
Introduction
Osteoarthritis (OA) is a chronic, debilitating joint disorder characterized by widespread morphological alterations, inflammation, and intolerable hyperalgesia. In 2019, China reported the highest number of OA cases globally, with 132.81 million individuals affected, representing a dramatic 156.58% increase since 1990.1 This growing prevalence constitutes a significant and escalating public health challenge, underscoring the urgent need for more effective clinical interventions.
Current clinical therapies for OA are primarily palliative because of the complex nature of its pathogenesis. Standard treatments are generally categorized into physiotherapies, pharmacological approaches, and surgical procedures, on the basis of the modality of intervention. Nonpharmacological strategies include weight management, exercise, assistive devices, and various physical therapies (e.g., therapeutic ultrasound, electrical stimulation, phototherapy, hydrotherapy, magnetotherapy, cryotherapy, and thermotherapy), along with acupuncture.2 Although these treatments can alleviate pain and improve joint mobility to some extent, they are predominantly symptomatic and fail to halt disease progression.
Pharmacological therapies, as outlined by international guidelines, typically involve oral nonsteroidal anti-inflammatory drugs (NSAIDs), intra-articular hyaluronic acid (HA) injections, and opioid administration.1,2 While these treatments may offer anti-inflammatory or analgesic benefits for certain patients, they often fall short of providing sustained symptomatic relief in those with moderate to advanced OA.3 Total joint replacement is considered the final therapeutic option when conservative treatments fail to yield improvements.4 Although clinical data demonstrate significant enhancements in joint function, pain relief, and quality of life following surgery,5 challenges remain—such as the emergence of catastrophizing pain at multiple sites during postsurgical rehabilitation and the limited lifespan of joint prostheses, which may require revision surgery.6 Consequently, novel, effective pharmacological therapies with less invasive administration and fewer adverse events have emerged as a growing clinical demand for OA treatment.
OA has a complicated and multifaceted pathology involving inflammation, hyperalgesia, and cartilage breakdown. Pain, a primary reason that OA patients seek medical treatment, is poorly managed by existing analgesics, highlighting the need for a deeper understanding of OA pain mechanisms. Disease pathogenesis is primarily attributed to pathological changes in three aspects: the extracellular matrix (ECM), chondrocytes, and homeostasis between osteoblasts and osteoclasts. Among these factors, inflammation driven by cytokines and disturbances in chondrocyte organelle function have been identified as key contributing factors. Recent research suggests that targeting cytokine signaling and organelle dysfunction in chondrocytes may offer promising therapeutic opportunities.
This review systematically examines the mechanisms underlying OA pain, along with the roles of cytokines and chondrocyte organelles in disease progression. On the basis of these insights, we discuss emerging therapeutic targets and corresponding pharmacological agents or drug delivery systems that may not only slow but also potentially reverse OA progression while simultaneously delivering meaningful symptomatic relief.
Epidemiology of osteoarthritis
Recent data from the Global Burden of Disease (GBD) study indicate that OA affected 7.6% of the global population—~595 million individuals—in 2020.7,8 The increasing prevalence of OA is partly attributed to geographic region, national economic status, and sociodemographic variables.7,9,10 Emerging evidence from global studies suggests that individuals in socioeconomically deprived areas experience higher rates of OA (hand, hip, and knee).11,12 Interestingly, countries with a high sociodemographic index (SDI), such as Australia, also report increased OA incidence, whereas middle-SDI countries such as China face a rapidly escalating disease burden.7,13
At the individual level, OA incidence demonstrates considerable heterogeneity across demographics. In 2020, the global age-standardized prevalence revealed marked sex disparities, with women exhibiting 8,058.9 cases per 100,000 (95% UI 7251.9–8867.9) compared with 5780.1 per 100,000 (95% UI 5217.8–6341.2) in men.7 Age further stratifies this disparity, with the prevalence increasing sharply among older populations and peaking between the ages of 55 and 64.14 Additionally, OA epidemiology is influenced by ethnic disparities.10,15 US-based research indicates that African Americans report more severe pain and disability than White Americans do (WOMAC SMD: 0.57, 95% CI: 0.54–0.61), whereas Asian Americans also report higher pain levels than their White counterparts do.10,16
