarina CCUG 71,370, and all Bacillus licheniformis and Micrococcus luteus strains. On the remaining strains, betalactone BGC was not predicted for any compounds. Most betalactone BGCs were predicted for fengycin (41 BGCs), and others were predicted for microansamycin (11 BGCs), glycopeptidolipid (3 BGCs), triacsin C (1 BGC), and corynecin III/corynecin I/corynecin II (1 BGC).
The highest cluster similarity was found with fengycin (53% with the most similar known cluster) in B. licheniformis strains (Supplementary Table S4). These predicted compounds for betalactone belonged to different types, according to antiSMASH. Fengycin and glycopeptidolipid are NRP types, microansamycin is a polyketide, and triacsin C and corynecin III/corynecin I/corynecin II are other types (a cluster containing a secondary metabolite-related protein that does not fit into any other category).
The second dominant BGC was NI-siderophore. In Vibrio campbellii, Alteromonas mediterranea, and Halomonas organivorans, the NI-siderophore was not predicted for any compounds in all strains. In contrast, in species like Photobacterium damselae and M. luteus, the NI-siderophore BGC was predicted in some strains and not in others. However, in M. luteus V017 and M. luteus MT1691313 the NI-siderophore BGC was predicted for FW0622 with 20% cluster similarity. Besides, in Microbulbifer agarilyticus GP101, the NI-siderophore BGC was predicted for putrebactin/avaroferrin with 40% similarity. In Vibrio harveyi, Vibrio alginolyticus, and some P. damselae strains, the NI-siderophore BGC was predicted for vibrioferrin with a similarity of 54%. However, in all B. licheniformis strains, the NI-siderophore BGC was predicted for schizokinen with 60% similarity. On the other hand, in Tenacibaculum singaporense DSM106434, T. mesophilum DSM 13,764, T. mesophilum bac2, and Tenacibaculum aiptasiae a4 strains, the NI-siderophore BGC was predicted for bisucaberin B with 100% similarity. All these compounds are referred to as others according to antiSMASH (Supplementary Table S4). The majority of these compounds are vibrioferrin and schizokinen. Besides, BGCs were cryptic, they were not predicted to any compounds, which could be explored for the production of novel bioactive compounds. Identification of cryptic and rare BGCs opens up avenues to explore hitherto unexploited secondary metabolite pathways.
Structural and genetic variability in Vibrioferrin-Producing BGCs
One of the focal points of the study was the diversity in siderophore BGCs, particularly those encoding vibrioferrin, a siderophore involved in iron acquisition. The investigation revealed significant variation in the amino acid sequences of the NI-siderophore BGCs, which were predicted to produce vibrioferrin among 58 strains belonging to Vibrio harveyi, Vibrio alginolyticus, and Photobacterium damselae, along with two reference genomes of V. alginolyticus and Vibrio parahaemolyticus, which is detailed in Supplementary Figure S5. The alignment of NI-siderophore BGC amino acid sequences (60 sequences in total) showed a low mean pairwise identity of 23.7%, despite all clusters being predicted to produce vibrioferrin based on antiSMASH. This diversity primarily reflects variability in accessory and tailoring genes, while core biosynthetic genes remain highly conserved, with 96.9% and 100% identity for PvsD and PvsB, respectively (Fig. 4). Alignment profiles (Supplementary Figure S5) further revealed additional conserved motifs across NRPS-like condensation domains and transport-associated proteins, indicating that functional cores are preserved even within highly divergent cluster backgrounds.

Amino acid sequence alignment of core NI-siderophore biosynthetic genes between Vibrio parahaemolyticus and Vibrio alginolyticus. (A) Alignment of PvsD (mean length 609 aa) shows 96.9% identity, with 590 identical sites across both sequences. (B) Alignment of PvsB (mean length 610 aa) shows 100% identity, with complete conservation of all residues. These results highlight the high conservation of essential biosynthetic enzymes within vibrioferrin pathways, despite overall variability among NI-siderophore BGCs.
