
Introduction
Citrus species, with a variety of cultivars in the world, are used in various industries. These species are grown in more than 140 countries, with a typical annual production of 161.8 million tons1, subject to the disruptive pathogens. The most damaging pathogens are three species of Candidatus Liberibacter, including Ca. Liberibacter asiaticus (CLas), Ca. Liberibacter africanus (CLaf), and Ca. Liberibacter americanus (Clam)2,3. This genus is the causative agent of Huanglongbing (HLB), or Citrus Greening disease. These gram-negative alpha-proteobacteria, belonging to the Rhizobiaceae family, are phloem-restricted and transmitted to citrus trees through grafting or by psyllid vectors, including the Asian citrus psyllid (Diaphorina citri) and the African citrus psyllid (Trioza erytreae)4. The HLB is a considerable threat to global citriculture, having reached epidemic levels in major citrus-producing regions and causing severe economic losses for the multi-billion-dollar citrus industry2,5.
Strategies to control this disease include the removal of infected trees, the use of disease-free seedlings6, the control of psyllid vectors7, and the use of antibiotics8. The lack of effective control of these methods due to the long period of the disease, widespread asymptomatic outbreaks of disease, and adverse effects on citrus species sustainability led researchers towards novel techniques5,8. To control the disease, emerging approaches, including the manipulation of transgenic lines, application of nanoparticles, antioxidants9, and gibberellins10 have been investigated. However, their effectiveness is limited due to high cost and low efficacy in field conditions.
This disease leads to occlusion of phloem, accumulation of starch in leaves, changes in the distribution of metabolites, imbalances in hormones, changes in the root extractions, and rhizosphere microbiome structure11,12. Some researchers have shown that the beneficial microbial species in the rhizosphere of infected hosts by these three species have been decreased, while some opportunistic pathogens have been increased13. To find more effective solutions, many researchers have studied the impact of HLB disease on the host rhizosphere microbiome community13. In addition, factors including the host’s immune system, environmental conditions, and microbe-microbe interactions affect the formation of microbial community structures that should be studied with novel approaches like next-generation sequencing (NGS)14. Given that the diversity and stability of the plant microbiome strongly affect plant growth, soil fertility, and ecosystem processes15. Microbial species are potentially ideal candidates for sustainable disease control, due to the close relationship of the microbiome with the host, their shared ecological niche with the pathogen, and the production of antimicrobial compounds against the pathogens16,17.
Inoculation of infected citrus with some beneficial bacterial species has shown promising results. For example, Azospirillum brasilense reduces CLas titers and increases nutrient uptake by improving root structure18. Bacillus amyloliquefaciens and Bacillus subtilis active Induced Systemic Resistance (ISR) by producing antimicrobial lipopeptides and restricting CLas infection in phloem tissues by restoring beneficial endophytes19,20. Paenibacillus polymyxa enhances the root structure and immune system by synthesizing bioactive metabolites and increasing the abundance of beneficial bacterial species associated with biofilm formation21,22.
Although some research has studied the communication of HLB disease and rhizosphere microbiome23, the important aspects of possible interactions have remained unknown. The HLB has been previously reported in southern Iran24. Therefore, we used a more powerful metagenome sequencing approach to investigate bacterial communities in the rhizosphere of CLas-infected sweet orange trees in this area. We identified interesting changes in some key genera involved in citrus health, the potential biological control agents that might be involved in approaches to disease management. These findings highlight the practical application of microbiome engineering of rhizosphere in CLas-infected sweet orange and movement towards HLB sustainable control.
Materials and methods
Sampling from the rizosphere of Citrus sinensis
To do sampling, three CLas-free sweet orange (Citrus sinensis) trees and three CLas-infected trees were identified from a 15-year-old orchard in Jahrom city (28.6248°N, 53.3043°E), Fars province, Iran, based on visual symptoms and PCR results. This PCR amplifies CLas 50 S ribosomal subunit protein L10 of CLas using primer pair A2: 5ʹ-TATAAAGGTTGACCTTTCGAGTTT-3ʹ and J5: 5ʹ-ACAAAAGCAGAAATAGCACGAACAA-3ʹ. We have already submitted it under the PQ589016 accession number (https://www.ncbi.nlm.nih.gov/nuccore/PQ589016.1/). Next, root samples (roots with adjacent soil layers of approximately 5 cm) were collected from the four corners of each tree at a distance of 50 cm away from the tree trunk and a depth of 30 cm, and were pooled together as a sample. The gloves were changed after each sampling, and the shovels were sterilized with 30% homemade bleach. After collection, the samples were immediately placed on dry ice and transferred to the laboratory. The soil adhering to the roots (rhizosphere soil) was carefully collected and placed in sterile Falcon tubes, then stored at −20 °C until further processing.
DNA extraction
To analyze the rhizosphere bacterial communities, DNA was extracted from rhizosphere soil following Basim’s protocol25. Briefly, 40 mL of sterile distilled water was added to 20 g of each soil sample, which was vortexed for 5 min and shaken for 30 min at 200 rpm. The samples were then centrifuged at 2000 rpm for 5 min; the supernatant was separated and centrifuged again at 10,000 rpm for 10 min. Sediments from both centrifugation stages were used separately for DNA extraction. To each sediment, 1.5 times the sample volume of homogenization buffer and 2.5 g of silica beads were added, followed by vigorous vortexing for 5 min and three freeze-thaw cycles. Each sample (soil sediment and supernatant sediment) was incubated overnight at 37 °C with 75 mg of lysozyme. For chemical lysis, lysis buffer (half the sample volume) and 50 µL of proteinase K were added, and the mixture was incubated for 2 h at 65 °C. Next, 20 mL of phenol/chloroform (50/50) was added, vortexed, and centrifuged at 10,000 rpm for 15 min. 15 mL of the supernatant was mixed with 15 mL of isopropanol and centrifuged at 10,000 rpm for 10 min to precipitate the DNA. The DNA was washed with 70% ethanol, centrifuged at 10,000 rpm for 10 min, and dissolved in 50 µL of sterilized distilled water. Finally, the DNA from both sediment sources was combined. The quantity and purity of the extracted DNA were assessed using PicoDrop, and DNA quality was evaluated via 1% agarose gel electrophoresis.
