
Introduction
Neurogenesis is a highly intricate process, vital for the formation of a fully functional nervous system. Any disruptions in this process can lead to severe neurodevelopmental and neurodegenerative disorders, such as Parkinson’s disease (PD)1.
PD is characterized by the gradual loss of dopaminergic neurons in the substantia nigra, accompanied by the build-up of misfolded proteins, which disrupt cellular function2. Central to the pathophysiology of PD are Lewy bodies, intracellular aggregates primarily composed of phosphorylated α-synuclein and ubiquitinated proteins, serving as harbingers of neuronal distress and dysfunction2,3. Understanding the molecular mechanisms governing protein aggregation and clearance, particularly by the ubiquitin–proteasome system (UPS), is imperative for elucidating the pathogenesis of PD and related neurodegenerative diseases.
UPS serves as critical cellular machinery responsible for maintaining protein homeostasis, regulating a plethora of cellular processes essential for cell survival and function4. Comprising a complex network of enzymes and regulatory proteins, the UPS operates in a finely orchestrated manner to identify, tag, and degrade proteins marked for disposal. The regulation of the proteasome is influenced by various factors that ensure its optimal function and adaptability to changing cellular conditions. Proteasome activity is modulated by several regulatory proteins and pathways, including the deubiquitinating enzymes (DUBs) that remove ubiquitin tags from substrates, thereby fine-tuning the degradation process5. Additionally, the assembly and disassembly of the proteasome itself are tightly controlled by chaperone proteins that assist in the proper folding and maintenance of the proteasome complex5,6. Cellular signalling pathways, such as those mediated by transcription factors and post-translational modifications, also play a crucial role in adjusting proteasome activity in response to cellular stress and changing metabolic demands7.
The Nfe2l1 gene (nuclear factor, erythroid 2-like 1, also known as NRF1) encodes a transcription factor pivotal for maintaining cellular proteostasis through its regulatory oversight of the UPS8,9. Nfe2l1 is activated in response to cellular stress and plays a crucial role in the adaptive response to proteotoxic stress10,11. By regulating the expression of various genes involved in the ubiquitin–proteasome pathway, Nfe2l1 enhances the cell’s ability to degrade misfolded and damaged proteins, thereby preventing their toxic accumulation8,10. Nfe2l1 operates by modulating the expression of several UPS components, including proteasome subunits and associated regulatory factors. It activates genes that encode DUBs, enhancing the cell’s capacity to recycle ubiquitin and regulate substrate degradation more precisely10. This regulation is particularly crucial in neurons, where the balance of protein synthesis and degradation must be meticulously maintained to support neuronal health and function8.
While the role of Nfe2l1 in orchestrating cellular responses to stress and maintaining proteostasis is well-documented, its specific functions within neuronal development and protein homeostasis are not fully understood. Several in vivo studies have suggested that Nfe2l1 plays crucial roles in the neuronal system, including its involvement in PD11,12,13,14. Our study advances this understanding by identifying several genes potentially regulated by Nfe2l1 and elucidating its role specifically in neuronal cells. This novel insight provides a more detailed view of the function of Nfe2l1 in neuronal physiology and pathology, complementing the existing evidence from in vivo studies.
This study aims to clarify the role of Nfe2l1 in the neuronal system, particularly in the context of protein aggregation, a prominent feature of neurodegenerative diseases like PD. We conducted experiments using P19 cells undergoing retinoic acid (RA)-induced neurogenesis (hereafter will be referred as P19 neuron) as an in vitro model of neuronal development and knockdown (KD) the Nfe2l1 gene using siRNA. Additionally, we induced proteotoxic stress using Mg132, a proteasome inhibitor. Through these experiments, we sought to uncover the specific contributions of Nfe2l1 to neuronal protein balance and its potential implications for neurodegenerative diseases such as PD. Our findings hold promise for the development of targeted therapeutic interventions aimed at mitigating the effects of PD and related disorders.
Results
The neural differentiation of P19 cells is accompanied by increased expression of Nfe2l1
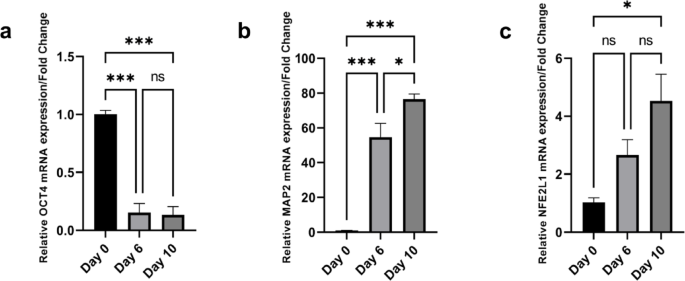
Following the initial neuronal differentiation period, cells were transferred to adherent culture dishes, and RNA was extracted at three distinct time points: day 0 (undifferentiated cells), day 6 (one day post-plating), and day 10 (final day of neuronal differentiation). The mRNA levels of Nfe2l1, Nfe2l2, Oct4 (pluripotency marker), and Map2 (neuronal marker) were measured using quantitative PCR (qPCR). Both Nfe2l1 (Fig. 1c) and Nfe2l2 (Supplementary Fig. 1) showed a significant increase during the course of neuronal differentiation. Nfe2l1 expression was upregulated on day 6, but not significantly. However, a significant increase was observed by day 10 (*p < 0.05). Similarly, Nfe2l2 expression also increased on day 6, but not significantly, and showed an even more pronounced increase by day 10 (**p < 0.01). Furthermore, the neuronal marker Map2 exhibited a substantial increase in expression at both day 6 and day 10 (Fig. 1b), with both time points showing a highly significant increase (***p < 0.001), confirming successful neuronal differentiation. Conversely, the pluripotency marker Oct4 showed a significant decrease in expression at days 6 and 10 (***p < 0.001) (Fig. 1a), indicating a loss of pluripotency as the cells differentiated into neurons. These results collectively indicate that the neuronal differentiation was effectively executed, as evidenced by the increased expression of the neuronal marker Map2 and decreased expression of the pluripotency marker Oct4. Additionally, the significant upregulation of Nfe2l1 and Nfe2l2 during the differentiation process suggests their potential involvement in neuronal differentiation and highlights the importance of exploring their roles further. Despite both genes being upregulated, we focused on Nfe2l1 as it plays a more critical role in the neuronal system and proteasome function14,15, making it the spotlighted member of the Nfe2l family in this setting.

RA-induced neurogenesis in P19 cells, assessed via qPCR analysis of (a) Oct4 and (b) Map2 mRNA levels, serving as pluripotent and neuronal markers, respectively. Additionally, (c) Nfe2l1 mRNA level was analysed to observe it’s expression changes during neurogenesis. Samples were collected at day 0 (WT cells), 6, and 10 post-cell plating and RA induction. Gapdh was used as the reference gene. Results are presented as fold change, with error bars indicating mean ± S.E.M. (n = 3 per group). Statistical significance denoted as ns (not significant), *p < 0.05, **p < 0.01, and ***p < 0.001, among groups.