Chemical structural differences in vibrioferrin variants
The reference genomes MIBiG (Minimum Information about a Biosynthetic Gene Cluster) accession numbers are BGC0000946 for Vibrio parahaemolyticus and BGC0000947 for Vibrio alginolyticus. Both genomes exhibit distinct structural features, which are visually compared in Fig. 5A and B, and 5C. The structural differences between compound A and compound B are primarily observed in their stereochemistry and functional groups. The stereochemical arrangement of the ring structures and substituents differs significantly between the two compounds, which could influence their ability to bind specific receptors and their overall biological activity. Moreover, a notable distinction lies in the functional groups present in the middle of their structures. Compound A contains an ester and an amide group separated by two carbons, while compound B features a carboxylic acid and an amide group separated by the same distance. This substitution of an ester group in compound A with a carboxylic acid in compound B is a critical variation that may affect their chemical reactivity, solubility, and potential interactions with iron or other biological molecules. These differences likely contribute to variations in their functional roles and properties, particularly in iron chelation and biological recognition.

Comparison of vibrioferrin biosynthetic features between Vibrio parahaemolyticus (BGC0000946) and Vibrio alginolyticus (BGC0000947). (A–B) Predicted chemical structures of vibrioferrin show conserved core scaffolds with minor side-chain modifications. (c) Amino acid sequence alignment of the entire NI-siderophore BGC reveals low overall conservation (mean length: 3,828 aa; 3,976 codons). Only 407 sites (11.3%) were identical, with a pairwise identity of 11.3% and a BLOSUM62-positive similarity of 20.1%. In the alignment, residues with 100% identity are shown in green, those with 30–99% identity in green–brown, and those below 30% identity in red. These results highlight both the conserved core biosynthetic framework and the extensive sequence divergence that underpins vibrioferrin structural variability.
Clustering analysis of Vibrioferrin-Producing NI-Siderophore BGCs
Clustering analysis of the 60 NI-siderophore BGCs was performed at two similarity cutoffs, 10% and 30%, to explore the diversity of biosynthetic gene clusters across Vibrio harveyi, Vibrio alginolyticus, Vibrio parahaemolyticus, and Photobacterium damselae. At the stricter 10% cutoff, the BGCs were grouped into 12 families across 3 Gene Cluster Families (GCFs). GCF 1 comprised six families (1, 3, 7, 8, 10, and 12), primarily associated with Vibrio alginolyticus. GCF 2 included four families (4, 5, 6, and 11), dominated by Vibrio harveyi. GCF 3 was exclusively represented by Family 0, linked to Photobacterium damselae (Fig. 6A). At this cutoff, the network contained 58 nodes and 396 edges, with a clustering coefficient of 0.862, indicating moderate connectivity and local clustering within the network.
At the more relaxed 30% cutoff (Fig. 6B), the BGCs were grouped into a single GCF consisting of 6 families: Family 0 (Photobacterium damselae), Families 1, 4, and 5 (Vibrio alginolyticus and Vibrio parahaemolyticus), and Families 2, 3, and 6 (Vibrio harveyi). The broader clustering at this cutoff resulted in a network containing 60 nodes and 789 edges, with a clustering coefficient of 0.937, highlighting strong connectivity and higher local clustering.

BiG-SCAPE clustering of NI-siderophore BGCs at two similarity thresholds. (A) At the stringent 10% cutoff, 58 vibrioferrin-producing clusters were divided into 12 families grouped within three distinct gene cluster families (GCFs), reflecting fine-scale diversity among strains. (B) At the broader 30% cutoff, all 60 clusters collapsed into a single GCF comprising six families, consistent with their shared function in vibrioferrin biosynthesis.
Each node represents a predicted NI-siderophore BGC, and edges indicate domain-sequence similarity. Differences between cutoffs illustrate how network resolution affects grouping of related clusters, highlighting both conserved biosynthetic roles and underlying sequence divergence.
Discussion
This study identified 29 different BGC types in 199 marine bacterial genomes, with NRPS, betalactone, NI-siderophore, and NRP-metallophore being the most prevalent. These findings align with previous studies showing that marine bacteria harbor a high diversity of BGCs, many of which contribute to the production of antibiotics, siderophores, and signaling molecules6. Recent studies have shown that marine actinobacteria, particularly those with larger genomes, harbor diverse biosynthetic gene clusters (BGCs), including rare ones, similar to well-known producers like Streptomyces and Salinispora, while certain taxa, such as those within the Micrococcineae suborder, exhibit fewer BGCs, highlighting the untapped potential of rare BGCs in marine bacteria for natural product discovery19.