Microbiome sequencing
The microbial DNA of the Rhizosphere was used to construct a metagenomic library targeting the 16 S rRNA. Two consecutive PCR reactions were carried out, and the obtained products were barcoded. In the first PCR, the hypervariable V4–V5 regions of the 16 S rRNA were amplified using primers 515 F (TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGGTGYCAGCMGCCGCGGTAA) and 926R (GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGCCGYCAATTYMTTTRAGTTT). The reaction mixture included 2.5 µL of PCR buffer, 1 µL of MgCl2, 0.5 µL of dNTP, 0.2 µL of Taq Polymerase enzyme, 2 µL of DNA, 1 µL of each primer, and 16.8 µL of deionized water, resulting in a final volume of 25 µL. The temperature protocol started with an initial denaturation at 95 °C for 5 min, followed by 25 cycles of denaturation at 95 °C for 30 s, annealing at 50 °C for 30 s, extension at 72 °C for 45 s, and a final extension at 72 °C for 10 min. The PCR product was verified using agarose gel electrophoresis. For the secondary PCR, 2 µL of the primary PCR product was used as a template, and exclusive Illumina primers were added to introduce 8-nucleotide barcodes and adapter sequences. The temperature protocol for the secondary PCR included an initial denaturation at 95 °C for 5 min and 15 cycles of denaturation at 95 °C for 30 s, annealing at 55 °C for 30 s, extension at 72 °C for 45 s, and a final extension at 72 °C for 10 min. The secondary PCR product was evaluated by agarose gel electrophoresis. The second PCR products were purified, quantified, and mixed at equimolar concentrations to construct the final library. Then, the final library was prepared for sequencing according to the Illumina protocol. The high-throughput metagenomic sequencing was performed on the Illumina NovaSeq 6000 platform (Novogene, UK) using 2 × 250 bp paired-end reads.
Bioinformatic analyses
The raw reads were processed and analyzed using the CLC Genomics Workbench (version 20, QIAGEN, Venlo, Netherlands). Briefly, the “ Prepare Sequencing Data ” workflow was employed. This stage involves two tools: the “QC for Sequencing Reads” tool, which assesses the data quality, sequencing depth, and read reliability, and the “Trim Reads” tool, which eliminates low-quality sequences defined as those shorter than 50 nucleotides, or containing more than two ambiguous bases, or including barcodes, adapters, and chimeric regions. Subsequently, paired-end reads with overlaps were merged into tags. To identify and analysis of Operational Taxonomic Units (OTUs), the “Microbial Genomics Module” in CLC was employed. All tags were clustered with the Greengenes reference database at 97% sequence similarity through the “Amplicon-Based Analysis” workflow and the “OTU Clustering” tool. The relative abundance of OTUs was observed through bar charts based on the levels of phylum, class, order, family, and genus.
Diversity of microbial communities
To measure biodiversity, a phylogenetic tree was constructed to illustrate the evolutionary relationships between OTUs. The phylogenetic tree was generated using the Maximum Likelihood approach based on a Multiple Sequence Alignment (MSA) by MUSCLE. For this purpose, taxa were aligned using the “Amplicon-Based Analysis” workflow and the “Align OTUs using MUSCLE” tool. Utilizing the obtained alignment file, the “Maximum Likelihood Phylogeny” tool and Jukes-Cantor substitution model were employed to draw the phylogenetic tree. To describe the diversity of species within the sample, an alpha diversity analysis was conducted. Various alpha diversity indices, including the Shannon index and Chao1 index, were calculated using the Analysis Abundance workflow and the Alpha Diversity tool. The diversity between the studied communities was assessed using the Bray-Curtis dissimilarity index and the UniFrac phylogenetic indices through the Beta Diversity tool. Principal Coordinate Analysis (PCoA) plots were used to visualize the results and analyze the correlation of CLas-free and CLas-infected samples.
Statistical analysis
The differential abundance analysis was performed by the DESeq2 R package in Bioconductor26,27,28. DESeq2 employs the mean count of every OTU, considering all samples, as its filtering criterion, and it excludes all OTUs with mean normalized counts below a given filtering threshold from multiple testing adjustments. This package was used to mine statistically significant differential OTUs based on the difference in count values between CLas-infected and CLas-uninfected samples. A log fold change of |1| and a P-value of ≤ 0.05 were used to define significant differential abundance. The box plot, heatmap, and PCA plots were considered to survey the analysis’s precision. To identify taxa shared among different sample groups, a Venn diagram analysis was performed using the online tool provided by the Good Calculators website (https://goodcalculators.com/venn-diagram-maker).