OA susceptibility is further influenced by key biomechanical and lifestyle factors. Obesity is one of the most significant modifiable risk factors, particularly for knee OA, with elevated body mass index (BMI) accounting for ~20% of incident cases.7,17 Previous traumatic joint injuries substantially increase OA risk; for example, an isolated anterior cruciate ligament (ACL) injury increases the likelihood of OA by 4.2 times (95% CI: 3.8–10.5), whereas an isolated meniscal injury increases the risk by 6.3-fold (95% CI: 4.9–8.3).18 Physical activity shows a biphasic association with OA risk: both sedentary lifestyles and frequent, high-intensity exercise are linked to greater incidence, whereas moderate activity appears protective.9 Furthermore, individuals in physically demanding occupations—such as construction workers, floor layers, bricklayers, fishermen, and farmers—are disproportionately affected by chronic mechanical stress on joints.19
Osteoarthritis prevention
OA prevention strategies are generally classified into primary and secondary prevention strategies. Primary prevention aims to reduce behaviors or exposures that lead to disease, thereby preventing its onset. In contrast, secondary prevention focuses on halting the progression of disease among individuals already exposed to risk factors.20,21
In OA, three key modifiable risk factors stand out: obesity, prior knee injury, and excessive musculoskeletal loading.22,23 Obesity is particularly important, with a normal BMI defined as 18.5–25 kg/m² and obesity defined as a BMI over 30 kg/m².24 Maintaining a BMI below 25 kg/m² is estimated to reduce the population-attributable risk of OA by 27–53%.22 Weight-reduction strategies center on healthy eating and consistent physical activity.23 While dietary restriction can aid in weight loss, macronutrients (protein, carbohydrate, and fat) have a limited impact unless an individual has obesity-related comorbidities such as diabetes or cardiovascular disease.21 Moreover, sustaining weight loss through diet alone proves challenging—studies have shown that up to 50% of individuals regain some weight loss within a year.25,26 Exercise alone is less effective for weight loss, but it is essential for weight maintenance.21,27 Therefore, a combined approach involving both diet and exercise is generally recommended. Other interventions include cognitive behavioral therapy, which is associated with substantial weight reduction, and bariatric surgery, which offers the most effective and sustained weight loss outcomes.21
Previous knee injuries are also strong predictors of OA.28 Epidemiological studies have revealed that one in two adults who suffer severe knee injury will develop OA in that joint during their lifetime. More than 50% of ACL-injured knees progress to OA within 10 years.29 However, consistent adherence to neuromuscular and proprioceptive training—typically 15–20 min per session, two to three times per week—has been shown to reduce the incidence of ACL injury by up to 50% in high-risk populations such as adolescents and adult athletes.21,22,30 Nevertheless, epidemiological data indicate that injury rates revert to baseline levels upon discontinuation of these programs.21
For individuals with preexisting risk factors such as knee injuries or physically demanding occupations, additional behavioral modifications—including the avoidance of occupational squatting, kneeling, and heavy lifting—can reduce OA risk by 15% to 30% in men.22 Notably, surgical repair of knee injuries does not mitigate the increased OA risk caused by initial trauma, as evidenced by longitudinal biomechanical studies.30,31 Current clinical guidelines emphasize secondary injury prevention and joint-protective exercise regimens as foundational therapeutic strategies.30
Osteoarthritis diagnosis