Nfe2l1 expression is augmented by proteasome inhibition
To investigate the role of Nfe2l1 in this context, we examined NFE2L1 protein levels and mRNA expression in P19 stem cells (Wild-type) and P19 neurons, under the proteasome inhibition using Mg132. To investigate the effect of proteasome inhibition on NFE2L1 protein levels, P19 WT cells and neuronal differentiated cells were treated with 25µM Mg132. Western blot analysis revealed a significant augmentation of NFE2L1 protein levels following Mg132 treatment in both WT and neuronal differentiated cells (Fig. 2a). This indicates that proteasomal degradation plays a crucial role in the regulation of NFE2L1 protein levels in P19 cells. In addition to protein level changes, we examined whether Mg132 treatment affected Nfe2l1 mRNA expression. qPCR analysis showed that Mg132 treatment not only increased NFE2L1 protein levels but also led to a significant upregulation of Nfe2l1 mRNA expression (Fig. 2b,c). This suggests that proteasome inhibition might also have transcriptional regulatory effects on Nfe2l1 expression. The significant increase in NFE2L1 protein and mRNA levels upon Mg132 treatment underscores the importance of proteasomal regulation in maintaining Nfe2l1 expression. The findings emphasize the potential impact of proteasome inhibitors on gene expression and protein stability, contributing to our understanding of cellular regulation and differentiation processes in neuronal development.

Levels of Nfe2l1 protein in P19 cells. Western blotting confirmed the level of NFE2L1 protein in both P19 undiffrentiated cells and P19 neurons. Treatment with the proteasome inhibitor Mg132 (25 µM) resulted in (a) increased protein levels, as well as (b, c) upregulation of mRNA expression. *p < 0.05, **p < 0.01, and ***p < 0.001 versus control.
RNA sequencing analysis: proteasome inhibition and neurodegeneration-related pathways
To understand the molecular effects of proteasome inhibition on neuronal differentiation, we employed RNA sequencing as a comprehensive approach to capture changes in gene expression. In our study, we used RNA sequencing to examine the impact of proteasome inhibition on neuronal-differentiated P19 cells. This technique enabled us to identify differentially expressed genes and elucidate the pathways affected by proteasome inhibition, particularly those relevant to neurodegenerative diseases.
When P19 cells were differentiated into neurons and treated with DMSO (control), uniquely 1549 genes exhibited altered expression levels. In contrast, treatment with the proteasome inhibitor Mg132 also uniquely resulted in changes in 1655 genes. Remarkably, a substantial overlap was observed between the two groups, with 8810 genes commonly affected (Fig. 3a). This overlap underscores the significant influence of neuronal differentiation on gene expression patterns and suggests a complex interplay between differentiation processes and proteasome activity in these P19 cells. To assess the impact of proteasome inhibition on gene expression profiles, RNA sequencing analysis was performed on both WT P19 cells and P19 neurons treated with Mg132. In Mg132-treated P19 stem cells, uniquely 1397 genes exhibited altered expression levels, while treatment of P19 neurons with Mg132 resulted in changes in uniquely 3953 genes. Notably, 2237 genes were commonly affected in both groups (Fig. 3b). Given the focus of our study on neuronal differentiation and neurons, particular attention was warranted on gene expression changes observed in the Mg132-treated P19 neurons, which may provide insights into the specific role of proteasome inhibition in neuronal differentiation processes.

RNA sequencing analysis shows neuronal differentiation and proteasome inhibition-dependent changes on gene expression profiles. Venn diagram illustrating the impact of (a) neuronal differentiation and (b) proteasome inhibition on gene expression profiles in P19 WT cells and P19 neurons. (c) KEGG pathway enrichment bubble chart depicting the top enriched pathways in Mg132-treated P19 neurons. PD is identified as the most enriched pathway.
Furthermore, in order to gain insight into the molecular mechanisms underlying the effects of proteasome inhibition, we conducted KEGG pathway enrichment analysis. The analysis revealed significant enrichment of pathways associated with neurodegenerative disorders, with Parkinson’s disease emerging as the top-ranked pathway (Fig. 3c). These findings suggest that proteasome inhibition may contribute to neurodegeneration-related pathways, highlighting the potential relevance of proteasome dysfunction in the context of neurodegenerative diseases.
To elucidate the association between proteasome inhibition and PD, we identified a set of PD-related genes that exhibited altered expression during proteasome inhibition in P19 neurons treated with Mg132. We cross-referenced these PD-associated genes with the differentially expressed genes obtained from RNA sequencing data. Our analysis revealed 27 upregulated (Supplementary Table 1) and 27 downregulated genes (Supplementary Table 2) among the PD-associated genes. Notably, 15 of the upregulated genes were found to be proteasome genes, while the remaining 12 belonged to other functional categories (Table 1).
For further investigation of the functional relevance of these identified genes, we selected the 12 non-proteasome genes (Table 1) that were upregulated during proteasome inhibition for downstream analysis. We aimed to evaluate whether the expression of these genes would be altered upon knockdown of Nfe2l1, a key regulator implicated in the compensatory response to proteasome inhibition, in P19 neurons treated with and without Mg132.
Nfe2l1 KD in neuronal cells
Following the validation of our system by RNA sequencing and the identification of key gene expression changes under proteasome inhibition, we further investigated the role of Nfe2l1 in regulating specific target genes. We focused on verifying the functional impact of Nfe2l1 KD in P19 neurons. This involved assessing how Nfe2l1 KD affected the expression of selected genes associated with PD and proteasome inhibition, as well as confirming the knockdown efficiency through protein analysis.
Transfection of P19 neurons with Nfe2l1 siRNA resulted in a significant reduction in NFE2L1 protein levels compared to cells transfected with universal control negative siRNA (siRNA negative control = siNC) in both undifferentiated and P19 neurons, as confirmed by western blot analysis (Fig. 4a,b).

NFE2L1 siRNA-mediated Knockdown. Transfection of (a) Undifferentiated and (b) P19 neurons with Nfe2l1 siRNA (si1 = siNEF2L1-1, si2 = siNEF2L1-2, and si3 = siNEF2L1-3) results in significant Knockdown of this gene. Error bars represent mean ± S.E.M. (n = 2 per group). Statistical significance is denoted as *p < 0.05, **p < 0.01, ***p < 0.001, and ns (not statistically significant).
These results demonstrate the successful knockdown of Nfe2l1 in both undifferentiated and P19 neurons using siRNA transfection, indicating the effectiveness of RNAi-mediated gene silencing in modulating Nfe2l1 expression.
Nfe2l1 KD in neuronal cells: MAP2 and ubiquitination regulation
Knockdown of Nfe2l1 significantly reduced both the mRNA expression and protein levels of the neuronal marker MAP2 compared to the siNC-treated control group in neuronal-differentiated P19 cells, as determined by western blotting and qPCR (Fig. 5a–c). Despite this reduction, MAP2 mRNA and protein levels remained significantly higher compared to undifferentiated cells, suggesting a partial disruption in neurogenesis.