Results and discussion
Identification of CLas-infected and CLas-uninfected trees and operational taxonomic units
The presence of the CLas was confirmed by specific PCR using A2/J5 primers targeting the β-operon of the ribosomal protein gene in the leaf and root of infected C. sinensis trees, while the CLas-free trees showed no signs of infection (Fig. S1). In addition, apparent signs such as yellowing of the tree crown and reduction of root mass were visible in the infected trees (Fig. S2, Fig. S3). Previous studies have reported a 30–50% reduction in root density before the onset of apparent symptoms29. DNA was extracted from rhizosphere soil samples of C. sinensis trees under two conditions (Fig. S4), then the bacterial 16 S rRNA was amplified, and PCR products were sequenced using the Illumina platform. The total number of 13,411,190 reads with a length of 250 bp was sequenced. Following the merging of paired-end sequences and the removal of redundant sequences, a total of 1,796,673 valid tags were generated (Table S1). All filtered tags were clustered in Operational Taxonomic Units (OTUs) with 97% similarity, resulting in 7,621 bacterial OTUs. Then, by classifying these OTUs, we identified a total of 40 bacterial phyla.
Taxonomic profiling of rhizosphere microbiota in CLas-Free and CLas-Infected trees
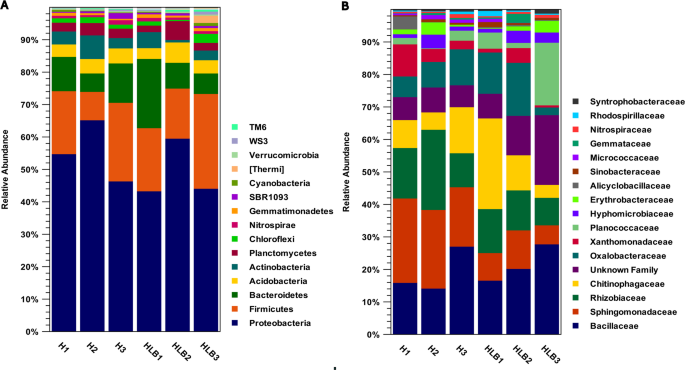
The dominant bacterial phyla in the rhizosphere of C. sinensis trees included Proteobacteria, Firmicutes, and Bacteroidetes, which constitute approximately 84% of the total microbial population in the rhizosphere of both CLas-free and CLas-infected trees (Fig. 1A, Table S2). The relative abundance of Firmicutes (P-value = 0.028) significantly increased in the infected trees (Table S2). According to our results, several studies have shown that the Firmicutes are associated with higher prevalence in CLas-infected citrus trees30,31. This might be associated with an increase in opportunistic pathogenic species32. This condition can lead to weakening of the root system in nutrient absorption and defense responses. In addition, some species of Firmicutes compete with pathogens for nutrients, restricting the pathogens from colonizing32. Some species produce antimicrobial compounds, inhibiting the growth of pathogens, and can trigger the ISR33,34. This change might be a defense response of the risosphere to the infection. Therefore, identifying species associated with these dual responses can provide practical solutions for improving rhizosphere engineering.
We discovered a significant decrease in Bacteroidetes (P-value = 0.0078) in the infected trees. These bacteria have shown a dominant role in suppressing pathogens35,36. Therefore, this decline may provide the opportunity for pathogens to be prevalent35,36. Field studies in geographical regions have identified Bacteroidetes and Proteobacteria as the dominant phyla in the rhizosphere microbiome of healthy citrus37. Therefore, the population of these bacteria could be an important biomarker of rhizosphere changes toward maintaining plant health or the beginning of disease. These shifts in the major bacterial phyla compositions support the dynamic nature of the microbiome15,38.
Another important phylum is Chlorobi (P-value = 0.0001, FC ≅ −2), which was dramatically decreased in the infected tree (Table S2). Chlorobi plays a significant role in both promoting plant health and suppressing plant diseases in the rhizosphere39. In healthy conditions, they contribute to nutrient cycling and improve plant growth. In disease-suppressive soils, they combat pathogens through various mechanisms39,40. Understanding how HLB alters the rhizosphere microbiome, including the Chlorobi, is crucial for developing strategies to manage the disease.
In addition, we identified other special phyla that might be useful to deal with HLB (Table S2). By understanding the specific roles of different microbial groups and how HLB affects them, researchers can potentially identify targets for biocontrol or develop strategies to promote beneficial microbial communities that mitigate the effects of HLB.

Taxonomic profile of the bacterial community in the C. sinensis rhizosphere. A stacked bar graph shows bacteria at the (A) phylum level and (B) family level. The X-axis indicates sample names, and the Y-axis represents relative abundance (%). H: CLas-free trees; HLB: CLas-infected trees. The dominant phyla in the C. sinensis rhizosphere include Proteobacteria, Firmicutes, and Bacteroidetes. The most abundant family in CLas-free trees is Sphingomonadaceae, and the most abundant family in CLas-infected trees is Bacillaceae.
In addition, we discovered helpful results at the family level (Table S3). Bacillaceae, Sphingomonadaceae, Rhizobiaceae, and Chitinophagaceae included approximately 30–50% of the total rhizosphere microbial community (Fig. 1B). The roles of these families in promoting plant growth, enhancing defense responses, and suppressing pathogens have been demonstrated41,42.
Sphingomonadaceae (P-value = 9.21E-05) was the most abundant family in CLas-free trees, while significantly decreased in the infected trees (FC = −1.43) (Table S3). Sphingomonadaceae produces antipathogenic metabolites, stimulates plant growth, and has shown control potential against HLB disease43,44. Some members are associated with healthy citrus trees and potentially contribute to suppressing HLB symptoms; others may be involved in the disease’s progression43,44. The decrease in the population of these bacteria can be an important biomarker of disease strengthening and rhizosphere weakening.