The phylogenetic analysis of 192 marine bacterial strains revealed two major groups, with one exclusively composed of B. licheniformis and the other containing diverse genera (Shewanella, Photobacterium, Vibrio, Tenacibaculum, etc.). Despite these phylogenetic distinctions, the presence of similar biosynthetic gene clusters (BGCs) across unrelated taxa suggests that horizontal gene transfer (HGT) contributes to BGC distribution. Previous studies have shown that environmental factors, such as nitrogen availability, can influence HGT rates and functional diversity in bacterial communities, potentially shaping the biosynthetic capabilities of marine bacteria20.
Based on the rpoB gene (bootstrap value 0.9), the phylogenetic grouping of the genera in the first section highlights their evolutionary relatedness; however, their distinct BGC profiles reveal functional divergence likely shaped by ecological adaptations. For instance, Halomonas and Microbulbifer harbor NI-siderophore and ectoine BGCs, essential for survival in nutrient-limited and osmotic-stress environments, respectively21,22. At the same time, Micrococcus exhibits a broader metabolic versatility with thiopeptides, RRE-containing, and terpenes BGCs. These differences reflect niche-specific selective pressures, consistent with studies showing that genomic adaptations in marine bacteria are often driven by their environmental characteristics and microbial interactions18.
The Tenacibaculum species (T. singaporense DSM 106434, T. mesophilum bac2, T. mesophilum DSM 13764, and T. aiptasiae a4), which cluster together with a bootstrap value of 1.0, exhibit both conserved and unique BGCs, as shown in Fig. 3B. All species share core BGCs, including NI-siderophore, NRPS, and terpene, reflecting their adaptation to nutrient-limited marine environments. However, T. singaporense DSM 106,434 uniquely harbors lassopeptide BGCs, while T. mesophilum strains possess arylpolyene and lanthipeptide Class II BGCs, indicating functional divergence and niche specialization. This variation highlights the influence of ecological pressures and horizontal gene transfer (HGT) in shaping the accessory genome of marine bacteria23.
The thirteen M. luteus strains, despite their close phylogenetic relationship (bootstrap 1.0), exhibit considerable variation in BGC content, reflecting functional divergence. Conserved BGCs like ectoine and NI-siderophore are widely shared, suggesting their critical role in osmotic stress tolerance and iron acquisition in marine environments. However, some strains uniquely harbor thiopeptides and RRE-containing clusters, indicating specialized secondary metabolite production for niche-specific interactions or microbial competition. Such variation, even among closely related strains, underscores the dynamic nature of the accessory genome, driven by horizontal gene transfer and ecological pressures, as observed in marine bacteria22. The high diversity across different genera and species regarding BGC content underscores the bioprospecting potential of marine bacteria for bioactive compound discovery.
Betalactone was found in 79% of the studied genomes, with most strains predicted to produce fengycin, microansamycin, and glycopeptidolipids. While fengycin is a well-characterized lipopeptide produced by Bacillus species with strong antifungal activity, betalactones are structurally distinct bioactive compounds known for their antimicrobial and cytotoxic properties. Although direct biosynthetic links between betalactones and fengycin or glycopeptidolipids remain unclear, their co-occurrence in many genomes suggests a complementary role in microbial defense and competitive interactions24.
The NI-siderophore BGC was highly variable across strains, with some species lacking it entirely, while others had functional predictions for vibrioferrin, schizokinen, or bisucaberin B. This variability suggests that marine bacteria employ diverse strategies for iron acquisition, influenced by ecological pressures and microbial competition.
The low pairwise identity (23.7%) observed among the 60 NI-siderophore BGCs suggests substantial diversification. This variability may be driven by multiple evolutionary processes. Horizontal gene transfer (HGT) has been shown to play a major role in siderophore and secondary metabolite gene cluster mobility in marine bacteria25. Recombination within modular biosynthetic genes can further increase diversity by shuffling catalytic domains25. Ecological specialization of host strains, particularly in iron-limited marine niches, can also drive diversification and retention of distinct siderophore systems26,27. Together, these processes likely contribute to the remarkable sequence and structural variability observed in vibrioferrin pathways.