Timely and accurate differentiation of symptomatic knee OA from other causes of knee pain is critical for ensuring appropriate patient management in clinical practice. When pain or discomfort lasts for weeks to months and is accompanied by the progressive worsening of symptoms—such as stiffness and limited mobility—with minimal pain-free intervals, this clinical pattern is typically indicative of early-stage knee OA.32 Clinical examination often reveals pain upon mobilization, joint-line tenderness, crepitus, or mild joint effusion, and these findings are supported by features such as older age, high BMI, a history of knee trauma, or a family history of OA.33 However, the elusive nature of typical radiographic features in early-stage OA has led to the absence of validated diagnostic criteria to date.34
The diagnosis of symptomatic OA is largely dependent on physical examination. In patients with knee OA, knee effusion is commonly absent or minimal and normothermic, accompanied by popliteal or “baker” cysts—extensions of synovial swelling that can be palpated at the posterior aspect of the knee—and may present with valgus or varus deformities. In contrast, patients with inflammatory, infectious, or crystalline arthritis typically present with a warm knee and easily palpable effusion.35 Additionally, the presence of Heberden’s and Bouchard’s nodes—swelling in the distal and proximal interphalangeal joints, respectively—is indicative of polyarticular disease and is linked to the progression of knee OA.34,36
Imaging modalities offer valuable information on structural changes in knee OA, enabling accurate diagnosis and clinical decision-making. Radiographic assessments are the most widely used and cost-effective imaging modality for depicting bony changes in knee OA, such as osteophyte formation, joint space narrowing, subchondral sclerosis, and subchondral cysts, which are widely applied radiographic criteria for establishing structural OA.35 However, radiography is unsuitable for early-stage OA diagnosis because of its inability to detect pathologic features before hallmark structural changes, such as osteophyte formation, occur.34,37 Magnetic resonance imaging (MRI) provides comprehensive visualization of pathologic changes, enabling the detection of early-stage OA structural alterations. However, its clinical application is hindered by the lack of reliable contrast agents to differentiate cartilage from surrounding synovial fluid, as well as the extended acquisition time (often exceeding 40 min) required to generate diagnostic-quality images.35,36,37 Computed tomography (CT) is not as commonly employed as radiography or MRI in clinical practice because of its inherently poor soft-tissue contrast; nevertheless, it remains a valuable tool for evaluating facet joint involvement in OA. It provides detailed visualization of cortical bone integrity and soft-tissue calcifications, thereby offering unique diagnostic insights into structural changes associated with the disease.38 Ultrasound imaging has emerged as a valuable modality for visualizing joint effusions, osteophytes, and other OA-related features. Its portability and cost-effectiveness have driven its widespread adoption for OA diagnosis in European and US centers. However, its utility in detecting joint space narrowing is less precise than that of MRI, and its detection depth is constrained by the operator’s expertise.37,39 Optical imaging, which provides quantitative information with high spatial resolution for structural OA diagnosis, faces significant limitations because of its shallow penetration depth, which is caused primarily by significant optical scattering within tissues.37 Photoacoustic imaging (PAI), an emerging optical imaging modality, leverages rich contrast, high spatial resolution, and deep penetration depth, thereby overcoming the limitations inherent in conventional optical imaging techniques and providing novel insights for OA diagnosis.40,41
Osteoarthritis pathogenesis
Emerging evidence suggests that OA pathogenesis arises from maladaptive crosstalk between sustained joint inflammation and intracellular organelle dysfunction. Proinflammatory cytokines, notably IL-1β, IL-6, and TNF-α, initiate inflammatory cascades in synovial tissue and chondrocytes. Concurrently, disturbances in mitochondrial bioenergetics, ER stress, and lysosomal destabilization within chondrocytes amplify oxidative stress and trigger apoptotic pathways. This synergy creates a feedforward loop that accelerates extracellular matrix degradation, impairs cartilage homeostasis, and promotes subchondral bone remodeling. The convergence of inflammatory signaling and organelle failure likely drives the transition from early chondrocyte senescence to irreversible tissue destruction, highlighting the potential of dual-targeting strategies to halt OA progression.
