
Introduction
A third of disease-causing variants are estimated to disrupt mRNA splicing [1, 2]. Splice-affecting variants are often missed and are under-ascertained in clinical variant databases, as these are not limited to canonical splice sites [3,4,5]. RNA testing is a complementary tool to DNA testing that provides functional evidence and identifies pathogenic events missed by traditional methods [6,7,8,9,10,11,12,13,14]. Within the UK or the Netherlands, some healthcare providers offer specialised RNA studies conducted via targeted reverse transcription PCR (RT-PCR) or RNA-sequencing (RNA-seq). RT-PCR is useful for genes with low expression (<1 transcripts per million [TPM]) and low-level splicing events [15]. However, RT-PCR is a bespoke test for each patient, and is inherently limited by gene annotation, amplicon lengths and prior assumptions on splicing abnormalities. In contrast, RNA-seq is independent of individual patients; it is agnostic to the abnormal transcript(s) and can aid in identifying a variety of events without a priori assumptions. Most RNA-seq studies rely on short-read (SR) RNA-seq, which, although it has some advantages over RT-PCR, cannot produce full-length transcripts, resolve complex regions, and identify certain types of aberrant splicing events such as long stretches of intron retention [16, 17].
As SR RNA-seq is integrated into clinical practice, it is essential to assess the potential benefits of long-read (LR) RNA-seq in this context. Full-length transcripts can improve the assessment of splicing and quantification of transcript abundance. Additionally, longer reads have sufficient genomic context to map challenging regions (high repetition, polymorphism, or low nucleotide diversity), therefore increasing coverage of genes that standard SR sequencing struggles to capture.
Long-read sequencing platforms, including Oxford Nanopore Technologies (ONT) and Pacific Biosciences (PacBio), have significantly improved read accuracy and scalability, although with lower throughput compared to short-read platforms [18]. PacBio’s Revio system claims 99.95% (Q33) read accuracy with read lengths of 15–20 kb, a yield 3-4x higher, and a 15x higher throughput than their previous Sequel Ile system. PacBio’s Kinnex kit, based on the MAS-seq method, concatenates smaller amplicons into larger fragment libraries for higher RNA sequencing throughput [19]. Similarly, ONT offers LR RNA-seq based on cDNA-converted transcripts and native RNA molecules, bypassing the need for reverse transcription or amplification, enabling the detection of full-length transcript isoforms, RNA modifications, and poly(A) tail length from the same molecule. Data storage requirements for LRs tend to be much higher than SRs for the same yield in gigabases. ONT generally requires greater raw data storage and offers lower read accuracy than PacBio at comparable yields.
Given limited published evidence demonstrating the ability of LR RNA-seq to identify pathogenic splicing events missed by short-read approaches [20,21,22], we investigated the utility of PacBio Kinnex RNA-seq (selected for its accuracy and reduced storage demands) to support clinical interpretation and characterise aberrant splicing in rare genetic disorders previously analysed by short-read RNA-seq.
Materials and methods
Patient cohort
Participants were enrolled into the University of Southampton’s Splicing and Disease study with appropriate ethical approval (REC 11/SC/0269, IRAS 49685, ERGO 23056). The sub-cohort used herein is comprised of 23 individuals with a suspected Mendelian disorder assessed by UK clinical genetics services in whom a candidate variant of uncertain significance (VUS) may have been identified through conventional DNA-based testing. SR RNA-seq results for six of these individuals were previously published [14]. Two participants examined at the Department of Clinical Genetics, Erasmus Medical Center (Rotterdam, Netherlands) for whom diagnostic RNA-seq was performed were also enroled (Institutional-review-board MEC-2012-387). SR RNA-seq results for one of these individuals were previously published [23]. Informed consent was obtained, and all individuals or their legal guardians provided written consent to share anonymised clinical and analysis data. Genotype and phenotype details are reported in Supplementary Table 1.
Short-read RNA-seq and analysis
Blood RNA extraction and sequencing were previously reported [14]. SR RNA-seq was generated at NovoGene (Hong Kong) with ≥70 million reads. FASTQ files were aligned to the human genome reference (GRCh38) with GENCODE annotation v38 [24] using STAR aligner v2.6.1c [25]. Gene counts were generated using Salmon v1.6.0 in alignment-based mode with the –gcBias flag [26].
Fibroblast cell culturing, cycloheximide (CHX) treatment, RNA extraction and sequencing were previously reported [13]. CHX is an inhibitor of nonsense mediated decay (NMD) and was employed to prevent degradation of transcripts containing a premature stop (potentially) associated with pathogenic variants or to demonstrate loss-of-function effect at mRNA level for selected variants. SR RNA-seq was generated at GenomeScan, Leiden, the Netherlands, with ≥40 million reads generated per sample. Trimmed reads were aligned to GRCh38 with HISAT2 v2.2.1 [27]. Transcripts per million were extracted using Stringtie v2.2.3 and annotations from GENCODE v38. Splicing junctions were quantified using regtools v1.0.0 [28].
LR sequencing and bioinformatics pipeline
Kinnex RNA-seq data extracted from blood were generated using a Revio system in two batches; each sequencing run lasted 24 h. The first batch of libraries was sequenced at PacBio Menlo Park, CA, while the second batch was sequenced at the PacBio EMEA headquarters in London.