Abundant families revealed significant differences between CLas-free and CLas-infected trees for Rhodospirillaceae (P-value = 0.019), Alicyclobacillaceae (P-value = 8.03E-06), Micrococcaceae (P-value = 3.10E-06), Planctomycetaceae (P-value = 0.0052), Clostridiaceae (P-value = 0.006), Pseudomonadaceae (P-value = 9.08E-08), and an unidentified family belonging to the Bacillales (P-value = 0.0014) (Table S3). The relative changes indicated that Micrococcaceae, Pseudomonadaceae, Planctomycetaceae, and Clostridiaceae were significantly induced (FCs = 2 to 6) in CLas-free trees.
The high relative abundance of Pseudomonadaceae in healthy trees and the production of important bioactive metabolites may limit the growth of pathogens and activate the plant’s immune system45. These changes in the family populations can serve as bioindicators to assess rhizosphere health and plant resistance to the HLB45. In addition, the presence of Micrococcaceae, which produce growth-promoting hormones and antimicrobial enzymes, and Clostridiaceae, which combat pathogens through antagonistic activity, can help support defense mechanisms and maintain microbial balance in the rhizosphere46,47.
In contrast, Rhodospirillaceae and Alicyclobacillaceae significantly increased in the rhizosphere of infected trees (Table S2). While not directly targeting the CLas, certain Rhodospirillaceae members influence the citrus microbiome, potentially promoting plant health and resilience against various stresses associated with HLB48. While some studies suggest a potential for biocontrol agents within Alicyclobacillaceae to suppress HLB symptoms, further investigation is needed to fully understand their interactions with the CLas, and the overall citrus microbiome49. Some members in the citrus rhizosphere exhibit traits like pectinase, cellulose, and chitinase production, which can be beneficial for plant health and potentially help control other root rot pathogens. Certain members trigger the ISR that which could make the plant more resilient to infection and reduce the severity of symptoms12,50. Finally, we identified many other novel beneficial families that can present practical clues for controlling HLB.
Key bacterial genera in the rhizosphere of CLas-Free and CLas-Infected trees
We considered the box plot (Fig. S5) and PCA plot (Fig. S6) during statistical analysis. The Heatmap shows that the data generated from the biological replications of CLas-free and infected rhizosphere samples are well separated, indicating the accuracy of sampling and data analysis (Fig. 2).

Heatmap of bacterial population based on genera in CLas-free and CLas-infected C. sinensis trees’ rhizosphere samples. H1-H3 show the three replications of CLas-free trees, and HLB1-HLB3 show the three replications of infected trees.
The results indicated the decreased abundance of Pseudomonas (P-value = 0.008, FC = −3.13), Chryseobacterium (P-value = 0.019, FC = −3.15), and an unclassified genus belonging to Aurantimonadaceae (P-value = 0.008, FC = −3.11), and increased abundance of Planococcus (P-value = 0.016, FC = 3.88) and an unclassified genus from the family Caulobacteraceae (P-value = 0.022, FC = 2.67) in the infected trees (Fig. 3, Table S4).
The Pseudomonas that is dramatically suppressed in the infected trees acts as the core of biological control, producing antimicrobial metabolites including DAPG, pyoluteorin, and pyrrolnitrin, and effectively competing through TonB receptor systems and siderophores, thereby directly inhibiting CLas51,52. The observed increase in the relative abundance of Chryseobacterium in the rhizosphere of healthy trees compared to infected trees indicates that this genus may play a complementary role in the synergistic functioning of the rhizosphere53.
This genus, through a combination of antagonistic capabilities, production of IAA, and solubilization of mineral phosphate, enhances the nutritional capacity and induces plant immunity54,55. Several studies have reported the synergistic interaction of Chryseobacterium with Bacillus, Pseudomonas, and Lysobacter against pathogens55,56. The results of Lee’s (2021) study on the citrus microbiome showed that Chryseobacterium increased in the rhizosphere of CLas-infected trees and is associated with host resistance57. This indicates that this genus plays a more significant role in the growth and development of plants under healthy conditions, whereas in infected trees, its activity shifts towards enhancing plant resistance.

Taxonomic profile of the bacterial community in the C. sinensis rhizosphere at the genus level. The X-axis indicates sample names, and the Y-axis represents relative abundance (%). H: CLas-free trees; HLB: CLas-infected trees. The genera Pseudomonas, Chryseobacterium, Planococcus, and unidentified genera belonging to Aurantimonadaceae and Caulobacteraceae were significantly different between CLas-free and CLas-infected trees.
While Aurantimonadaceae, which is intensely suppressed in the infected tree, is a bacterial family found in the citrus rhizosphere, its direct role in combating HLB, also known as citrus greening, is not definitively established. Our research suggests that some members of this family may be beneficial, while others could potentially be detrimental, and more research is needed to fully understand their impact.
The increased abundance of Planococcus may play a beneficial role in managing HLB by influencing the rhizosphere microbiome and enhancing its resistance to the disease. While not directly targeting the HLB pathogen, this genus promotes plant health and potentially creates an environment less susceptible to HLB58. Some strains can produce auxins and gibberellins, which can enhance root development and nutrient uptake, leading to a more robust and resilient host59. Some strains produce antimicrobial compounds that inhibit the growth of other bacteria, including those associated with HLB58. The precise role of this genus in interaction with the CLas requires more functional studies in the future.
The abundance increase of an unidentified novel genus from the Caulobacteraceae family may also indicate its involvement in altering the microbial community structure under disease pressure. Members of this family are known for their endophytic colonization capabilities and utilization of competitive mechanisms such as contact-dependent killing systems and the production of antibacterial compounds to eliminate competitors31.