MAP2 expression is altered via NFE2L1 KD in P19 neurons. (a) Western blot analysis shows a significant reduction in protein levels of the neuronal marker MAP2 in NFE2L1 KD cells compared to siNC-treated control cells. (b) Quantification of western blotting shows a significant decrease in MAP2 protein levels in differentiated NFE2L1 siRNA-treated cells compared to siNC-treated control cells, with undifferentiated cells showing the lowest levels. Data are presented as mean ± SEM, n = 3 per group. Statistical significance was determined using one-way ANOVA with post hoc tests (***p < 0.001, *p < 0.05). (c) qPCR results demonstrate a similar trend in MAP2 mRNA expression, with levels remaining significantly higher (**p < 0.01) in KD cells compared to undifferentiated cells, indicating a disruption in neurogenesis. Data are presented as mean ± SEM, (n = 3 per group). (d) Immunocytochemistry (ICC) confirms the presence of neuronal projections in control cells (siNC), while these connections are markedly absent in NFE2L1 KD cells. Although some signalling is detected in approximately half of the KD cells, the lack of established connections highlights the detrimental impact of NFE2L1 KD on neuronal differentiation and connectivity. Scale bar = 20 µm. (e) Quantification of MAP2 expression reveals a significant reduction in the corrected MAP2 intensity in NFE2L1 KD cells compared to control cells (***p < 0.001, unpaired t-test, n = 6 per group). Data represent mean ± SEM.
Moreover, immunocytochemistry analysis confirmed the presence of neuronal projections in control conditions, but a significant absence of these connections in Nfe2l1 knockdown cells. Notably, while some signaling was still detected in approximately half of the cells, the lack of established connections underscores the impact of Nfe2l1 depletion on neuronal differentiation and connectivity (Fig. 5d,e).
Additionally, the mRNA levels of the neural progenitor marker Pax6, which is crucial for neurogenesis and differentiation, were significantly reduced (*p < 0.05) following Nfe2l1 knockdown (Supplementary Fig. 2), indicating impaired neuronal differentiation. Western blot analysis of ubiquitin levels showed that polyubiquitinated proteins were present in both control groups, with higher levels in Mg132-treated cells compared to DMSO-treated cells (Fig. 6a). In the NFE2L1 KD group treated with Mg132, an even greater accumulation of ubiquitinated proteins was observed compared to the Mg132-treated control group. ICC results corroborated these findings, showing increased levels of ubiquitinated proteins in the NFE2L1 KD groups (Fig. 6c) compared to control (Fig. 6b). This indicates that ubiquitination is a normal physiological process enhanced under proteasome inhibition by Mg132, and that NFE2L1 KD further stimulates ubiquitin accumulation (Fig. 6d).

Further ubiquitination is triggered within NFE2L1 KD. In P19 neurons, western blot analysis (a) shows that poly-ubiquitinated proteins are present in both control groups, with higher levels observed in Mg132-treated cells compared to DMSO-treated cells. In the NFE2L1 KD group treated with Mg132, there is a more pronounced accumulation of ubiquitinated proteins compared to the Mg132-treated control group. ICC corroborates these findings, demonstrating increased levels of ubiquitinated proteins in (c) NFE2L1 KD cells relative to (b) control cells, scale bar = 20 µm. (d) Quantification of ubiquitin aggregates in ICC, reveals a significant increase in the number of aggregates in NFE2L1 KD cells compared to control cells (siNC). Data were analysed using One-way ANOVA with Tukey’s post-hoc test, and are presented as mean ± SEM. The number of ubiquitin aggregates per cell was counted for all 4 groups, based on 10 images taken from 2 biological replicates (5 images per replicate), with 100 cells counted per group. *p < 0.05, **p < 0.01.
These observations align with findings from other studies across different systems16,17, reinforcing the notion that Nfe2l1 regulates ubiquitination processes within the neuronal system. Altogether, our data indicate that the absence of Nfe2l1 disrupts neuronal differentiation, as evidenced by reduced Map2 levels, and leads to increased ubiquitin accumulation, which is further enhanced by proteasome inhibition.
Nfe2l1 KD in neuronal cells: selective mRNA expression changes
To assess the effects of Nfe2l1 KD on the expression of 12 selected genes associated with PD and proteasome inhibition, qPCR analysis was performed in P19 neurons. The cells were transfected with either Nfe2l1 siRNA or siNC, followed by treatment with either DMSO or Mg132. mRNA levels were compared among four experimental groups: siNC with DMSO treatment, siNC with Mg132 treatment, Nfe2l1 KD with DMSO treatment, and Nfe2l1 KD with Mg132 treatment. Among the 12 selected genes, Nfe2l1 KD (siNFE2L1) resulted in significantly decreased mRNA levels of Atf6 (**p < 0.01), Camk2d (**p < 0.01), and Sod1 (**p < 0.01) (Fig. 7a–c) compared to control cells transfected with siNC, in the presence of Mg132. Six genes showed significant upregulation in response to Mg132 treatment, regardless of Nfe2l1 KD. These genes include Keap1 (**p < 0.01), Ubb (**p < 0.01), Casp3 (**p < 0.01), Eif2ak3 (**p < 0.01), Tuba8 (*p < 0.05), and Tuba4a (***p < 0.001) (Supplementary Fig. 3a–f). The mRNA levels of Tubb2a, Daxx, and Sic39a9 were unaffected by both Nfe2l1 KD and Mg132 treatment (Supplementary Fig. 4a–c). These results indicate complex regulatory interactions between Nfe2l1, Mg132 treatment, and the expression of genes associated with PD and proteasome inhibition in P19 neurons. The observed changes in gene expression suggest potential mechanisms underlying the compensatory response to proteasome inhibition and warrant further investigation into the functional significance of these alterations.

Impact of Nfe2l1 KD on PD-related gene expression in P19 neurons. qPCR analysis of P19 neurons shows that, among the 12 selected genes associated with PD and proteasome obtained from RNA sequencing, the Knockdown of Nfe2l1 significantly decreased mRNA levels of (a) Atf6 (**p < 0.01), (b) Camk2d (**p < 0.01), and (c) Sod1 (**p < 0.01) in the presence of Mg132. Data are mean ± SEM, n = 3 per group, one-way ANOVA with post hoc tests.