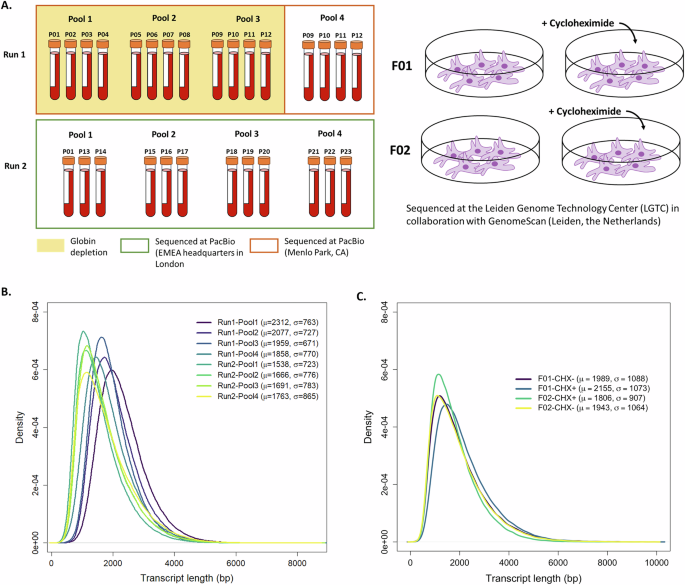
Run 1 was comprised of 16 libraries split across four pools (SMRTcells), sequencing 12 unique samples. Pools 1–3 consisted of four samples per SMRTcell and were globin-depleted. Pool 4 sequenced the same four samples in Pool 3 but with no globin depletion (Fig. 1A). Comparison of transcriptome profiles in Run 1 showed depletion reduced diversity, so the depletion step was omitted in Run 2 (Supplementary Fig. 1). To compensate for the reads that would be taken up by globin genes, the 12 libraries in Run 2 were split across four SMRTcells instead of three. Splicing and expression results for 22 of the 23 patients are reported. One patient was excluded due to lack of VUS.

A Sample preparation and sequencing overview. Left: Blood derived RNA samples were processed across two sequencing runs. Pools 1–3 in Run 1 underwent globin depletion, while Pool 4 in Run 1 and Pools 1–4 in Run 2 did not. Right: Fibroblast cell lines were treated with cycloheximide (+CHX) to stabilise transcripts subject to nonsense-mediated decay. B, C Distribution of read lengths across pools/SMRTcell in blood and fibroblasts, respectively. Mean and standard deviation are shown on the top right.
HiFi reads were processed using the Read Segmentation and Iso-Seq workflow from SMRT Link v13.1. Kinnex arrays were segmented into their constituent cDNA reads using skera v1.2.0. Barcode removal and demultiplexing were performed with Lima v2.10.0. The Iso-Seq bioinformatics toolkit v4.1.2 was used to remove polyA tails, identify artefactual concatamers and cluster sequences, which were mapped against GRCh38_no_alt_analysis_set using pbmm2 1.14.0 in its specialised Iso-Seq mode. Remaining reads were collapsed using the PacBio Iso-Seq toolkit and then classified and filtered using Pigeon v1.2.0.
Fibroblast Kinnex RNA-seq data were generated using a Revio system at the Leiden Genome Technology Center (LGTC) in collaboration with GenomeScan (Leiden, the Netherlands), with sequencing runs of 24 h, and ≥10 million reads per sample. Two libraries were generated per individual, one library from fibroblasts treated with CHX+ and one CHX-, for a total of four libraries across one SMRTcell. Sequencing reads were processed with the Iso-Seq pipeline, specifically Iso-Seq v4.2.0, pbmm2 v1.16.0, and Pigeon v1.3.0, with the same reference and annotation used for the SR data.
RStudio v.4.4.2 was used for data visualisation and statistical analyses [29]. Enrichment analyses were also carried out in RStudio using EnrichR v3.4 [30].
Assessment of aberrant splicing
To determine the functional consequence at a transcript level for each variant, both SR and LR RNA-seq data were loaded into the Integrative Genomics Viewer (IGV) [31], and each variant was examined as previously detailed [14]. Sashimi plots were generated with ggsashimi v.1.1.5 [32] following modification of the CIGAR strings in the aligned bams to ensure compatibility using an in-house script (https://github.com/f-ferraro/Kinnex-ggsashimi).
Aberrant splicing events from short- and long-read RNA-seq were collated into a comprehensive list and cross-checked to determine whether they were detected by the other platform. This approach allowed for a direct comparison of the sensitivity of each technology in identifying aberrant events.
Results
Quality assessment of Kinnex LR RNA-seq data
Blood
Sequencing depth varied across pools and runs, where Run 1 (n = 16 libraries; 4 per SMRTcell) had higher variability compared to Run 2 (n = 12 libraries; 3 per SMRTcell). Each library had ~13.2 million full length non-chimeric (FLNC) reads (range 4.9–24.3 million). Transcript length distributions had a consistent shape across runs and pools with slight shifts in peak positions. Notably, Run 1 pools tended to have higher mean transcript lengths compared to Run 2, with lower means and greater variability (Fig. 1B).
Globin depletion enhances detection of transcripts with lower expression in SR RNA-seq, as it reduces the representation of globin mRNA (~30% of transcripts), freeing up sequencing reads for more relevant transcripts [33,34,35]. To assess its utility on Kinnex data, we sequenced a pool of the same biological samples (n = 4) with and without globin depletion. Along with depletion of globin genes, genome-wide transcript diversity was negatively impacted by this procedure. Saturation curves of known genes and isoforms consistently showed higher number of detected genes and isoforms at the same depth for the undepleted libraries (Supplementary Fig. 1).
Iso-Seq is prone to overestimation of transcript diversity. To mitigate this, we performed Pigeon filtering to remove low-confidence isoforms, resulting in ~30% reduction of detected isoforms, in particular genic and intergenic sections of transcript (Supplementary Fig. 2A). We further validated junction support from LR-RNAseq using SR RNA-seq. On average 51% of novel junctions found within novel in-catalogue and not-in-catalogue transcripts (supported by ≥5 FLNC reads) were supported by the paired SR data (Fig. 2A). Additionally, 54% of novel junctions were present in at least two libraries, where 17,846 of novel junctions were unique to a single library and 508 were shared across all 28 (Fig. 2B).