It is noteworthy that numerous genera, including Bacillus, Ensifer, Sphingobium, Sphingomonas, Pseudoxanthomonas, and Sphingopyxis, did not show significant differences between the infected and uninfected trees. According to previous studies, these genera overlap with those reported as the core microbiome of citrus in various research13,37. Such overlap suggests that these genera are likely part of the core rhizosphere microbiome of citrus and are primarily unaffected by disease conditions due to their key roles in maintaining microbial ecosystem functioning12,60. These genera enhance plant metabolism through pathways such as nitrogen fixation and nutritional activities, production of growth-promoting compounds, and secondary metabolites, thereby providing a suitable environment for the efficient operation of biocontrol agents61,62. In addition, we have identified other new genera that are of great importance for HLB control and are a valuable research area for the future.
Diversity and structure of rhizosphere bacterial communities in CLas-Free and CLas-Infected trees
Bacterial diversity in the rhizosphere of CLas-free and CLas-infected trees was evaluated using the Chao1 and Shannon, indices (Fig. 4). The Chao1 index, which estimates microbial richness, was higher in infected trees compared to CLas-free ones, suggesting that the microbial population in infected trees is richer, potentially as a microbiome-mediated response to pathogen invasion or the entry of opportunistic species in disease conditions. Accordingly, the Shannon index, indicating the number of species in each sample, was slightly higher in infected trees, reflecting greater diversity. The lower Shannon index values observed in CLas-free trees may be attributed to a reduction in species richness or the dominance of one or a few species.

Alpha diversity indices of the bacterial communities in the C. sinensis rhizosphere of CLas- free and CLas-infected trees. (A) Chao1 index; (B) Shannon index. The x-axis represents the number of sequencing reads, and the y-axis shows the numerical values obtained for each sample based on the index. H: CLas-free trees; HLB: CLas-infected trees. CLas-infected trees showed higher values for both indices than CLas-free trees.
Based on the Bray-Curtis index, a distinct clustering was observed between CLas-free and CLas-infected trees (Fig. 5). Although the visual separation is not absolute, the low overlap in the data indicates that the composition of the bacterial community under the two conditions is somewhat different, which could be due to the impact of the disease. To examine the differences in the microbial community structure under the two conditions, PCoA based on the weighted UniFrac index was used. Weighted UniFrac, as a phylogenetic-based metric, allows for the assessment of bacterial community structure by considering the evolutionary relationships between species and their relative abundances (Fig. 5). The resulting PCoA plot showed that the first principal coordinate (PC1) accounts for approximately 58% of the total variance, while the second coordinate (PC2) explains about 23%, together encompassing more than 81% of the total variance. The separation of CLas-free and CLas-infected samples into distinct clusters on the plot indicates a significant change in the structure of the microbial community associated with CLas infection. This separation may indicate a changed abundance of specific bacterial species in response to the disease. It is noteworthy that the CLas-free samples were very close to each other, indicating the uniformity of the microbial community. According to the unweighted UniFrac index, which only considers the presence or absence of species and their relationships, the distance between the CLas-free and CLas-infected samples has increased. Analysis of these indices showed that CLas infection leads to significant changes in the microbial structure of the rhizosphere (D-0.5 UniFrac and unweighted UniFrac are shown in Fig. S7).

PCoA of microbial communities in the rhizosphere of C. sinensis trees. Beta diversity was estimated using (A) Bray-Curtis dissimilarity and (B) weighted UniFrac distance. The x-axis represents the first principal component, and the percentage represents its contribution to sample differences; the y-axis represents the second principal component, and the percentage represents its contribution to sample differences. H: CLas-free trees; HLB: CLas-infected trees. Based on the Bray-Curtis index, the composition of the bacterial community in CLas-free and CLas-infected trees was somewhat different. Based on the UniFrac weighted index, the bacterial community structure differs between CLas-free and CLas-infected trees.
The consistency of this pattern with previous reports indicates that the pathogen CLas can shift the microbial community structure from a uniform and stable state in healthy trees to a dispersed and heterogeneous state in infected trees, a condition that is consistent with the concept of “dysbiosis”13. In one study, simultaneous changes in the diversity and composition of the community under pathogen pressure led to a decrease in microbial network stability63. It has also been reported that even with a relatively stable alpha diversity, significant changes in beta diversity and community structure can be associated with the severity of HLB symptoms13.
This overlap with the current results, in which species richness showed a modest increase and community composition changed, suggests that richness-only indices are insufficient to predict host health, as compositional and phylogenetic changes also play a determining role. The discrepancy with reports that indicate a decrease in alpha diversity in advanced stages of the disease31,42 is likely due to differences in environmental conditions, the physiological status of the trees, sequencing depth, and analytical approaches30. On the other hand, the findings of Yang et al., who reported an increase in diversity in hosts infected with Phytophthora disease64, support the notion that this pattern may be a result of the introduction of opportunistic taxa or community reassembly as a compensatory mechanism. Overall, the findings of this study suggest that simultaneous analysis of richness, dispersion, and phylogenetic indices is essential for explaining microbial responses to HLB.
Distribution of rhizosphere bacteria in CLas-Free and CLas-Infected trees
To identify the shared and unique OTUs of the samples, a Venn diagram was used (Fig. 6). A total of 811 shared OTUs (238 genera) were identified among all samples. Most of these species belong to the phyla Proteobacteria, Acidobacteria, Actinobacteria, Firmicutes, and Planctomycetes, and most of them can play an essential role in plant health. Examination of unique OTUs in the samples showed that the CLas-free tree samples of H1, H2, and H3 showed 299, 496, and 365 unique OTUs, respectively, some of which may have a protective role in the plant. In contrast, the CLas-infected samples (HLB1, HLB2, HLB3) showed a different number of unique OTUs, so that the HLB1 sample showed 823 OTUs, while in the HLB2 sample, this number decreased to 582 OTUs, which may indicate dysbiosis.