Recent studies have shown that coral-associated bacteria can degrade siderophores produced by other microbes, reshaping microbial community dynamics and iron availability28. This underscores the adaptive nature of siderophore biosynthesis, where genetic variability in NI-siderophore BGCs may reflect an evolutionary response to fluctuating iron conditions and microbial interactions in marine environments.
The contrasting results between the 10% and 30% cutoffs underscore the impact of similarity thresholds in delineating biosynthetic diversity. While the 30% cutoff captures conserved core pathways, the 10% cutoff reveals finer substructures that reflect species-specific variations and functional adaptations. These findings highlight the conserved nature of vibrioferrin biosynthesis while emphasizing the role of accessory genes in driving chemical diversity among closely related species. Such diversity aligns with thresholds used in BiG-SCAPE, where clusters sharing less than 30% sequence similarity are often classified as distinct29, reflecting the tool’s threshold for delineating unique clusters30.
Interestingly, the differences in clustering at the 10% cutoff align with slight variations in vibrioferrin chemical structures observed among the analyzed BGCs. These structural differences may stem from variations in accessory genes within the biosynthetic pathways, which could influence the final siderophore product. Such functional diversity may reflect ecological or adaptive significance, enabling species to thrive in varying environmental conditions or competitive niches27. These results reflect finer resolution in the clustering, capturing potential interspecies variations within vibrioferrin biosynthesis pathways. The distinct clustering of P. damselae in GCF 3 suggests unique adaptations in its siderophore biosynthesis. These results emphasize the high conservation of core genes involved in vibrioferrin biosynthesis, consistent with the siderophore’s functional importance in iron acquisition across diverse marine environments. However, the inclusion of multiple species within single families suggests shared biosynthetic features, possibly due to evolutionary convergence or horizontal gene transfer.
The significant differences in amino acid sequences could lead to variations in the Fe-binding affinity and overall functionality of the vibrioferrin produced. Changes in specific amino acids of vibrioferrin can significantly alter its iron-chelating efficiency by modifying key functional groups critical for Fe(III) binding27. These changes may also impact solubility, as polar residues enhance aqueous dispersibility, while nonpolar substitutions reduce it32. Stability under varying pH or oxidative conditions may shift due to differences in the reactivity of the substituted amino acids32. Additionally, amino acid modifications can disrupt recognition by bacterial siderophore transport receptors, affecting uptake efficiency33. Finally, overall biological activity, such as competition with other siderophores, can diminish if structural changes impair iron sequestration or transport mechanisms34. Vibrio species are well-known pathogens of fish, where siderophores such as vibrioferrin play a key role in virulence by facilitating iron acquisition from host environments34,35.
The observed structural diversity of vibrioferrin clusters may therefore reflect not only adaptation to environmental iron limitation but also selective pressures imposed by host–pathogen interactions in aquaculture and natural marine systems.
This study has several limitations. First, the dataset was restricted to marine bacteria isolated from Sea of Oman sediments, which may bias the observed BGC diversity compared with broader ocean microbiomes. Second, biosynthetic gene cluster predictions rely on antiSMASH 7.0, which can misclassify or miss cryptic and hybrid clusters, particularly in draft assemblies. Third, the phylogenetic reconstruction was based on the rpoB gene alone. While this marker provides useful resolution for the taxa included, multilocus or genome-scale phylogenies would offer greater robustness. These constraints do not undermine the main findings but should guide cautious interpretation and motivate future validation with expanded datasets and complementary methods.