A Fraction of novel junctions (supported by at least 5 FLNC reads) supported by paired short-read data in blood. Orange shows the number of junctions present in short reads (chr:start:end identical in both datasets). B Novel junctions shared across blood libraries. Each bar shows how many junctions are shared by exactly N libraries (supported by at least 5 FLNC reads). 17,846 junctions unique to a single library, and 508 junctions are shared across all 28. C Isoform distribution across structural categories in blood samples. D Isoform count per gene in blood libraries. Calculation includes only full-splice match isoforms and only those supported by at least 5 FLNC reads. E Novel junctions shared across fibroblast libraries. Each bar shows how many junctions are shared by exactly N libraries (supported by at least 5 FLNC reads). F Fraction of novel junctions (supported by at least 5 FLNC reads) supported by paired short-read data in fibroblasts. Orange shows the number of junctions present in short reads (chr:start:end identical in both datasets). G Distribution of isoforms across structural categories in CHX- and CHX+ fibroblasts. H Isoform count per gene in CHX- and CHX+ fibroblasts. Calculation includes only full-splice match isoforms and only those supported by at least 5 FLNC reads. CHX cycloheximide.
Fibroblasts
Libraries from two patient samples were sequenced on a single SMRTcell (two libraries per sample: cycloheximide CHX+/CHX−) and obtained ~11.7 million FLNC reads per sample (range 11–12.6 million). Transcript-length distributions appeared uniform across the four samples (Fig. 1C), comparable to that observed for blood (Fig. 1B). Similarly, filtering of the detected isoforms by Pigeon resulted in the greatest reduction of intergenic, genic, and antisense categories (Supplementary Fig. 2B). Splice junction validation revealed that 45% of novel junctions (supported by ≥5 FLNC) were shared across at least two libraries (Fig. 2E). Paired SR RNA-seq libraries supported, on average, 82% and 75% of junctions in CHX- and CHX+ fibroblasts, respectively (Fig. 2F).
High coverage and consistent quantification across short- and long-read RNA-seq
Blood
The Iso-Seq pipeline detected a minimum of 13,826 genes (31,132 isoforms) and a maximum of 18,642 genes (52,128 isoforms) annotated in GENCODE (Supplementary Fig. 3A, B) across the 28 libraries. To evaluate the diagnostic potential of LR RNA-seq, disease gene pick-up rate was also assessed, and at least 50% of both OMIM [36] and PanelApp [37] genes were detected across all libraries (Supplementary Fig. 3C, D). Transcript abundance estimates derived from SR and LR sequencing show strong concordance with a Pearson coefficient of 0.86 (Supplementary Fig. 3E), indicating high agreement across platforms.
Fibroblasts
In untreated fibroblasts, the Iso-Seq pipeline detected a minimum of 14,495 annotated genes, increasing to 15,994 in CHX-treated fibroblasts (Supplementary Fig. 4A, B). We observed that 64.6% of the genes detected in one untreated cell line were also detected in the other cell line, and this percentage increased to 69.4% in CHX+ cells. Biotype annotation of the 3541 genes detected only in either of the CHX+ fibroblast lines showed that these genes were mainly lncRNAs (36.1%), followed by protein coding (29.1%) and readthrough transcripts (24.4%) (Supplementary Fig. 4C). Further enrichment analysis in Enrichr, did not reveal a statistically significant enrichment in any of the sets included in the three Gene Ontology classes (cellular component, molecular function, biological process), Human phenotype ontology, nor OMIM.
Detected transcripts also increased after CHX treatment, likely related to transcripts normally undergoing nonsense mediated mRNA decay (Supplementary Fig. 4D, E). Importantly, the number of genes/transcripts discoverable appeared to be already saturated at the achieved read depth (Supplementary Fig. 4A, B, D, E). At equal sequencing depth, blood samples express, on average, a higher number of GENCODE-annotated genes and transcripts (Supplementary Fig. 5A, B). However, fibroblasts encompass a greater number of disease-relevant genes compared to blood [13] (Supplementary Fig. 4F). Correlation between level of expression estimated with short- and long-reads in fibroblasts was lower than what was observed in blood but still high with a Pearson coefficient of 0.646 and 0.669 in CHX+ and CHX- lines, respectively (Supplementary Fig. 4G).
Assessment of identified transcripts
Blood
Using isoforms supported by ≥5 FLNC reads, the largest structural category annotated by Pigeon was incomplete-splice match (ISM), followed by full-splice match (FSM) isoforms. Among FSM isoforms, just over 30% of detected genes were represented by a single isoform and another 30% by 2–3 isoforms. Approximately 15% of genes showed high isoform diversity represented by six or more isoforms (Fig. 2C, D).
Fibroblasts
Most of the isoforms detected in the CHX- fibroblasts were annotated as FSM and ISM categories, similar to blood (Fig. 2G). CHX treatment led to a higher number of transcripts classified as novel not in catalogue, i.e. transcripts that use novel donors and/or acceptors not present in GENCODE annotation (Fig. 2G). Across the four cell lines, most genes had 2–3 distinct isoforms, while a similar proportion of genes were represented by a single or ≥6 isoforms (Fig. 2H).
LR RNA-seq improves resolution and variant interpretation in select cases
Across SR and LR RNA-seq, 27 distinct splicing outcomes were identified across all patient samples. LR RNA-seq detected: 18 aberrant splicing events linked to a variant, three normal splicing outcomes, two splicing events supported by a low number of reads and failed to capture four outcomes (Table 1). Three of the four events missed by LR were due to low/no coverage of the gene (PHF8, COX7B and KIAA0825); PHF8 was likely attributed to a poor sequencing run. The transcript abundance of COX7B decreased approximately 14-fold in blood and 5-fold in fibroblast from SR to LR sequencing, however, the underlying reason remains unclear. The remaining two events were not detected, likely due to their inherently low expression levels.
Where gene coverage was sufficient, LRS confirmed aberrant splicing found previously, and in eight cases (P01, P02, P09, P13, P15, P20, P23), LRS detected additional effects on transcripts, enhancing interpretation of variants and either helping resolve pathogenicity or providing additional biological insights into the variant’s effect. This included identification of intron retention events with more confidence (P01, P13), i.e. did not need to validate with RT-PCR or intronic reads not present in other samples, phasing variants of interest (P15, F01), and quantifying both known and novel transcripts (P02, P09, P20, P23, F01). Detailed figures/results for all samples with aberrant splicing are detailed in Supplemental Results, and we highlight a few examples in the next sections.