Venn diagram of OTUs among rhizosphere bacterial communities of C. sinensis. H: CLas-free trees; HLB: CLas-infected trees. According to the Venn diagram, there are 811 shared taxa in all samples.
These results suggest that CLas infection can have variable effects on the microbial diversity of trees. On the other hand, 81 shared OTUs were observed among HLB samples, indicating the adaptability of some species under disease conditions. These species belong to the genera Novosphingobium, Sphingomonas, Devosia, Agrobacterium, Aminobacter, Rhodoplanes, Paenibacillus, Steroidobacter, Arenimonas, Lysobacter, Chitinophaga, Flavobacterium, Bdellovrio, and some strains of the genus Bacillus. In contrast, CLas-free samples contained 16 shared OTUs belonging to the genera Paenibacillus, Agromyces, Catellatospora, Azospirillum, Flavobacterium, Aquicella, Streptomyces, Pseudomonas, and unidentified genera of Chitinophagaceae, Sinobacteraceae, Sphingomonadaceae, Rhodospirillaceae, and Chitinophagaceae. Although the abundance of unique species was very low, they may play an important role in citrus health and therefore deserve further investigation. These results show that microbial diversity and the presence of specific bacterial species are related to the health status of the trees, such that the difference in bacterial composition can indicate the impact of infection on the microbial community.
Conclusion
In this study, we provided a comprehensive description of the rhizosphere bacterial communities in CLas-free and CLas-infected C. sinensis trees. The indicator bacteria that showed statistically significant differences included Pseudomonas, Chryseobacterium, Planococcus, and an unidentified genus belonging to the Aurantimonadaceae and Caulobacteraceae families. The relative abundance of Pseudomonas, Chryseobacterium, and the unidentified genus of Aurantimonadaceae was induced in healthy trees, whereas Planococcus and the unidentified genus of Caulobacteraceae were more dominant in infected trees. Furthermore, genera such as Ensifer, Sphingopyxis, Sphingobium, Sphingomonas, and Bacillus, which are known to be part of the core microbiome, did not show statistically significant differences. The microbial population associated with CLas-infected trees is more dispersed compared to CLas-free trees, and its overall abundance was suppressed, which may indicate dysbiosis caused by CLas infection. These results highlight the important role of the rhizosphere microbiome in plant health and may lead to the identification of potential microbial markers for HLB disease management. In the future, a more detailed examination of the species and strains of these genera and elucidating their functional roles could pave the way for targeted microbiome engineering and the development of sustainable HLB management strategies.
Data availability
All data generated or analyzed during this study are included in this published article (and its Supplementary Information files). In addition, the datasets generated during and/or analyzed during the current study are available in the NCBI repository. This direct link “https://dataview.ncbi.nlm.nih.gov/object/PRJNA1134754?reviewer=3juf9lha7miu1pfp2dud5nbjdt” is accessible to the data. POINT OF CONTACT: Please contact the corresponding author, Ali Moghadam, if someone wants to request the data from this study.
References
-
Gonzatto, M. P. & Santos, J. S. Introductory Chapter: World Citrus Production and research, in Citrus Research-Horticultural and Human Health Aspects (IntechOpen, 2023).
-
Gottwald, T. R. Current epidemiological Understanding of citrus Huanglongbing. Annu. Rev. Phytopathol. 48, 119–139 (2010).
-
Bové, J. M. Huanglongbing: a destructive, newly-emerging, century-old disease of citrus. J. plant. Pathol. 81 (1), 7–37. (2006).
-
Yan, H. et al. Comparative Analysis of Bacterial and Fungal Endophytes Responses To Candidatus Liberibacter Asiaticus Infection in Leaf Midribs of Citrus Reticulata cv. Shatangju113p. 101590 (Physiological and Molecular Plant Pathology, 2021).
-
Hu, B. et al. Molecular signatures between citrus and candidatus liberibacter Asiaticus. PLoS Pathog. 17 (12), e1010071 (2021).
-
Coletta-Filho, H. D. et al. Temporal progression of ‘Candidatus liberibacter asiaticus’ infection in citrus and acquisition efficiency by diaphorina citri. Phytopathology 104 (4), 416–421 (2014).
-
Tomaseto, A. F. et al. Orange jasmine as a trap crop to control Diaphorina citri. Scientific Reports, 9 (1): p. 2070. (2019).
-
Blaustein, R. A., Lorca, G. L. & Teplitski, M. Challenges for managing candidatus liberibacter spp.(Huanglongbing disease pathogen): current control measures and future directions. Phytopathology 108 (4), 424–435 (2018).
-
Taghizadeh, M. S. et al. Novel bioactive peptides of Achillea eriophora show anticancer and antioxidant activities. Bioorg. Chem. 110, 104777 (2021).
-
Ma, W. et al. Citrus Huanglongbing is a pathogen-triggered immune disease that can be mitigated with antioxidants and Gibberellin. Nat. Commun. 13 (1), 529 (2022).
-
Nehela, Y. & Killiny, N. Revisiting the complex pathosystem of huanglongbing: Deciphering the role of citrus metabolites in symptom development. Metabolites 10 (10), 409 (2020).