Summary
This study revealed significant biosynthetic gene cluster (BGC) diversity in 199 marine bacterial genomes, with NRPS, betalactone, NI-siderophore, and NRP-metallophore being the most prevalent. The phylogenetic analysis showed that some BGCs are highly conserved within genera, while others vary across strains, suggesting that horizontal gene transfer and environmental pressures influence their distribution. A major focus was on vibrioferrin-producing NI-siderophore BGCs, which exhibited high variability in accessory genes despite conserved core genes (PvsD, PvsB). Structural analysis showed that small chemical modifications, such as ester vs. carboxyl group substitutions, could impact iron-chelation efficiency and ecological interactions. Clustering analysis indicated that at a 10% similarity cutoff, vibrioferrin BGCs formed species-specific GCFs, while at 30%, they merged into a single GCF, reflecting a shared biosynthetic framework with species-level variations.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information files. Should raw data files be needed in another format, they are available from the corresponding author upon reasonable request.
References
-
Sridhar, K., Usmani, Z. & Sharma, M. Bioactive formulations in Agri-Food-Pharma: source and applications. Bioengineering 10, 191 (2023).
-
Wang, Y., Shi, Y. N., Xiang, H. & Shi, Y. M. Exploring nature’s battlefield: organismic interactions in the discovery of bioactive natural products. Nat. Prod. Rep. 41, 1630–1651 (2024).
-
Xie, C. L., Xia, J. M., Wang, J. S., Lin, D. H. & Yang, X. W. Metabolomic investigations on Nesterenkonia Flava revealed significant differences between marine and terrestrial actinomycetes. Mar. Drugs. 16, 356 (2018).
-
Barzkar, N., Sukhikh, S. & Babich, O. Study of marine microorganism metabolites: new resources for bioactive natural products. Front. Microbiol. 14, 1285902 (2024).
-
Romsdahl, J. & Wang, C. C. C. Recent advances in the genome mining of Aspergillus secondary metabolites (covering 2012–2018). MedChemComm 10, 840–866 (2019).
-
Chen, J. et al. Global marine microbial diversity and its potential in bioprospecting. Nature 633, 371–379 (2024).
-
Kaari, M., Manikkam, R. & Baskaran, A. Exploring newer biosynthetic gene clusters in marine microbial prospecting. Mar. Biotechnol. N Y N. 24, 448–467 (2022).
-
Tanabe, T. et al. Analysis of the vibrioferrin biosynthetic pathway of vibrio parahaemolyticus. BioMetals https://doi.org/10.1007/s10534-023-00566-x (2023).
-
Challis, G. L. & Hopwood, D. A. Synergy and contingency as driving forces for the evolution of multiple secondary metabolite production by Streptomyces species. Proc. Natl. Acad. Sci. U. S. A. 100, 14555–14561 (2003).
-
Paul, A. & Dubey, R. Characterization of protein involved in nitrogen fixation and Estimation of Co-Factor. Int. J. Curr. Res. Biosci. Plant. Biol. 2, 89–97 (2015).
-
Chen, J. et al. Chemistry and biology of siderophores from marine microbes. Mar. Drugs. 17, 562 (2019).
-
Amin, S. A., Green, D. H., Küpper, F. C. & Carrano, C. J. Vibrioferrin, an unusual marine siderophore: iron Binding, Photochemistry, and biological implications. Inorg. Chem. 48, 11451–11458 (2009).
-
Zhang, X., Baars, O. & Morel, F. M. M. Genetic, structural, and functional diversity of low and high-affinity siderophores in strains of nitrogen fixing azotobacter chroococcum†. Metallomics 11, 201–212 (2019).
-
Beck, C. et al. Activation and identification of a Griseusin cluster in streptomyces sp. CA-256286 by employing transcriptional regulators and Multi-Omics methods. Molecules 26, 6580 (2021).
-
Kloosterman, A. M., Medema, M. H. & van Wezel, G. P. Omics-based strategies to discover novel classes of RiPP natural products. Curr. Opin. Biotechnol. 69, 60–67 (2021).
-
Blin, K. et al. AntiSMASH 7.0: new and improved predictions for detection, regulation, chemical structures and visualisation. Nucleic Acids Res. 51, W46–W50 (2023).
-
Adékambi, T., Drancourt, M. & Raoult, D. The RpoB gene as a tool for clinical microbiologists. Trends Microbiol. 17, 37–45 (2009).
-
Navarro-Muñoz, J. C. et al. A computational framework to explore large-scale biosynthetic diversity. Nat. Chem. Biol. 16, 60–68 (2020).