Regulation of lncRNAs by proteasome activity and Nfe2l1 in neuronal differentiated cells
Given the emerging role of long non-coding RNAs (lncRNAs) in neurodegenerative diseases and the intricate interplay between proteasome function and neuronal homeostasis18,19, we sought to investigate the potential impact of proteasome inhibition on the expression of lncRNAs in P19 WT cells and P19 neurons. Building upon existing literature implicating lncRNAs such as Malat1 and Neat1 in neurodegenerative processes20,21,22,23, we aimed to elucidate their response to proteasome inhibition and explore their putative involvement in cellular stress responses and neurodegeneration. We examined the expression levels of two prominent lncRNAs, Malat1 and Neat1, in P19 WT cells and P19 neurons treated with the proteasome inhibitor Mg132 compared to control cells treated with DMSO by qPCR. Our results revealed a significant upregulation of both Neat1 and Malat1 RNA levels in Mg132-treated cells compared to control cells (Fig. 8a,b). These findings indicate that proteasome inhibition leads to the upregulation of Malat1 and Neat1 lncRNAs in both P19 WT cells and P19 neurons. Furthermore, to investigate the effect of Nfe2l1 KD on the expression of Neat1 and Malat1, P19 WT cells and neurons were transfected with Nfe2l1 siRNA and siNCs in the presence and absence of Mg132 and subjected to qPCR. The results showed that Nfe2l1 KD led to a significant reduction in Neat1 RNA levels (**p < 0.01) (Fig. 8c) compared to cells transfected with siNC in presence of Mg132. However, no significant change in Malat1 RNA levels was observed following Nfe2l1 KD (Fig. 8d). These findings support the previous findings regarding a specific regulatory role of Nfe2l1 in Neat1 transcription, highlighting its potential involvement in neuronal development and PD pathogenesis. Further studies are warranted to elucidate the underlying mechanisms and functional significance of this regulatory interaction.

Impact of Proteasome inhibition and Nfe2l1 KD on lncRNA expression. This Figure demonstrates the impact of proteasome inhibition on the expression of Malat1 and Neat1 lncRNAs, in undifferentiated and P19 neurons (neuronal diffrentiated). Our findings indicate a significant upregulation of both (a) Neat1 (***p < 0.001) and (b) Malat1 (***p < 0.001, **p < 0.01) in Mg132-treated cells compared to control cells treated with DMSO. Additionally, Nfe2l1 KD resulted in a significant reduction in (c) Neat1 RNA levels (**p < 0.01), but no significant change was observed in (d) Malat1 RNA levels (ns). Error bars represent mean ± S.E.M. (n = 3 per group).
Discussion
This study explored the molecular mechanisms underlying ubiquitinated protein accumulation induced by Nfe2l1 KD in neuronal-differentiated P19 cells, shedding light on its implications for neurodegenerative diseases such as PD. Neurodegenerative disorders are characterized by protein aggregation and neuronal dysfunction, making neuronal differentiation models crucial for understanding their pathophysiology. Our findings highlight the pivotal role of Nfe2l1 in maintaining neuronal proteostasis. Knockdown of Nfe2l1 during retinoic acid-induced neurogenesis in P19 cells resulted in exacerbated ubiquitinated protein accumulation, a hallmark of neurodegeneration. This observation aligns with existing literature linking impaired protein degradation pathways to PD pathogenesis2,24. Furthermore, transcriptomic analysis revealed significant alterations in gene expression profiles upon Nfe2l1 KD, particularly affecting Atf6, Camk2d, Sod1, and lncRNA Neat1.
The activity of Nfe2l1 is tightly regulated through various post-translational modifications and cellular localization mechanisms. Under normal conditions, Nfe2l1 is retained in the endoplasmic reticulum (ER) membrane and undergoes a series of modifications, including glycosylation and proteolytic cleavage, which are critical for its activation and nuclear translocation and upon cellular stress or proteasome inhibition, Nfe2l1 is cleaved and translocated to the nucleus, where it activates the transcription of target genes involved in proteasome synthesis and other aspects of proteostasis9,10,14,15. This finely-tuned regulatory mechanism ensures that NFE2L1 can swiftly respond to changes in the cellular environment, particularly during conditions of proteotoxic stress, to restore homeostasis and prevent neuronal damage8. By understanding the specific roles and regulatory mechanisms of NFE2L1, we can gain insights into how neurons manage proteotoxic stress and maintain proteostasis, providing potential therapeutic targets for enhancing neuronal resilience in neurodegenerative diseases such as PD. In vivo studies have highlighted the critical role of NFE2L1 in neuronal health and function. For instance, research has demonstrated that NFE2L1 is essential for protecting neurons against oxidative stress and neurodegenerative processes by regulating the expression of genes involved in the cellular response to stress11,14. These studies also suggest that NFE2L1 contributes to maintaining neuronal protein homeostasis and preventing neuronal dysfunction, which are key factors in neurodegenerative diseases such as PD11,14,15. Furthermore, the role of Nfe2l1 extends to the regulation of neuroinflammation and neuronal survival. Evidence indicates that Nfe2l1 modulates the expression of neuroprotective factors and participates in cellular mechanisms that counteract neurotoxic conditions, thereby supporting neuronal resilience11,12,13. This regulatory capacity is crucial for maintaining neuronal integrity and function in the context of neurodegenerative diseases, where dysregulation can lead to significant neuronal damage and loss.
Our RNA sequencing analysis revealed significant changes in gene expression associated with proteasome inhibition, particularly in the context of neuronal differentiation. In neuronal-differentiated P19 cells treated with the proteasome inhibitor Mg132, we observed a substantial alteration in 3952 genes, with 2237 of these being commonly affected in both undifferentiated and differentiated states. This differential gene expression underscores the intricate interplay between proteasome activity and neuronal differentiation, highlighting pathways that are crucial for maintaining neuronal health. KEGG pathway enrichment analysis identified neurodegeneration-related pathways, particularly PD, as significantly impacted by proteasome inhibition. These findings suggest that the disruption of proteasome function may contribute to neurodegenerative processes by influencing key regulatory pathways. Importantly, many of the genes associated with these pathways, including those involved in oxidative stress responses and neuronal differentiation, showed altered expression in response to proteasome inhibition, reinforcing the role of Nfe2l1 as a neuroprotective factor. Our findings suggest that Nfe2l1 may play a significant role in regulating the expression of genes involved in oxidative stress management and protein quality control, such as Atf6, Camk2d, and Sod1. These regulatory functions indicate that Nfe2l1 could be an important modulator in protecting neurons from degeneration. Our data are consistent with previous studies, which also highlight the potential significance of Nfe2l1 in maintaining neuronal homeostasis11,14,15,25. This positions Nfe2l1 as a potential target for future therapeutic strategies aimed at mitigating neurodegenerative diseases.