NM_001011.4(RPS7):c.507+3A > G skews expression toward unannotated intron retained transcript
RPS7 encodes a ribosomal protein essential for ribosome biogenesis and function; variants in this gene have been associated with Diamond-Blackfan anaemia [38, 39], consistent with the patient’s (P09) phenotype. SR sequencing suggested potential intron 6 retention; however, due to presence of intronic reads in controls results were inconclusive (Fig. 3A). Proband P09 had also previously undergone RT-PCR, which yielded normal results [15]. Salmon was used to quantify the transcript abundance of the MANE Select transcript (ENST00000645674.2/NM_001011.4) in SR RNA-seq. When compared to 87 unrelated samples, quantification of ENST00000645674.2/NM_001031710.3 in SRs suggested a slight increase in expression of this transcript (Fig. 3B). LR RNA-seq confirmed the variant did lead to intron 6 retention, however, intron 6 retention was also observed in controls, explaining RT-PCR results. Both patient and control samples exhibited two transcript isoforms: one retaining intron 6 and the MANE Select transcript (Fig. 3A, C, D). In controls, the intron-retained isoform was present at the same or lower levels than the MANE Select transcript. In the patient this intron retained transcript was ~40 times more abundant. This significant shift in transcript ratio could lead to RPS7 protein deficiency, likely due to competition of the aberrant isoform. While SRs were able to pick up the potential intron retention, the difference in isoform usage was only made clear with the long-read data.

A Sashimi plot comparing splicing patterns between proband and controls. Both short-read (SR) and long-read (LR) data are shown. B TPM values of ENST00000645674.2 (RPS7) calculated by Salmon in short read data across 88 samples. Transcripts per million (TPM) value for proband highlighted in red. C Proportion of MANE select transcript (blue) vs intron 6 – retained (orange) transcript across blood libraries. Red arrow points to bar reflecting the proband. D Visualisation of transcripts identified in proband (red), P06 as control (green) and annotated reference (blue).
Homozygous NM_001031710.3(KLHL7):c.936+3_936+22del variant causes leaky splicing
Biallelic KLHL7 variants are known to cause PERCHING syndrome, a rare multisystemic developmental disorder [40, 41]. Previous RT-PCR assay identified exon skipping caused by NM_001031710.3:c.936+3_936+22del variant, leading to variant reclassification. LR RNA-seq confirmed these results but also identified a complete splice match to the MANE Select transcript (NM_001031710.3), see Supplemental Results Fig. 2. While a low-level event, no normal transcripts were expected as this is a homozygous variant, indicating leaky splicing. This patient has most of the constituent features of PERCHING syndrome, as previously described [41] (patient 6), not consistent with an attenuated phenotype. However, this patient is ambulant and still living at age 17 and perhaps not as profoundly disabled as some of the more severe cases.
LRS facilitates variant phasing
Proband P15 was initially referred for splicing assessment of variant NM_001378452.1:c.1712A > G in exon 17 of ITPR1. Both SR and LR RNA-seq showed the presence of an alternative donor site in intron 17, leading to an out-of-frame insertion (Supplemental Results – Fig. 10). After receiving the LR results, the referring diagnostic team contacted us to investigate phasing for a second variant in exon 23 (NM_001378452.1:c.2659C > T), previously unreported to us. LR RNA-seq successfully provided phasing information, revealing that the second variant was in trans with the initial variant. This was a complex clinical case where the clinical team ultimately reported the two variants as VUSs, mainly because there is currently insufficient evidence to support that null alleles are a mechanism of disease in autosomal recessive ITPR1-related disorders.
Retrotransposon-induced isoform switch in TCOF1
Loss-of-function variants in TCOF1 are the most common cause of Treacher-Collins syndrome [42], and recently, a retrotransposon insertion as novel pathogenic mechanism in this gene was reported [23]. SR RNA-seq outlier-analysis suggested an isoform switch from the canonical TCOF1 transcript (ENST00000643257.2/NM_001371623.1) to a shorter transcript (ENST00000513538.2, Fig. 4A, B), lacking a nucleolar localisation signal expected to impair ribosome biosynthesis [43]. SR RNA-seq suggested skewing of one allele (allele frequency 0.33, based on four heterozygous SNVs), partially restored by CHX (allele frequency 0.39). However, no pathogenic event was identified. Characterization by ONT LR genome sequencing and direct RNA-seq [23] showed insertion of a SINE-VNTR-Alu (SVA) retrotransposon into TCOF1 intron 17. The SVA is partially exonized, inducing an early termination codon leading to nonsense-mediated mRNA decay.

A Z-score plot showing the relative expression level of the exons of TCOF1 estimated from SR RNA-seq. B Sashimi plot showing level of expression of exons and splicing junctions observed in SR and LR RNA-seq data from F01 and an unrelated control. CHX+ and CHX- have been summed up. C Phased FLNC data from F01. The SVA-carrying allele shows preferential expression of the shorter TCOF1 isoform compared to the reference allele. Inset show exonized SVA insertion in the longer transcripts. D Schematic of genomic coordinates and transcripts in the locus, and of the SVA insertion.
We performed Kinnex LR RNA-seq (Fig. 4B–D) in fibroblasts from patient F01, carrying this pathogenic event. Based on full-splice and incomplete-splice match transcripts, TCOF1 expression was TPM 12.7 in CHX- and TPM 8.3 in CHX+ (compared to TPM 13.4 and 6.36 in CHX- and CHX+ fibroblasts from F02, respectively). Manual inspection of the FLNC of CHX- fibroblasts from F01 and phasing of the reads by heterozygous SNVs, showed the isoform switch induced by the SVA: while the reference allele produced mostly long TCOF1 isoforms (TPM 8.7 long isoform vs 0.9 short isoform, similarly to what was observed in the two haplotypes in F02 average TPM 2.7 vs 0.5), the allele carrying the SVA produced mostly shorter TCOF1 isoforms (TPM 1 vs 2.1) (Fig. 4C). This was confirmed by the data from the CHX+ fibroblast RNA (TPM 6 vs 0.24; SVA-allele TPM 1.1 vs 0.9), where partial exonization of the SVA in 12 of the 16 reads supporting a long isoform were also detected (Fig. 4D). Notably, no SVA-containing isoform was reconstructed by the Iso-Seq pipeline, although the event was clearly evident in the raw sequencing data. While limitations in the bioinformatic analysis of LR RNA-seq still exist, the raw data completely resolved and phased full-length transcripts that included the SVA insertion that went undetected using SR RNA-seq.