-
Srivastava, A. K. et al. Bioprospecting Microbiome for soil and plant health management amidst Huanglongbing threat in citrus: A review. Front. Plant Sci. 13, 858842 (2022).
-
Ginnan, N. A. et al. Disease-induced microbial shifts in citrus indicate microbiome-derived responses to Huanglongbing across the disease severity spectrum. Phytobiomes J. 4 (4), 375–387 (2020).
-
Naylor, D. & Coleman-Derr, D. Drought stress and root-associated bacterial communities. Front. Plant Sci. 8, 303756 (2018).
-
Zhang, Y. et al. The citrus Microbiome: from structure and function to Microbiome engineering and beyond. Phytobiomes J. 5 (3), 249–262 (2021).
-
Eljounaidi, K., Lee, S. K. & Bae, H. Bacterial endophytes as potential biocontrol agents of vascular wilt diseases–review and future prospects. Biol. Control. 103, 62–68 (2016).
-
Soltani, Z., Moghadam, A. & Shamekh, M. Comparative meta-analysis of barely transcriptome: pathogen type determines host preference. PloS One. 20 (6), e0320708 (2025).
-
Rafael Trinidad-Cruz, J. et al. Inductors of Plant Resistance in the Control of Candidatus Liberibacter Asiaticus in Mexican Lemon (Citrus aurantifolia) Trees37 (Revista Mexicana de Fitopatología, 2019). 2.
-
Tang, J. et al. Transcriptome sequencing and ITRAQ reveal the detoxification mechanism of Bacillus GJ1, a potential biocontrol agent for Huanglongbing. PLoS One. 13 (8), e0200427 (2018).
-
Munir, S. et al. Defeating Huanglongbing pathogen candidatus liberibacter Asiaticus with Indigenous citrus endophyte Bacillus subtilis L1-21. Front. Plant Sci. 12, 789065 (2022).
-
Yang, Y. et al. Inhibition of citrus Huanglongbing disease by Paenibacillus polymyx KN-03 and analysis with transcriptome and microflora. Agronomy 13 (12), 2958 (2023).
-
Moghadam, A. et al. System network analysis of Rosmarinus officinalis transcriptome and metabolome—Key genes in biosynthesis of secondary metabolites. PLoS One. 18 (3), e0282316 (2023).
-
Kapourchali, S. S. et al. Microbiota dynamic communities in sweet orange infected by Huanglongbing in iran: microbiota associated with sweet orange infected by Huanglongbing in Iran. Phytopathologia Mediterranea. 64 (1), 129–143 (2025).
-
Salehi, M. et al. Distribution of citrus Huanglongbing disease and its vector in Southern Iran. Iran. J. Plant. Pathol. 48 (2), 195–208 (2012).
-
Basim, Y. et al. Comparison of performance and efficiency of four methods to extract genomic DNA from oil contaminated soils in Southwestern of Iran. J. Environ. Health Sci. Eng. 18, 463–468 (2020).
-
Xia, Y. Statistical normalization methods in Microbiome data with application to Microbiome cancer research. Gut Microbes. 15 (2), 2244139 (2023).
-
Nixon, M. P., Gloor, G. B. & Silverman, J. D. Incorporating scale uncertainty in Microbiome and gene expression analysis as an extension of normalization. Genome Biol. 26 (1), 139 (2025).
-
Segata, N. et al. Metagenomic biomarker discovery and explanation. Genome Biol. 12 (6), R60 (2011).
-
Johnson, E. et al. Association of ‘C andidatus L iberibacter asiaticus’ root infection, but not phloem plugging with root loss on huanglongbing-affected trees prior to appearance of foliar symptoms. Plant. Pathol. 63 (2), 290–298 (2014).
-
Liu, H. Q. et al. Accurate prediction of Huanglongbing occurrence in citrus plants by machine learning-based analysis of symbiotic bacteria. Front. Plant Sci. 14, 1129508 (2023).
-
Trivedi, P. et al. Huanglongbing alters the structure and functional diversity of microbial communities associated with citrus rhizosphere. ISME J. 6 (2), 363–383 (2012).
-
Idbella, M. et al. Windstorm disturbance sets off plant species invasion, microbiota shift, and soilborne pathogens spread in an urban mediterranean forest. For. Ecol. Manag. 540, 121058 (2023).
-
Igiehon, B. C., Babalola, O. O. & Hassen, A. I. Rhizosphere competence and applications of plant growth-promoting rhizobacteria in food production–A review. Sci. Afr. 23, e02081 (2024).
-
Moghadam, A. et al. Exploring novel insights: Methyl jasmonate treatment reveals novel lncRNA-mediated regulation of secondary metabolite biosynthesis pathways in Echinacea purpurea. Food Bioscience. 57, 103457 (2024).
-
Song, L. et al. Variation and stability of rhizosphere bacterial communities of cucumis crops in association with root-knot nematodes infestation. Front. Plant Sci. 14, 1163271 (2023).
-
Pan, X., Raaijmakers, J. M. & Carrión, V. J. Importance of bacteroidetes in host–microbe interactions and ecosystem functioning. Trends Microbiol. 31 (9), 959–971 (2023).
-
Xu, J. et al. The structure and function of the global citrus rhizosphere Microbiome. Nat. Commun. 9 (1), 4894 (2018).
-
Berendsen, R. L., Pieterse, C. M. & Bakker, P. A. The rhizosphere Microbiome and plant health. Trends Plant Sci. 17 (8), 478–486 (2012).
-
Thepbandit, W. & Athinuwat, D. Rhizosphere microorganisms supply availability of soil nutrients and induce plant defense. Microorganisms 12 (3), 558 (2024).