As mentioned before, we found that Nfe2l1 KD in neurons leads to downregulation of Atf6, a key regulator of the unfolded protein response (UPR), which is crucial for managing ER stress and protein quality control in neurons. ATF6 is crucial for managing ER stress, and its impaired signalling, as seen with α-synuclein in Parkinson’s disease and other stressors in Alzheimer’s disease, significantly contributes to the progression of neurodegenerative diseases by disrupting protein quality control and increasing pro-apoptotic signalling26. In 2012, Hashida and colleagues demonstrated that ATF6A plays a pivotal role in protecting dopaminergic neurons from degeneration in Parkinson’s disease by regulating critical neurotrophic factors such as brain-derived neurotrophic factor (BDNF) and oxidative stress responses, including heme oxygenase-1 (HO-1) and xCT. Their study revealed that ATF6A-deficient mice have accelerated neuronal loss, along with significantly reduced levels of BDNF, HO-1, and xCT following MPTP/probenecid injections. This highlights the crucial role of ATF6A in modulating neurotrophic support and mitigating oxidative stress, which are essential for neuronal survival and function in the context of neurodegeneration27. This observation suggests a potential link between Nfe2l1 and UPR pathways in managing ER stress, which aligns with the broader understanding that disturbances in ER stress are implicated in neurodegenerative diseases such as Parkinson’s and Alzheimer’s28. This finding highlights the potential role of Nfe2l1 in the cellular mechanisms underlying these disorders and may provide novel insights into disease mechanisms and therapeutic targets.
Another gene crucial for neuronal signalling and synaptic plasticity is CAMK2D, a calcium/calmodulin-dependent protein kinase29,30. Its decreased expression upon NFE2L1 KD suggests a regulatory role of Nfe2l1 in neuronal calcium signalling pathways, which are implicated in PD pathophysiology31,32. Our finding that Nfe2l1 KD leads to decreased Camk2d expression aligns with other studies showing that oxidative stress disrupts CAMKII function and its interaction with calmodulin in PD31,33,34. This disruption, characterized by the formation of disulfide bridges that impair CAMKII–calmodulin binding, highlights the critical role of calcium signalling in PD pathology33,34,35. The involvement of Nfe2l1 in regulating CAMK2D implies it may influence both oxidative stress responses and calcium signalling pathways essential for maintaining dopaminergic neuron function. These insights suggest that targeting Nfe2l1 could help mitigate oxidative stress effects on calcium signalling and advance our understanding of neurodegenerative processes in PD.
SOD1, a crucial antioxidant enzyme, defends neurons against oxidative stress by converting superoxide radicals into hydrogen peroxide and molecular oxygen36. The downregulation of Sod1 observed in Nfe2l1-deficient cells may intensify oxidative stress, thereby contributing to neuronal dysfunction seen in PD37. Our finding that Nfe2l1 knockdown leads to decreased mRNA levels of Sod1 in neuronal cells builds on similar observations in mouse embryonic fibroblast cells, where Nfe2l1-/- also resulted in reduced Sod1 expression38. Although the previous studies were conducted in fibroblasts, they support the role of Nfe2l1 in regulating Sod1. In the context of neurons, reduced SOD1 levels due to Nfe2l1 deficiency could further exacerbate oxidative stress and increase neuronal vulnerability. This underscores the essential role of Nfe2l1 in neuroprotection and suggests that its dysfunction may significantly contribute to the pathogenesis of neurodegenerative diseases.
Recent studies have highlighted the critical role of long non-coding RNAs (lncRNAs) in regulating cellular homeostasis and disease pathology within the neuronal system39,40,41. lncRNAs are pivotal regulators of gene expression, chromatin dynamics, and cellular stress responses, and are crucial for biological processes such as stem cell pluripotency and neurogenesis42,43. Their regulatory roles extend across transcriptional, epigenetic, and post-transcriptional levels. Additionally, lncRNAs are implicated in the pathogenesis of neurological disorders, including PD44,45. Despite the recognized individual roles of lncRNAs and Nfe2l1, their interaction particularly under stress conditions during neuronal development remains poorly understood.
The lncRNA Neat1 has emerged as a critical regulator in neuronal systems46, especially in response to proteasome inhibition and neurodegenerative disorders47,48,49. Recent research underscores its involvement in stress responses and neuroprotection, particularly where its RNA levels are significantly upregulated under proteasome inhibition50,51. Our current observation of increased Neat1 expression in response to proteasome inhibition highlights its dynamic regulation in proteotoxic stress conditions within differentiated neurons. Specifically, in certain studies the lncRNA Neat1 has been identified as a significant regulator of α-synuclein expression and aggregation. Neat1 has been reported not only to promote the transcription of α-synuclein but also to modulate apoptotic pathways by enhancing the BAX/BCL ratio and CASP3 activity, suggesting its critical role in α-synuclein-associated neuronal apoptosis in PD51,52. Furthermore, dysregulation of Neat1 has been associated with various neurodegenerative disorders, impacting neuronal viability and function through mechanisms involving RNA–protein interactions and epigenetic modifications22,23. In our study, we observed a novel finding where Nfe2l1 KD resulted in reduced Neat1 RNA levels during proteasome inhibition in neuronal-differentiated P19 cells. This suggests a potential regulatory role of Nfe2l1 in modulating Neat1 expression under conditions of cellular stress and protein degradation. The interaction between Nfe2l1 and Neat1 warrants further investigation to elucidate the underlying molecular mechanisms. It is plausible that Nfe2l1 may directly or indirectly modulate Neat1 expression through transcriptional regulation or by influencing pathways involved in RNA stability and processing. This potential interaction underscores the complex network of regulatory pathways involved in neuronal proteostasis and their relevance to neurodegenerative diseases. Given the crucial role of Nfe2l1 in regulating proteostasis, its modulation presents a promising therapeutic strategy for PD. Enhancing Nfe2l1 activity could improve the cellular ability to manage proteotoxic stress, thereby reducing protein aggregation and neuronal dysfunction. Developing small molecules or gene therapies to enhance Nfe2l1 function or mimic its activity could offer novel treatments for PD. Screening for Nfe2l1 activity and its downstream effects could also serve as a valuable diagnostic tool to identify early stages of PD or monitor disease progression.
In conclusion, our study integrates the roles of Nfe2l1 in the regulation of Atf6, Camk2d, Sod1, and Neat1 in neuronal proteostasis, emphasizing its significant implications for neurodegenerative disorders like PD. By modulating protein degradation pathways, influencing the expression of key neuroprotective genes, and regulating noncoding RNA dynamics, NFE2L1 emerges as a pivotal regulator in neurodegenerative diseases characterized by protein aggregation. Additionally, we highlight the role of Nfe2l1 in neuronal differentiation, as demonstrated by the reduction of neuronal markers Map2 and Pax6 following Nfe2l1 KD in RA-induced P19 cells. This suggests that Nfe2l1 is not only crucial for maintaining neuronal homeostasis but also plays a significant role in the differentiation process of neurons.
Future research should focus on unravelling the specific mechanisms linking Nfe2l1 function to these regulatory elements and evaluating their therapeutic potential in restoring neuronal homeostasis, enhancing neuronal differentiation, and mitigating protein aggregation in PD.
Methods
Cell culture and neuronal differentiation
P19 embryonal carcinoma cells were cultured in Maintenance Medium (DMEM High Glucose w/ L-Glutamine w/ Sodium Pyruvate supplemented with 10% FBS, 100 units/mL penicillin and 100 units/mL streptomycin), and maintained at 37 °C in a humidified atmosphere with 5% CO2.