Role of proinflammatory cytokines in osteoarthritis
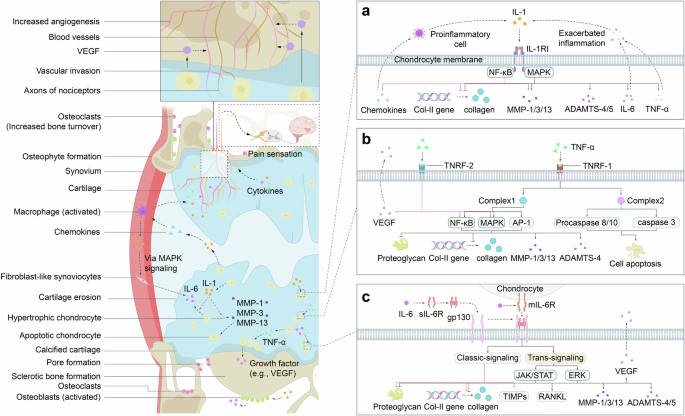
Proinflammatory cytokines play crucial roles in OA pathogenesis, promoting a self-sustaining inflammatory environment within the knee joint. These cytokines perpetuate a vicious cycle through their own signaling pathways, intensifying pain sensation and disrupting joint metabolic balance. They enhance osteoclast activity, leading to bone resorption, and promote the secretion of catabolic enzymes and chondrocyte apoptosis,42 all of which contribute to detrimental alterations in joint morphology. Clinical trials have revealed elevated levels of IL-1β, IL-6, and TNF-α in the synovial fluid, synovial membrane, subchondral bone, and cartilage of patients with OA,43,44 supporting their role as key contributors to catabolic processes in OA pathogenesis (Fig. 1). A comprehensive understanding of their roles offers promising avenues for developing targeted therapies that may surpass the effectiveness of traditional anti-inflammatory treatments.

Schematic representation of the inflammatory signaling pathways and their impact on joint tissues in OA. The left panel illustrates the pathological changes within the joint, emphasizing the role of inflammatory cells and the degradation of the extracellular matrix. The right side delves into the molecular mechanisms underlying these changes, revealing the pivotal role of IL-1, TNF-α, and IL-6 in modulating the activity of key signaling pathways that drive ECM degradation and inflammation. a IL-1 signaling role in osteoarthritis progression. b TNF-α signaling role in osteoarthritis progression. c IL-6 signaling role in osteoarthritis progression. These findings underscore the complex feedback loops involving these cytokines and their downstream effectors, which contribute to the chronic inflammation and tissue remodeling characteristic of OA. The figure also incorporates the vascular aspects of OA, highlighting the role of VEGF in angiogenesis and its influence on chondrocyte survival and function. This schematic provides a visual summary of the current understanding of OA pathogenesis, emphasizing the multifactorial nature of the disease and the potential therapeutic targets within the inflammatory signaling cascades
IL-1
The IL-1 family comprises IL-1α, IL-1β, and the IL-1 receptor antagonist (IL-1Ra), with IL-1α and IL-1β acting as agonists. Both IL-1α and IL-1β are critical proinflammatory cytokines involved in OA etiology, causing inflammation and ECM degradation in articular cartilage, as evidenced in clinical trials,44,45,46 ultimately leading to cartilage destruction. These cytokines are synthesized primarily as precursor peptides, pro-IL-1α and pro-IL-1β, in monocytes and macrophages. Pro-IL-1α is biologically active and can be cleaved by the cytosolic cysteine protease calpain to generate mature IL-1α, which has a greater binding affinity to IL-1 receptors.47,48 Interestingly, IL-1α is rarely detected in healthy human tissues but is substantially elevated in disease states, such as OA, suggesting that it may be released from lysed cells as an alarmin signal of cellular damage or necrosis.48,49,50 In contrast, pro-IL-1β is biologically inactive and requires cleavage by caspase-1 (also known as ICE) to become active IL-1β.48,49,51 IL-1β is produced by various cell types in the knee joint, including chondrocytes, mononuclear cells, osteoblasts, activated macrophages, and synovial tissues.44,45,46 It is found at elevated levels in the synovial fluid, synovial membrane, cartilage, and subchondral tissue of OA patients.44,52 Both IL-1α and IL-1β mediate inflammation by binding to type I IL-1 receptor I (IL-1RI), a membrane protein expressed in various cells, such as chondrocytes, synovial fibroblasts, osteoblasts, osteoclasts, and macrophages.53,54,55 IL-1RI contains three extracellular immunoglobulin-like domains and an intracellular Toll/IL-1R (TIR) domain.48,56 The interaction between IL-1 and IL-1RI activates NF-κB, leading to inflammatory responses.50,56,57