Deletion of YY1 distal exons induces transcript readthrough
Loss-of-function YY1 variants cause autosomal dominant Gabriele-de Vries syndrome [44]. SR RNA-seq identified YY1 as an expression outlier in individual F02, presenting with phenotype consistent with the Gabriele-de Vries syndrome. Manual inspection of the SR RNA-seq data indicated read-through transcripts involving the locus of two genes downstream of YY1, i.e., SLC25A29 and SLC25A47 (Supplemental Results Fig. 18A, B). Reanalysis of exome data identified a heterozygous deletion of the last 3 exons of YY1 (NM_003403.5) and exon 2 and 3 of the neighbouring gene SLC25A29 (NM_001039355.3) (Supplemental Results Fig. 18C). LR RNA-seq (CHX+ and CHX- fibroblasts) from this patient, confirmed the presence of YY1 transcripts including portions of one of the SLC genes or both (Supplemental Results Fig. 18B). LR data also confirmed partial intron retention around the annotated exons SLC25A29 included in the read-through transcripts. Iso-Seq analysis showed reduced YY1 expression in CHX-untreated fibroblasts from F02 (22.2 TPM) compared to F01 (51.7 TPM). It also reconstructed a YY1 read-through into the SLC25A29 locus in both samples but did not detect isoforms involving SLC25A47, as these occurred in fewer than two FLNC reads.
Discussion
We applied PacBio full-length Kinnex RNA-seq in blood (±globin depletion) and fibroblasts (±cycloheximide) to assess throughput, compare its performance to SR technology, and evaluate its utility for clinical interpretation of variants referred for RNA-seq studies. Kinnex RNA-seq confirmed previous SR RNA-seq findings in blood and fibroblast samples. This new technology enhanced characterisation and facilitated interpretation of events that were only partially detectable by SR RNA-seq. In some cases, reanalysis of the SR RNA-seq using insights gained from LR sequencing can also improve interpretation.
Limitations of LR RNA-seq
While our study demonstrates the potential of PacBio Kinnex LR RNA-seq for variant interpretation, several limitations remain. Despite a strong correlation between short- and long-reads (Pearson coefficients of 0.86 in blood; 0.66 in fibroblasts), coverage was lower for a subset of genes. In blood, pathway analysis revealed that these genes were predominantly immune-related, including T-cell receptor variable and immunoglobulin genes. For example, PHF8 and COX7B had TPM values of 7.5 and 4.4, respectively, in SR data, compared to 3.2 and 0.3 in blood LR data, impairing variant assessment. In contrast, fibroblasts provided better resolution for these genes, with median TPMs of 30 and 27 in SR data, and 8 and 11 in LR data, respectively.
Interestingly, discrepancies in the predicted expression values were observed across both tissues. These may be attributed to differences in read depth, suggesting deeper sequencing (achievable with longer sequencing times) might be required to fully capture transcript diversity. Additionally, some transcripts may be subject to nonsense-mediated decay, further reducing their detectability and requiring more sensitive approaches, as might have been the case with the SETD5 canonical splice site variant (P19).
Current bioinformatics pipelines for quantifying aberrant transcripts and identifying novel events are not yet optimised for LR RNA-seq [45]. Given the abundance of novel transcripts detected, distinguishing disease-relevant events from those arising from transcript diversity yet to be characterised or artefacts remains a challenge. We demonstrate that stricter filtering strategies (e.g. FLNC support) on top of specialised tools (e.g. Pigeon) can mitigate this issue. While half of novel junctions were supported by SR RNA-seq, the remaining junctions may still represent biologically relevant events, particularly in well-expressed genes where short-read mapping is difficult. Future work integrating proteomic data will be essential to assess which isoforms are translated and potentially functional. Additionally, validation across tissues could help distinguish ubiquitous transcripts from tissue-specific or disease-relevant splicing events. These complementary approaches will be key to realising the full diagnostic potential of LR RNA-seq.
Technical considerations
Although our assessment of globin depletion and cycloheximide treatment was based on eight and four libraries, respectively, our data offer valuable insights into optimising experimental design. These include selecting appropriate library prep, tissue types, and determining the number of samples per SMRTcell to best adapt to the needs of the study. Strikingly, while globin depletion in blood is associated with higher transcript diversity in SR RNA-seq [33, 46], the opposite was observed with the Kinnex protocol. Variability in coverage was evident in the first run, and although we cannot attribute the variability to the depletion process, the non-depleted pools performed better. In fibroblasts, cycloheximide treatment enhances transcript and gene diversity using Kinnex, in line with observations in SR.
Enhanced transcript resolution with LR RNA-seq
LR RNA-seq successfully identified additional events and provided a clearer understanding of aberrant splicing and isoform usage. Compared to SR RNA-seq, LR RNA-seq can better detect intron retention events with less noise and events that span multiple exons. Moreover, it enables full-length transcript detection, quantification of transcript diversity, and allele-specific expression quantification.
At least 10% of detected transcripts were novel, not in the catalogue, and this could have significant implications when standard annotations are used to quantify gene expression. This was exemplified with the quantification of RPS7 by Salmon in proband P09, where no annotated transcripts include the retention of intron 6. This demonstrated how relying solely on SR RNA-seq quantification of specific isoforms can be misleading. There is large variability in the quantification of this transcript using SR RNA-seq, and only with the LR RNA-seq is the differential isoform usage apparent.