For neuronal differentiation, P19 cells were induced with 0.5 μM retinoic acid (Cat. #R2625, Sigma-Aldrich, St. Louis, MO, USA) in a Differentiation Medium (same components as the Maintenance Medium, but supplemented with 2% FBS instead of the standard 10%), for 4 days in a non-adherent culture dish (Cat. #633,180; Greiner bio-one GmbH, Kremsmünster, Austria) to promote the formation of embryoid bodies (EBs). Subsequently, EBs were plated onto adherent culture dishes and allowed to further differentiate in Maintenance Medium for an additional 6 days53,54. This approach ensures optimal conditions for neuronal differentiation while modulating the FBS concentration to facilitate the formation and maturation of embryoid bodies and subsequent neuronal differentiation54.
siRNA transfection and Nfe2l1 KD
Nfe2l1 KD was achieved using specific small interfering RNA (siRNA). P19 cells were transfected with three different Nfe2l1 siRNAs (siNFE2L1-1 Sense: GCCUGUAGAAGAAUUCAAU, Antisense: AUUGAAUUCUUCUACAGGC, siNFE2L1-2 Sense: CUUUGGCUCUACCAACCUA, Antisense: UAGGUUGGUAGAGCCAAAG, siNFE2L1-3 Sense: CAACCUGCCUGUAGAAGAA, Antisense: UUCUUCUACAGGCAGGUUG) or MISSION® siRNA Universal Negative Control (siNC) (Cat. #SIC-001, Sigma-Aldrich, Saint Louis, MO, USA) using Lipofectamine™ RNAiMAX Transfection Reagent (Cat. #13,778,075, Thermo Fisher Scientific, Waltham, MA, USA) and Opti-MEM™ (Cat.# 51,985,034, Thermo Fisher Scientific, Waltham, MA, USA) following the protocol outlined in our previous publication53 with siRNA final concentration of 20 nM. Transfection efficiency and KD levels were confirmed by western blotting (Fig. 4A,B).
Proteasome inhibition
Proteasome inhibition was induced using Mg132 (Cat. #C2211-5MG, Sigma-Aldrich, Saint Louis, MO, USA), a potent proteasome inhibitor. Differentiated P19 cells were treated with 25 μM Mg132 for 2 h. Control group was treated with Dimethyl sulfoxide (DMSO), the solvent of Mg132. Both siNFE2L1 and siNC groups were subjected to Mg132 treatment to investigate the effects of Nfe2l1 KD under proteotoxic stress.
Quantitative PCR
Total RNA was extracted using NucleoSpin RNA, Mini kit for RNA purification (Cat. #74,095,550; MACHEREY–NAGEL GmbH & Co. KG, Düren, Germany) according to the manufacturer’s instructions. cDNA was synthesized using a NG dART cDNA Reverse Transcription Kit (Cat. #E0801-02, EURx, Gdansk, Poland). qPCR was performed using SYBR Green Master Mix (Cat. #2017-100HS, A&A Biotechnology,) on the Roche LightCyclerⓇ 480 Real-Time PCR System under the following conditions: incubation at 95 °C for 5 min, followed by 40 cycles of 95 °C for 10s, 60 °C for 10s, and 72 °C for 10s. Relative expression levels of target genes were normalized to Gapdh and analysed using the 2(−ΔΔCt) method. Primer sequences for target genes, including Nfe2l1 can be found in Table 2.
Western blotting
Protein samples were extracted using T-PER buffer with added protease and phosphatase inhibitors (Cat. #78,510, Thermo Fisher Scientific, Waltham, MA, USA). Protein concentration was determined using a BCA assay (Cat. #23,225, Thermo Fisher Scientific, Waltham, MA, USA). Equal amounts of protein (20 µg per lane) were separated by SDS-PAGE and transferred to PVDF membranes (Cat. #IPFL00010, Merck KGaA, Cork, Ireland). The membranes were then blocked with 5% non-fat dry milk and incubated overnight at 4 °C with primary antibodies against NFE2L1 (Cat. #8052, Cell Signalling Technology, Danvers, MA, USA), MAP2 (Cat. #PA5-17,646, Thermo Fisher Scientific, Waltham, MA, USA, diluted 1:1000), ubiquitin (Cat. #sc-8017, Santa Cruz Biotechnology, Dallas, TX, USA, diluted 1:1000), OCT-4 (Cat. #2840, Cell Signaling Technology, Danvers, MA, USA, diluted 1:1000) and β-Actin (Cat. #4970, Cell Signalling Technology, Danvers, MA, USA) as a loading control. Following primary antibody incubation, membranes were incubated with HRP-conjugated secondary antibodies at room temperature for 1 h: anti-rabbit IgG for NFE2L1, MAP2, OCT-4 and β-ACTIN (Cat. #A6154, Sigma-Aldrich, Saint Louis, MO, USA, diluted 1:5000) and anti-mouse IgG for ubiquitin (Cat. #A5278, Sigma-Aldrich, Saint Louis, MO, USA, diluted 1:5000). The signals were then detected using an ECL substrate (Cat. #RPN2235, GE Healthcare, Amersham Place, UK) and visualized with Bio-RAD chemiluminescence imaging system.
Immunostaining
For immunostaining, differentiated P19 cells were fixed with 4% paraformaldehyde (Sigma-Aldrich, Saint Louis, MO, USA) for 10–15 min at room temperature, followed by permeabilization with 0.5% Triton X-100 (Sigma-Aldrich, Saint Louis, MO, USA) in PBS (PBST) for 10 min. Cells were then blocked with 5% non-fat dry milk in 0.1% PBST (0.1% Triton X-100 in PBS) for 30 min and incubated with primary antibodies (diluted in 1% non-fat dry milk in 0.1% PBST) against ubiquitin (Cat. #sc-8017, Santa Cruz Biotechnology, Dallas, USA, diluted 1:200) or MAP2 (Cat. #13–1500, Thermo Fisher Scientific, Waltham, MA, USA, diluted 1:400) overnight at 4 °C. After washing with 0.1% PBST, cells were incubated with fluorescently labelled secondary antibodies. For MAP2, a goat anti-rabbit IgG secondary antibody, namely, Alexa Fluor Plus 488 (Cat. # A-11008, Thermo Fisher Scientific, Waltham, MA, USA, diluted 1:500) was used, and in the case of ubiquitin, Alexa Fluor™ 546, Goat anti-Mouse IgG (Cat. #A11030, Thermo Fisher Scientific, Waltham, MA, USA, diluted 1:500) was used as the secondary antibody, for 1 h at room temperature in the dark (to protect from photobleaching). Nuclei were counterstained with DAPI (Cat. #D9564, Sigma-Aldrich, Saint Louis, MO, USA), diluted 1:1000 in PBS, and incubated for 10 min at room temperature. Afterward, cells were washed briefly with PBST. Observations were carried out under a confocal microscope (Nikon A1R, Japan) equipped with 10x, 20x, 40x, and 60 × lenses; Nomarski’s DIC contrast; Hoffman’s modulation contrast; 405 nm, 488 nm, 561 nm, and 640 nm lasers; a hybrid scanner and a resonance scanner. The workstation was equipped with Nikon’s Confocal NIS-Elements package (Nikon, Japan). A computer workstation with advanced image reconstruction and analysis software, including NIS Elements (Nikon) and Imaris Cell (Bitplane), was used for detailed image analysis. Signal intensity quantification was performed using ImageJ software to assess the fluorescence intensity of the specific markers.