-
Schorn, M. A. et al. Sequencing rare marine actinomycete genomes reveals high density of unique natural product biosynthetic gene clusters. Microbiology 162, 2075–2086 (2016).
-
Gene horizontal transfers. And functional diversity negatively correlated with bacterial taxonomic diversity along a nitrogen gradient | Npj biofilms and microbiomes. https://www.nature.com/articles/s41522-024-00588-4?utm_source=chatgpt.com(2024)
-
Gao, H., Bian, X. & Editorial Microbial siderophores: Biosynthesis, Regulation, and physiological and ecological impacts. Front Microbiol 13, 1-3 (2022).
-
Penesyan, A., Paulsen, I. T., Kjelleberg, S. & Gillings, M. R. Three faces of biofilms: a microbial lifestyle, a nascent multicellular organism, and an incubator for diversity. NPJ Biofilms Microbiomes. 7, 80 (2021).
-
Medema, M. H. et al. Minimum information about a biosynthetic gene cluster. Nat. Chem. Biol. 11, 625–631 (2015).
-
Yin, Y., Wang, X., Zhang, P., Wang, P. & Wen, J. Strategies for improving Fengycin production: a review. Microb. Cell. Factories. 23, 144 (2024).
-
Medema, M. H. & Osbourn, A. Computational genomic identification and functional reconstitution of plant natural product biosynthetic pathways. Nat. Prod. Rep. 33, 951–962 (2016).
-
D’Onofrio, A. et al. Siderophores from neighboring organisms promote the growth of uncultured bacteria. Chem. Biol. 17, 254–264 (2010).
-
Hider, R. C. & Kong, X. Chemistry and biology of siderophores. Nat. Prod. Rep. 27, 637–657 (2010).
-
Monge-Loría, M., Zhong, W., Abrahamse, N. H., Hartter, S. & Garg, N. Discovery of peptidic siderophore degradation by screening natural product profiles in Marine-Derived bacterial Mono- and cocultures. Biochemistry 64, 634–654 (2025).
-
Sánchez-Hidalgo, M., García, M. J., González, I., Oves-Costales, D. & Genilloud, O. Complete Genome Sequence Analysis of Kribbella sp. CA-293567 and Identification of the Kribbellichelins A & B and Sandramycin Biosynthetic Gene Clusters. Microorganisms 11, 265 (2023).
-
Kim, H., Ahn, J., Kim, J. & Kang, H. S. Metagenomic insights and biosynthetic potential of candidatus Entotheonella symbiont associated with halichondria marine sponges. Microbiol. Spectr. 0, e02355–e02324 (2024).
-
Miethke, M. & Marahiel, M. A. Siderophore-Based iron acquisition and pathogen control. Microbiol. Mol. Biol. Rev. 71, 413–451 (2007).
-
Ahmed, E. & Holmström, S. J. M. Siderophores in environmental research: roles and applications. Microb. Biotechnol. 7, 196–208 (2014).
-
Wandersman, C. & Delepelaire, P. Bacterial iron sources: from siderophores to hemophores. Annu. Rev. Microbiol. 58, 611–647 (2004).
-
Lemos, M. L. & Balado, M. Iron uptake mechanisms as key virulence factors in bacterial fish pathogens. J. Appl. Microbiol. 129, 104–115 (2020).
-
Conrad, R. A., Evenhuis, J. P., Lipscomb, R. S., Birkett, C. & McBride, M. J. Siderophores produced by the fish pathogen flavobacterium columnare strain MS-FC-4 are not essential for its virulence. Appl Environ. Microbiol 88, e00948–e00922 (2022) .
Funding
This research was funded by the Ministry of Higher Education, Research and Innovation, Oman, under the project code BFP/GRG/EBR/23/027. The authors gratefully acknowledge their support.
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
This article contains no studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Al-Siyabi, N., Al-Ansari, A., Abri, M.A.A. et al. Genomic insights into biosynthetic gene cluster diversity and structural variability in marine bacteria. Sci Rep 15, 37644 (2025). https://doi.org/10.1038/s41598-025-21523-3
-
Received:
-
Accepted:
-
Published:
-
DOI: https://doi.org/10.1038/s41598-025-21523-3