LR RNA-seq also revealed leaky splicing in KLHL7. While not altering clinical interpretation of the variant, leaky splicing could explain variability across disorders caused by variants in the same gene. Similarly, LR data for the MLH1 variant (Supplemental Results – Fig. 15) also detected additional exon 9–10 skipping at similar ratios to the exon 9 skipping initially detected in SR data. These observations underscore the added value of LR RNA-seq in uncovering subtle splicing events that may be contributing to disease heterogeneity [47, 48].
Furthermore, having full-length transcripts, or at least very long reads, enables transcript phasing as evidenced in probands P15 and F01. Therefore, broadening access to testing for patients where parental DNA/RNA is unavailable, where we can still assess cis/trans expression of variants, if variants are not spliced out.
Clinical integration and future directions
LR RNA-seq is a powerful and increasingly accessible tool for detecting aberrant splicing and gene expression in rare disease diagnostics. Recent advances have significantly improved throughput, though costs remain higher than SR sequencing, which also benefits from well-established pipelines. For cases with a variant of uncertain significance in a well-expressed gene (TPM ≥ 5) and outside repetitive regions, SR RNA-seq remains a practical first-line test. However, LR RNA-seq provides critical transcript-level resolution and can refine or validate SR findings. Alternatively, targeted deep long-read sequencing may also be applied to specific genes with a known VUS [49], though throughput is a consideration.
In the UK, SR RNA-seq is only just being introduced into clinical practice, so it will take time before LR RNA-seq becomes part of the NHS diagnostic framework. Similarly, in the Netherlands, diagnostic SR RNA-seq is available in only a few clinical centres. The next step is to develop LR methods for identifying relevant events in patients without a VUS, already being explored with SR RNA-seq [6,7,8, 13, 14], helping to determine which patients and samples would benefit most from this technology.
While LR RNA-seq has clear potential in rare disease diagnostics, its integration into clinical workflows is actively progressing and will benefit from continued optimisation of analytical pipelines, cost reduction and standardisation.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Code availability
We made use of publicly available tools listed in the methods with default parameters. We recommend using the latest update of these tools.
References
-
Baralle D, Lucassen A, Buratti E. Missed threads. The impact of pre-mRNA splicing defects on clinical practice. EMBO Rep. 2009;10:810–6. https://doi.org/10.1038/embor.2009.170.
-
Lim KH, Ferraris L, Filloux ME, Raphael BJ, Fairbrother WG. Using positional distribution to identify splicing elements and predict pre-mRNA processing defects in human genes. Proc Natl Acad Sci USA. 2011;108:11093–8. https://doi.org/10.1073/pnas.1101135108.
-
Blakes AJM, Wai HA, Davies I, Moledina HE, Ruiz A, Thomas T, et al. A systematic analysis of splicing variants identifies new diagnoses in the 100,000 Genomes Project. Genome Med. 2022;14:79. https://doi.org/10.1186/s13073-022-01087-x.
-
Sullivan PJ, Quinn JMW, Ajuyah P, Pinese M, Davis RL, Cowley MJ. Data-driven insights to inform splice-altering variant assessment. Am J Hum Genet. 2025;112:764–78. https://doi.org/10.1016/j.ajhg.2025.02.012.
-
Ellingford JM, Ahn JW, Bagnall RD, Baralle D, Barton S, Campbell C, et al. Recommendations for clinical interpretation of variants found in non-coding regions of the genome. Genome Med. 2022;14:73. https://doi.org/10.1186/s13073-022-01073-3.
-
Cummings BB, Marshall JL, Tukiainen T, Lek M, Donkervoort S, Foley AR, et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci Transl Med. 2017;9:eaal5209. https://doi.org/10.1126/scitranslmed.aal5209.
-
Kremer LS, Bader DM, Mertes C, Kopajtich R, Pichler G, Iuso A, et al. Genetic diagnosis of Mendelian disorders via RNA sequencing. Nat Commun. 2017;8:15824. https://doi.org/10.1038/ncomms15824.
-
Frésard L, Smail C, Ferraro NM, Teran NA, Li X, Smith KS, et al. Identification of rare-disease genes using blood transcriptome sequencing and large control cohorts. Nat Med. 2019;25:911–9. https://doi.org/10.1038/s41591-019-0457-8.
-
Murdock DR, Dai H, Burrage LC, Rosenfeld JA, Ketkar S, Müller MF, et al. Transcriptome-directed analysis for Mendelian disease diagnosis overcomes limitations of conventional genomic testing. J Clin Investig. 131:e141500. https://doi.org/10.1172/JCI141500.
-
Gonorazky HD, Naumenko S, Ramani AK, Nelakuditi V, Mashouri P, Wang P, et al. Expanding the boundaries of RNA sequencing as a diagnostic tool for rare Mendelian disease. Am J Hum Genet. 2019;104:466–83. https://doi.org/10.1016/j.ajhg.2019.01.012.
-
Lee H, Huang AY, Wang L-K, Yoon AJ, Renteria G, Eskin A, et al. Diagnostic utility of transcriptome sequencing for rare Mendelian diseases. Genet Med. 2020;22:490–9. https://doi.org/10.1038/s41436-019-0672-1.
-
Yépez VA, Gusic M, Kopajtich R, Mertes C, Smith NH, Alston CL, et al. Clinical implementation of RNA sequencing for Mendelian disease diagnostics. Genome Med. 2022;14:38. https://doi.org/10.1186/s13073-022-01019-9.
-
Dekker J, Schot R, Bongaerts M, de Valk WG, van Veghel-Plandsoen MM, Monfils K, et al. Web-accessible application for identifying pathogenic transcripts with RNA-seq: increased sensitivity in diagnosis of neurodevelopmental disorders. Am J Hum Genet. 2023;110:251–72. https://doi.org/10.1016/j.ajhg.2022.12.015.