RNA sequencing and data analysis
Total RNA from Mg132 and DMSO treated neurons and WT groups was extracted and sent for high-throughput RNA sequencing performed by BGI (BGI Genomics, Shenzhen, China). Multi-omics integration analysis, visualization, and interpretation were conducted using Dr. Tom, a web-based solution provided by BGI. Sequencing was performed on a DNB-SEQ platform. Differentially expressed genes (DEGs) were identified using DESeq2. Pathway enrichment analysis of DEGs was performed by BGI software, Dr. Tom.
Statistical analysis
Data are presented as mean ± standard error of the mean (SEM) from at least three independent experiments. Statistical significance was determined using Student’s t-test or one-way ANOVA with Tukey’s post-hoc test where applicable. A *p-value < 0.05 was considered as statistically significant.
Data availability
The datasets generated and/or analyzed during the current study are publicly available in the NCBI Sequence Read Archive (SRA) under the BioProject accession number PRJNA1196185.
References
-
Taoufik, E., Kouroupi, G., Zygogianni, O. & Matsas, R. Synaptic dysfunction in neurodegenerative and neurodevelopmental diseases: An overview of induced pluripotent stem-cell-based disease models. Open Biol. 8, 180138 (2018).
-
Morris, H. R., Spillantini, M. G., Sue, C. M. & Williams-Gray, C. H. The pathogenesis of Parkinson’s disease. The Lancet 403, 293–304 (2024).
-
Behl, T. et al. Exploring the role of ubiquitin–proteasome system in Parkinson’s disease. Mol. Neurobiol. 59, 4257–4273 (2022).
-
Nandi, D., Tahiliani, P., Kumar, A. & Chandu, D. The ubiquitin-proteasome system. J. Biosci. 31, 137–155 (2006).
-
Rousseau, A. & Bertolotti, A. Regulation of proteasome assembly and activity in health and disease. Nat. Rev. Mol. Cell Biol. 19, 697–712 (2018).
-
Shiber, A. & Ravid, T. Chaperoning proteins for destruction: Diverse roles of Hsp70 chaperones and their co-chaperones in targeting misfolded proteins to the proteasome. Biomolecules 4, 704–724 (2014).
-
Filtz, T. M., Vogel, W. K. & Leid, M. Regulation of transcription factor activity by interconnected post-translational modifications. Trends Pharmacol. Sci. 35, 76–85 (2014).
-
Chandran, A., Oliver, H. J. & Rochet, J.-C. Role of NFE2L1 in the regulation of proteostasis: Implications for aging and neurodegenerative diseases. Biology 12, 1169 (2023).
-
Kim, H. M., Han, J. W. & Chan, J. Y. Nuclear factor erythroid-2 like 1 (NFE2L1): Structure, function and regulation. Gene 584, 17–25 (2016).
-
Liu, X., Xu, C., Xiao, W. & Yan, N. Unravelling the role of NFE2L1 in stress responses and related diseases. Redox Biol. 65, 102819 (2023).
-
Taniguchi, H. et al. Possible roles of the transcription factor Nrf1 (NFE2L1) in neural homeostasis by regulating the gene expression of deubiquitinating enzymes. Biochem. Biophys. Res. Commun. 484, 176–183 (2017).
-
Villaescusa, J. C. et al. A PBX1 transcriptional network controls dopaminergic neuron development and is impaired in Parkinson’s disease. EMBO J. 35, 1963–1978 (2016).
-
Lee, C. S. et al. Loss of nuclear factor E2-related factor 1 in the brain leads to dysregulation of proteasome gene expression and neurodegeneration. Proc. Natl. Acad. Sci. 108, 8408–8413 (2011).
-
Łuczyńska, K., Zhang, Z., Pietras, T., Zhang, Y. & Taniguchi, H. NFE2L1/Nrf1 serves as a potential therapeutical target for neurodegenerative diseases. Redox Biol. 69, 103003 (2024).
-
Khodadadi, H., Łuczyńska, K., Winiarczyk, D., Leszczyński, P. & Taniguchi, H. NFE2L1 as a central regulator of proteostasis in neurodegenerative diseases: Interplay with autophagy, ferroptosis, and the proteasome. Front. Mol. Neurosci. 18, 1551571 (2025).
-
Ofoghi, A. et al. Activating the NFE2L1-ubiquitin-proteasome system by DDI2 protects from ferroptosis. Cell Death Differ., 1–8 (2024).
-
Hu, X. et al. UBE4A catalyzes NRF1 ubiquitination and facilitates DDI2-mediated NRF1 cleavage. Biochimica et Biophysica Acta (BBA)- Gene Regulatory Mech. 1866, 194937 (2023).
-
Riva, P., Ratti, A. & Venturin, M. The long non-coding RNAs in neurodegenerative diseases: Novel mechanisms of pathogenesis. Curr. Alzheimer Res. 13, 1219–1231 (2016).
-
Wan, P., Su, W. & Zhuo, Y. The role of long noncoding RNAs in neurodegenerative diseases. Mol. Neurobiol. 54, 2012–2021 (2017).
-
Yao, J. et al. Long non-coding RNA MALAT 1 regulates retinal neurodegeneration through CREB signaling. EMBO Mol. Med. 8, 346–362 (2016).
-
Abrishamdar, M., Jalali, M. & Rashno, M. MALAT1 lncRNA and Parkinson’s Disease: The role in the pathophysiology and significance for diagnostic and therapeutic approaches. Mol. Neurobiol. 59, 5253–5262 (2022).
-
Li, K. & Wang, Z. lncRNA NEAT1: Key player in neurodegenerative diseases. Ageing Res. Rev. 86, 101878 (2023).
-
An, H., Williams, N. G. & Shelkovnikova, T. A. NEAT1 and paraspeckles in neurodegenerative diseases: A missing lnc found?. Non-coding RNA Res. 3, 243–252 (2018).
-
Ebrahimi-Fakhari, D., Wahlster, L. & McLean, P. J. Protein degradation pathways in Parkinson’s disease: Curse or blessing. Acta Neuropathol. 124, 153–172 (2012).
-
Li, F., Gao, B., Dong, H., Shi, J. & Fang, D. Icariin induces synoviolin expression through NFE2L1 to protect neurons from ER stress-induced apoptosis. PLoS ONE 10, e0119955 (2015).