-
Kaviani, E. et al. Phytoremediation of Pb-contaminated soil by salicornia iranica: key physiological and molecular mechanisms involved in Pb detoxification. CLEAN–Soil Air Water. 45 (5), 1500964 (2017).
-
Gouda, S. et al. Revitalization of plant growth promoting rhizobacteria for sustainable development in agriculture. Microbiol. Res. 206, 131–140 (2018).
-
Blaustein, R. A. et al. Defining the core citrus leaf-and root-associated microbiota: factors associated with community structure and implications for managing Huanglongbing (citrus greening) disease. Appl. Environ. Microbiol. 83 (11), e00210–e00217 (2017).
-
Harirchi, S. et al. Bacillales: from taxonomy to biotechnological and industrial perspectives. Microorganisms 10 (12), 2355 (2022).
-
Asaf, S. et al. Sphingomonas: from diversity and genomics to functional role in environmental remediation and plant growth. Crit. Rev. Biotechnol. 40 (2), 138–152 (2020).
-
Meliani, A. et al. Plant growth-promotion and IAA secretion with Pseudomonas fluorescens and Pseudomonas Putida. Res. Reviews: J. Bot. Sci. 6 (2), 16–24 (2017).
-
Rivera-Hernández, G. et al. Evaluation of functional plant growth-promoting activities of culturable rhizobacteria associated to tunicate maize (Zea Mays var. Tunicata A. St. Hil), a Mexican exotic landrace grown in traditional agroecosystems. Front. Microbiol. 15, 1478807 (2024).
-
Shirane, S. et al. Fungicidal activity of caproate produced by clostridium sp. strain E801, a bacterium isolated from Cocopeat medium subjected to anaerobic soil disinfestation. Agronomy 13 (3), 747 (2023).
-
Esiobu, N. et al. Rhizosphere microbiomes of citrus plants in historically undisturbed 100-Year-Old groves appear to mitigate susceptibility to citrus greening disease. Microorganisms 13 (4), 763 (2025).
-
Zhang, Y. et al. Controlling citrus Huanglongbing based on soil remediation and biocontrol. Eur. J. Plant Pathol. 169 (2), 379–393 (2024).
-
Amiri, F. et al. Identification of key genes involved in secondary metabolite biosynthesis in digitalis purpurea. PloS One. 18 (3), e0277293 (2023).
-
Alquézar, B. et al. Engineering of citrus to obtain Huanglongbing resistance. Curr. Opin. Biotechnol. 70, 196–203 (2021).
-
Sah, S., Krishnani, S. & Singh, R. Pseudomonas mediated nutritional and growth promotional activities for sustainable food security. Curr. Res. Microb. Sci. 2, 100084 (2021).
-
Park, I., Seo, Y. S. & Mannaa, M. Recruitment of the rhizo-microbiome army: assembly determinants and engineering of the rhizosphere Microbiome as a key to unlocking plant potential. Front. Microbiol. 14, 1163832 (2023).
-
Sang, M. et al. Growth promotion and root colonisation in pepper plants by phosphate-solubilising Chryseobacterium sp. strain ISE14 that suppresses phytophthora blight. Ann. Appl. Biol. 172 (2), 208–223 (2018).
-
Wang, T. et al. Effect of plant-derived microbial soil legacy in a grafting system—a turn for the better. Microbiome, 12(1): p. 234. (2024).
-
Mülner, P. et al. Microbiota associated with sclerotia of soilborne fungal pathogens–a novel source of biocontrol agents producing bioactive volatiles. Phytobiomes J. 3 (2), 125–136 (2019).
-
Li, H. et al. Microbiome and metagenome analysis reveals Huanglongbing affects the abundance of citrus rhizosphere bacteria associated with resistance and energy metabolism. Horticulturae 7 (6), 151 (2021).
-
Zhang, Y. et al. Huanglongbing impairs the rhizosphere-to-rhizoplane enrichment process of the citrus root-associated Microbiome. Microbiome 5 (1), 97 (2017).
-
Soltani, Z. et al. Integrative systems biology analysis of barley transcriptome hormonal signaling against biotic stress. Plos One. 18 (4), e0281470 (2023).
-
Trivedi, P. et al. Enabling sustainable agriculture through Understanding and enhancement of microbiomes. New Phytol. 230 (6), 2129–2147 (2021).
-
Dong, Z. H. et al. Association between plant nutrients, the development of Huanglongbing and abnormal growth symptoms in navel orange. Plant Biol. 23 (6), 1167–1176 (2021).
-
Xia, K. et al. Potential functions of the shared bacterial taxa in the citrus leaf midribs determine the symptoms of Huanglongbing. Front. Plant Sci. 14, 1270929 (2023).
-
Trivedi, P., Duan, Y. & Wang, N. Huanglongbing, a systemic disease, restructures the bacterial community associated with citrus roots. Appl. Environ. Microbiol. 76 (11), 3427–3436 (2010).
-
Yang, C. H., Crowley, D. & Menge, J. 16S rDNA fingerprinting of rhizosphere bacterial communities associated with healthy and Phytophthora infected avocado roots. FEMS microbiology ecology, 35(2): pp. 129–136. (2001).
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Arjmand, E., Moghadam, A., Afsharifar, A. et al. Metagenome analysis of Citrus sinensis rhizosphere infected with Candidatus liberibacter asiaticus reveals distinct structure in bacterial communities. Sci Rep 15, 37987 (2025). https://doi.org/10.1038/s41598-025-21973-9
-
Received:
-
Accepted:
-
Published:
-
DOI: https://doi.org/10.1038/s41598-025-21973-9