-
Jaramillo Oquendo C, Wai HA, Rich WI, Bunyan DJ, Thomas NS, Hunt D, et al. Identification of diagnostic candidates in Mendelian disorders using an RNA sequencing-centric approach. Genome Med. 2024;16:110. https://doi.org/10.1186/s13073-024-01381-w.
-
Wai HA, Constable M, Drewes C, Davies IC, Svobodova E, Dempsey E, et al. Short amplicon reverse transcription-polymerase chain reaction detects aberrant splicing in genes with low expression in blood missed by ribonucleic acid sequencing analysis for clinical diagnosis. Hum Mutat. 2022;43:963–70. https://doi.org/10.1002/humu.24378.
-
Broseus L, Ritchie W. Challenges in detecting and quantifying intron retention from next generation sequencing data. Comput Struct Biotechnol J. 2020;18:501–8. https://doi.org/10.1016/j.csbj.2020.02.010.
-
David JK, Maden SK, Wood MA, Thompson RF, Nellore A. Retained introns in long RNA-seq reads are not reliably detected in sample-matched short reads. Genome Biol. 2022;23:240. https://doi.org/10.1186/s13059-022-02789-6.
-
Kovaka S, Ou S, Jenike KM, Schatz MC. Approaching complete genomes, transcriptomes and epi-omes with accurate long-read sequencing. Nat Methods. 2023;20:12–6. https://doi.org/10.1038/s41592-022-01716-8.
-
Al’Khafaji AM, Smith JT, Garimella KV, Babadi M, Popic V, Sade-Feldman M, et al. High-throughput RNA isoform sequencing using programmed cDNA concatenation. Nat Biotechnol. 2024;42:582–6. https://doi.org/10.1038/s41587-023-01815-7.
-
Dainis A, Tseng E, Clark TA, Hon T, Wheeler M, Ashley E. Targeted long-read RNA sequencing demonstrates transcriptional diversity driven by splice-site variation in MYBPC3. Circ Genom Precis Med. 2019;12:e002464. https://doi.org/10.1161/CIRCGEN.119.002464.
-
Sedaghat-Hamedani F, Rebs S, Kayvanpour E, Zhu C, Amr A, Müller M, et al. Genotype complements the phenotype: identification of the pathogenicity of an LMNA splice variant by nanopore long-read sequencing in a large DCM family. Int J Mol Sci. 2022;23:12230 https://doi.org/10.3390/ijms232012230.
-
Kawakami R, Hiraide T, Watanabe K, Miyamoto S, Hira K, Komatsu K, et al. RNA sequencing and target long-read sequencing reveal an intronic transposon insertion causing aberrant splicing. J Hum Genet. 2024;69:91–9. https://doi.org/10.1038/s10038-023-01211-8.
-
Ferraro F, Kühn N, Rots D, van der Linde HC, Mohseni B, van Unen L, et al. Long-read DNA and RNA sequencing reveal an intronic retrotransposon insertion in TCOF1 causing Treacher Collins syndrome. 2025. https://doi.org/10.1101/2025.04.24.25326319.
-
Frankish A, Carbonell-Sala S, Diekhans M, Jungreis I, Loveland JE, Mudge JM, et al. GENCODE: reference annotation for the human and mouse genomes in 2023. Nucleic Acids Res. 2023;51:D942–9. https://doi.org/10.1093/nar/gkac1071.
-
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. https://doi.org/10.1093/bioinformatics/bts635.
-
Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 2017;14:417–9. https://doi.org/10.1038/nmeth.4197.
-
Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37:907–15. https://doi.org/10.1038/s41587-019-0201-4.
-
Cotto KC, Feng Y-Y, Ramu A, Richters M, Freshour SL, Skidmore ZL, et al. Integrated analysis of genomic and transcriptomic data for the discovery of splice-associated variants in cancer. Nat Commun. 2023;14:1589. https://doi.org/10.1038/s41467-023-37266-6.
-
Posit team. RStudio: Integrated Development Environment for R. Posit Software, PBC; 2025.
-
Xie Z, Bailey A, Kuleshov MV, Clarke DJB, Evangelista JE, Jenkins SL, et al. Gene set knowledge discovery with EnrichR. Curr Protoc. 2021;1:e90. https://doi.org/10.1002/cpz1.90.
-
Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–6. https://doi.org/10.1038/nbt.1754.
-
Garrido-Martín D, Palumbo E, Guigó R, Breschi A. ggsashimi: sashimi plot revised for browser- and annotation-independent splicing visualization. PLoS Comput Biol. 2018;14:e1006360 https://doi.org/10.1371/journal.pcbi.1006360.
-
Mastrokolias A, den Dunnen JT, van Ommen GB, t Hoen PA, van Roon-Mom WM. Increased sensitivity of next generation sequencing-based expression profiling after globin reduction in human blood RNA. BMC Genom. 2012;13:28. https://doi.org/10.1186/1471-2164-13-28.
-
Harrington CA, Fei SS, Minnier J, Carbone L, Searles R, Davis BA, et al. RNA-Seq of human whole blood: evaluation of globin RNA depletion on Ribo-Zero library method. Sci Rep. 2020;10:6271. https://doi.org/10.1038/s41598-020-62801-6.
-
Sheerin D, Lakay F, Esmail H, Kinnear C, Sansom B, Glanzmann B, et al. Identification and control for the effects of bioinformatic globin depletion on human RNA-seq differential expression analysis. Sci Rep. 2023;13:1859. https://doi.org/10.1038/s41598-023-28218-7.
-
Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015;43:D789–798. https://doi.org/10.1093/nar/gku1205.
-
Martin AR, Williams E, Foulger RE, Leigh S, Daugherty LC, Niblock O, et al. PanelApp crowdsources expert knowledge to establish consensus diagnostic gene panels. Nat Genet. 2019;51:1560–5. https://doi.org/10.1038/s41588-019-0528-2.
-
Volejnikova J, Vojta P, Urbankova H, Mojzíkova R, Horvathova M, Hochova I, et al. Czech and Slovak Diamond-Blackfan Anemia (DBA) Registry update: clinical data and novel causative genetic lesions. Blood Cells Mol Dis. 2020;81:102380. https://doi.org/10.1016/j.bcmd.2019.102380.