Additionally, IL-1 promotes the secretion of proinflammatory cytokines and mediators, including IL-6, TNF-α, nitric oxide (NO), cyclooxygenase-2 (COX-2), and PGE2.58 It also enhances the expression of matrix-degrading enzymes,59 particularly matrix metalloproteinases (MMPs) and disintegrin-like and metalloproteinase with thrombospondin motifs (ADAMTS), which mediate the degradation of ECM proteins, such as MMP-1, MMP-3, MMP-13, ADAMTS-4, and ADAMTS-5.44,45,53,60 IL-1 also impairs proteoglycan aggrecan and type II collagen synthesis by suppressing SRY-box transcription factor 9 (SOX-9),60 a critical gene whose protein product directs the transcription of genes encoding type II collagen and the proteoglycan aggrecan via the MAPK signaling pathway,61 further aggravating the inflammatory microenvironment initiated by IL-1.44,53
Recent studies have highlighted the critical role of IL-1 in activating the NF-κB signaling pathway, which exacerbates OA progression. NF-κB activation induces the expression of MMPs (MMP-1, MMP-2, MMP-3, MMP-7, MMP-8, MMP-9, and MMP-13) and ADAMTS family members (ADAMTS-4 and ADAMTS-5), as well as inflammatory mediators, including COX-2, PGE2, NO, inducible nitric oxide synthase (iNOS), and hypoxia-inducible factor-2α, all of which have been validated both in vitro (e.g., chondrocyte culture) and in vivo (e.g., murine OA models), and further enhances MMP expression.53,62,63 Additionally, NF-κB activation increases the secretion of cytokines and chemokines such as TNF-α, IL-1β, IL-8, monocyte chemoattractant protein-1 (MCP-1/CCL2), CCL5, macrophage inflammatory protein-1α (MIP-1α), and receptor activator of NF-κB ligand (RANKL).53,63 These chemokines recruit inflammatory cells, such as activated macrophages, which are a major source of IL-1β in the synovium, thereby contributing to a vicious cycle of IL-1-mediated catabolic events that accelerate OA progression (Fig. 1a).53
This IL-1-mediated signaling is subject to reversible regulation by the naturally occurring inhibitors IL-1Ra and IL-1RII. IL-1Ra, which is synthesized in the Golgi apparatus, is primarily secreted from the same cells that release IL-1β.57 Similar to IL-1α and IL-1β, IL-1Ra competitively binds to the same site on IL-1RI, exhibiting near-equal avidity, thus inactivating IL-1 signaling.44,49,53,64 Complete inhibition of IL-1 signaling requires a 100–1000-fold molar excess of IL-1Ra to generate IL-1, as even minimal engagement of IL-1 with IL-1RI is sufficient to initiate cellular inflammatory responses.48,50 IL-1RII, a member of the IL-1 receptor family, shares structural similarities with IL-1RI but lacks the TIR domain, making it a decoy receptor that does not initiate signal transduction.48 Additionally, IL-1RII exists in both membrane-bound and soluble forms, both of which exhibit a greater affinity for IL-1β than for IL-1α or IL-1Ra, which bind almost irreversibly to IL-1β.48,56
TNF-α
TNF-α is a pivotal proinflammatory cytokine that plays a central role in driving numerous inflammatory responses. Elevated TNF-α secretion, in combination with IL-1β, contributes to deleterious morphological changes in joints during OA progression by modulating gene expression, apoptosis, differentiation, proliferation, and inflammatory pathways.53,65,66 TNF-α is initially synthesized as a transmembrane form (tmTNF-α) by the same cell types and tissues that produce IL-1β, including chondrocytes, mononuclear cells, osteoblasts, and synovial tissues.44,65 This tmTNF-α, a stable homotrimeric type II transmembrane protein, can be cleaved by the metalloprotease TNF-α-converting enzyme (TACE) into its soluble form (sTNF-α), which is then released into the extracellular environment.65
TNF-α exerts its effects by binding to two cognate receptors, tumor necrosis factor receptor 1 (TNFR-1) and TNFR-2 (Fig. 1b). Recent studies indicate that TNFR-1 is primarily responsible for TNF-α-mediated signaling, as it can interact with both tmTNF-α and sTNF-α and is expressed in almost all cell types. In contrast, TNFR-2 preferentially binds to tmTNF-α and is expressed in a more limited subset of cells.53,65 These two receptors initiate distinct signal transduction pathways, largely due to differences in their intracellular domains. TNFR-1 contains a death domain (DD) that is absent in TNFR-2.65 Under resting conditions, TNFR-1 remains inactive as the silencer of the death domain (SODD) binds to