-
Credle, J. J. et al. α-Synuclein-mediated inhibition of ATF6 processing into COPII vesicles disrupts UPR signaling in Parkinson’s disease. Neurobiol. Dis. 76, 112–125 (2015).
-
Hashida, K. et al. ATF6alpha promotes astroglial activation and neuronal survival in a chronic mouse model of Parkinson’s disease. PLoS ONE 7, e47950 (2012).
-
Lindholm, D., Wootz, H. & Korhonen, L. ER stress and neurodegenerative diseases. Cell Death Differ. 13, 385–392 (2006).
-
Rigter, P. M. et al. Role of CAMK2D in neurodevelopment and associated conditions. Am. J. Hum. Genet. 111, 364–382 (2024).
-
Kool, M. J. et al. CAMK2-dependent signaling in neurons is essential for survival. J. Neurosci. 39, 5424–5439 (2019).
-
Bohush, A., Leśniak, W., Weis, S. & Filipek, A. Calmodulin and its binding proteins in Parkinson’s disease. Int. J. Mol. Sci. 22, 3016 (2021).
-
Zhang, S., Xie, C., Wang, Q. & Liu, Z. Interactions of CaMKII with dopamine D2 receptors: Roles in levodopa-induced dyskinesia in 6-hydroxydopamine lesioned Parkinson’s rats. Sci. Rep. 4, 6811 (2014).
-
Di Maio, R. et al. Disulfide bridge formation prevents CaMKII/Calmodulin interaction in Parkinson’s disease. BioRxiv, 2020.2002. 2014.947960 (2020).
-
Pullara, F. et al. NADPH oxidase 2 activity disrupts Calmodulin/CaMKIIα complex via redox modifications of CaMKIIα-contained Cys30 and Cys 289: Implications in Parkinson’s disease. Redox Biol. 75, 103254 (2024).
-
Moriguchi, S., Yabuki, Y. & Fukunaga, K. Reduced calcium/calmodulin-dependent protein kinase II activity in the hippocampus is associated with impaired cognitive function in MPTP-treated mice. J. Neurochem. 120, 541–551 (2012).
-
Eleutherio, E. C. A., Magalhães, R. S. S., de Araújo Brasil, A., Neto, J. R. M. & de Holanda Paranhos, L. SOD1, more than just an antioxidant. Arch. Biochem. Biophys. 697, 108701 (2021).
-
Trist, B. G. et al. Amyotrophic lateral sclerosis-like superoxide dismutase 1 proteinopathy is associated with neuronal loss in Parkinson’s disease brain. Acta Neuropathol. 134, 113–127 (2017).
-
Zhang, S. et al. Synergism and antagonism of two distinct, but confused, Nrf1 factors in integral regulation of the nuclear-to-mitochondrial respiratory and antioxidant transcription networks. Oxid. Med. Cell. Longev. 2020, 5097109 (2020).
-
Srinivas, T., Mathias, C., Oliveira-Mateos, C. & Guil, S. Roles of lncRNAs in brain development and pathogenesis: Emerging therapeutic opportunities. Mol. Ther. 31, 1550–1561 (2023).
-
Aliperti, V., Skonieczna, J. & Cerase, A. Long non-coding RNA (lncRNA) roles in cell biology, neurodevelopment and neurological disorders. Non-coding RNA 7, 36 (2021).
-
Zhao, Y., Liu, H., Zhang, Q. & Zhang, Y. The functions of long non-coding RNAs in neural stem cell proliferation and differentiation. Cell Biosci. 10, 1–10 (2020).
-
Kung, J. T., Colognori, D. & Lee, J. T. Long noncoding RNAs: Past, present, and future. Genetics 193, 651–669 (2013).
-
Ng, S. Y., Johnson, R. & Stanton, L. W. Human long non-coding RNAs promote pluripotency and neuronal differentiation by association with chromatin modifiers and transcription factors. EMBO J. 31, 522–533 (2012).
-
Lyu, Y., Bai, L. & Qin, C. Long noncoding RNAs in neurodevelopment and Parkinson’s disease. Anim. Models Exp. Med. 2, 239–251 (2019).
-
Xin, C. & Liu, J. Long non-coding RNAs in Parkinson’s disease. Neurochem. Res. 46, 1031–1042 (2021).
-
Barry, G. et al. The long non-coding RNA NEAT1 is responsive to neuronal activity and is associated with hyperexcitability states. Sci. Rep. 7, 40127 (2017).
-
Hirose, T. et al. NEAT1 long noncoding RNA regulates transcription via protein sequestration within subnuclear bodies. Mol. Biol. Cell 25, 169–183 (2014).
-
Boros, F. A., Vécsei, L. & Klivényi, P. NEAT1 on the field of Parkinson’s disease: Offense, defense, or a player on the bench?. J. Parkinsons Dis. 11, 123–138 (2021).
-
Yan, W., Chen, Z.-Y., Chen, J.-Q. & Chen, H.-M. LncRNA NEAT1 promotes autophagy in MPTP-induced Parkinson’s disease through stabilizing PINK1 protein. Biochem. Biophys. Res. Commun. 496, 1019–1024 (2018).
-
Adriaens, C. et al. p53 induces formation of NEAT1 lncRNA-containing paraspeckles that modulate replication stress response and chemosensitivity. Nat. Med. 22, 861–868 (2016).
-
Simchovitz, A. et al. NEAT1 is overexpressed in Parkinson’s disease substantia Nigra and confers drug-inducible neuroprotection from oxidative stress. FASEB J. 33, 11223 (2019).
-
Liu, Y. & Lu, Z. Long non-coding RNA NEAT 1 mediates the toxic of Parkinson’s disease induced by MPTP/MPP+ via regulation of gene expression. Clin. Exp. Pharmacol. Physiol. 45, 841–848 (2018).
-
Khodadadi, H. & Taniguchi, H. A method for siRNA-mediated knockdown of target genes in RA-induced neurogenesis using P19 cells. MethodsX 14, 103177 (2025).
-
Leszczyński, P. et al. Neurogenesis using P19 embryonal carcinoma cells. JoVE (Journal of Visualized Experiments), e58225 (2019).
Acknowledgements
The authors express their gratitude to Mrs. Parniansadat Khalafi, Prof. Robert Viger and Dr. Marie France Bouchard for their invaluable support and guidance. This research was conducted at the Department of Experimental Embryology, Institute of Genetics and Animal Biotechnology, Polish Academy of Sciences, with facilities and resources provided.
Ethics declarations
Competing interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Khodadadi, H., Winiarczyk, D., Łuczyńska, K. et al. Nfe2l1 dysfunction alters Parkinson’s disease-related gene expression and impairs neuronal differentiation under ubiquitin stress in neuronal differentiated P19 Cells. Sci Rep 15, 30542 (2025). https://doi.org/10.1038/s41598-025-08204-x
-
Received:
-
Accepted:
-
Published:
-
DOI: https://doi.org/10.1038/s41598-025-08204-x