-
Smetanina NS, Mersiyanova IV, Kurnikova MA, Ovsyannikova GS, Hachatryan LA, Bobrynina VO, et al. Clinical and genomic heterogeneity of Diamond Blackfan anemia in the Russian Federation. Pediatr Blood Cancer. 2015;62:1597–600. https://doi.org/10.1002/pbc.25534.
-
Angius A, Uva P, Buers I, Oppo M, Puddu A, Onano S, et al. Bi-allelic mutations in KLHL7 cause a crisponi/CISS1-like phenotype associated with early-onset retinitis pigmentosa. Am J Hum Genet. 2016;99:236–45. https://doi.org/10.1016/j.ajhg.2016.05.026.
-
Bruel A-L, Bigoni S, Kennedy J, Whiteford M, Buxton C, Parmeggiani G, et al. Expanding the clinical spectrum of recessive truncating mutations of KLHL7 to a Bohring-Opitz-like phenotype. J Med Genet. 2017;54:830–5. https://doi.org/10.1136/jmedgenet-2017-104748.
-
Ulhaq ZS, Nurputra DK, Soraya GV, Kurniawati S, Istifiani LA, Pamungkas SA, et al. A systematic review on Treacher Collins syndrome: correlation between molecular genetic findings and clinical severity. Clin Genet. 2023;103:146–55. https://doi.org/10.1111/cge.14243.
-
Ciccia A, Huang J-W, Izhar L, Sowa ME, Harper JW, Elledge SJ. Treacher Collins syndrome TCOF1 protein cooperates with NBS1 in the DNA damage response. Proc Natl Acad Sci USA. 2014;111:18631–6. https://doi.org/10.1073/pnas.1422488112.
-
Gabriele M, Vulto-van Silfhout AT, Germain P-L, Vitriolo A, Kumar R, Douglas E, et al. YY1 haploinsufficiency causes an intellectual disability syndrome featuring transcriptional and chromatin dysfunction. Am J Hum Genet. 2017;100:907–25. https://doi.org/10.1016/j.ajhg.2017.05.006.
-
Pardo-Palacios FJ, Wang D, Reese F, Diekhans M, Carbonell-Sala S, Williams B, et al. Systematic assessment of long-read RNA-seq methods for transcript identification and quantification. Nat Methods. 2024;21:1349–63. https://doi.org/10.1038/s41592-024-02298-3.
-
Shin H, Shannon CP, Fishbane N, Ruan J, Zhou M, Balshaw R, et al. Variation in RNA-seq transcriptome profiles of peripheral whole blood from healthy individuals with and without globin depletion. PLoS ONE. 2014;9:e91041 https://doi.org/10.1371/journal.pone.0091041.
-
Elasal MA, Khateb S, Panneman DM, Roosing S, Cremers FPM, Banin E, et al. A leaky deep intronic splice variant in CLRN1 is associated with non-syndromic retinitis pigmentosa. Genes. 2024;15:1363 https://doi.org/10.3390/genes15111363.
-
Nakagama Y, Hamanaka K, Mimaki M, Shintaku H, Miyatake S, Matsumoto N, et al. Leaky splicing variant in sepiapterin reductase deficiency. Neurol Genet. 2019;5:e319. https://doi.org/10.1212/NXG.0000000000000319.
-
Walsh T, Casadei S, Munson KM, Eng M, Mandell JB, Gulsuner S, et al. CRISPR-Cas9/long-read sequencing approach to identify cryptic mutations in BRCA1 and other tumour suppressor genes. J Med Genet. 2021;58:850–2. https://doi.org/10.1136/jmedgenet-2020-107320.
Acknowledgements
The authors thank all patients and families taking part in this research and all clinicians involved. We thank all staff from regional genetics services who recruited patients: SanSan Htun, Tessy Thomas, Andrew Douglas, Claire G Salter (https://orcid.org/0000-0002-2494-1644), Lucy Side (https://orcid.org/0000-0003-4476-8735), Mary O’Driscoll (https://orcid.org/0000-0002-7119-7571), Mark Hamilton, Dr Nayana Lahiri, Sahar Mansour, Stephanie Grenville-Heygate (https://orcid.org/0000-0003-1516-3016), M Suri, Ed Blair, Nicola Foulds (https://orcid.org/0000-0002-5779-0096), Jessica Radley (https://orcid.org/0000-0002-0776-0091), Helen Stewart, Caroline Pottinger, Vivienne McConnell, Ajoy Sarkar. The authors acknowledge the use of the IRIDIS High Performance Computing Facility and associated support services at the University of Southampton in the completion of this work. Preparation of this manuscript was supported by C.J.O attending the University of Southampton Faculty of Medicine/Faculty of Environmental and Life Sciences Writing Retreat, July 2024.
Funding
The D.B. Laboratory is supported by the National Institute for Health and Care Research Professorship (RP-2016-07-011) and the NIHR Senior Investigator Award NIHR303895.
Ethics declarations
Competing interests
L.T., H.D., S.H., and J.Z. are current or former employees and shareholders of Pacific Biosciences stock.
Ethical approval
Informed consent was obtained, and all individuals or their legal guardians provided written consent to share anonymized clinical and analysis data. Participants were enroled into the University of Southampton’s Splicing and Disease study with appropriate ethical approval (REC 11/SC/0269, IRAS 49685, ERGO 23056). Use of genome-wide technologies for diagnostic purposes was previously approved for individuals from the Erasmus MC (Institutional-review-board MEC-2012-387).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jaramillo Oquendo, C., Ferraro, F., Wai, H.A. et al. HiFi long-read RNA sequencing enhances clinical diagnostics in rare disorders. Eur J Hum Genet (2026). https://doi.org/10.1038/s41431-026-02042-9
-
Received:
-
Revised:
-
Accepted:
-
Published:
-
Version of record:
-
DOI: https://doi.org/10.1038/s41431-026-02042-9