the DD.67 Upon TNF-α binding, SODD dissociates from the DD, leading to TNFR-1 activation, which then recruits TNF receptor-associated death domain (TRADD) and other adapter proteins, such as receptor-interacting protein-1, TNFR-associated factor 2 (TRAF2), cellular inhibitor of apoptosis proteins 1 and 2 (cIAP1, cIAP2), and the linear ubiquitin chain assembly complex, to form complex I, which activates the NF-κB signaling pathway.66,68 Additionally, the MAPK signaling pathway is activated via TRAF2, leading to the nuclear translocation of activated JNK, which subsequently induces transcription factors such as activator protein 1 (AP-1).69,70 These combined pathways drive the degradation of the proteoglycan aggrecan, inhibit type II collagen expression, and upregulate MMP and ADAMTS secretion, as demonstrated both in vitro and in vivo.44,53 Under certain circumstances, the failure of complex I to activate the NF-κB pathway results in the inhibition of cFLIP, an NF-κB-induced protein that prevents the recruitment and activation of pro-caspase 8, protecting cells from apoptosis.71 Instead, the components of complex I (TRADD-RIP1-TRAF2) dissociate and bind to FAS-associated death domain proteins, forming complex II in the cytosol. Complex II recruits and activates pro-caspase 8, which activates caspase 3 to induce chondrocyte apoptosis.66,67,70 This disruption of the ECM repair process leads to a failure to maintain cartilage homeostasis, contributing to OA pathogenesis.72
The signaling mechanism of TNFR-2 is less well understood, but it is believed to support cell activation, migration, and proliferation.68 The binding of tmTNF-α to TNFR-2 facilitates the recruitment of TRAF2, TRAF1, cIAP1, and cIAP2, which assemble to activate both the NF-κB and MAPK signaling pathways. These pathways, as previously described, are implicated in driving inflammatory responses. Interestingly, studies by Takahito et al. revealed that ablation of TNFR-2 in TNF∆ARE mice exacerbated joint inflammation, suggesting that TNFR-2 may act as a crucial anti-inflammatory mediator in OA progression.73
Healthy cartilage is an avascular tissue with low oxygen tension (1–5%), supporting the relatively slow metabolic activity of chondrocytes. However, as OA progresses, the microvasculature from the subchondral bone invades the calcified cartilage, increasing oxygen tension and promoting chondrocyte apoptosis.74 Angiogenesis, largely driven by chondrocyte-produced vascular endothelial growth factor, is further stimulated by TNF-α.74,75 Additionally, experimental studies in OA chondrocytes have demonstrated that TNF-α enhances the synthesis of various chemokines and inflammatory mediators, including IL-8, CCL5 (also known as RANTES), iNOS, COX-2, and PGE2,53,65 all of which exacerbate the inflammatory state within the joint. In conjunction with IL-1β-induced inflammatory responses, this further amplifies the production of TNF-α, creating a positive feedback loop that accelerates OA progression.
IL-6
IL-6 has long been recognized as a proinflammatory cytokine that plays a pivotal role in the catabolic processes of OA progression. However, emerging evidence suggests that IL-6 has a complex biological function in OA pathogenesis, contributing to both protective and detrimental effects. IL-6 is produced primarily by chondrocytes, osteoblasts, synovial fibroblasts, and the infrapatellar fat pad in OA.76 Notably, the infrapatellar fat pad is a major source of adipose tissue within the knee joint, implicating obesity as a significant risk factor for IL-6-driven OA.77
IL-6 exerts its effects by binding to two distinct forms of nonsignaling IL-6 receptor (IL-6R): the membrane-bound IL-6 receptor (mIL-6R) and the soluble IL-6 receptor (sIL-6R) (Fig. 1c). sIL-6R results either from the cleavage of mIL-6R by metalloproteases, including ADAM10 and ADAM17, or, to a lesser extent, from alternative splicing of the same mRNA encoding IL-6R.78,79 The binding of IL-6 to IL-6R results in the formation of a complex that subsequently associates with glycoprotein 130 (gp130), a ubiquitously expressed transmembrane protein responsible for IL-6 signal transduction.77,80 The signaling pathway involving mIL-6R is termed the “classic-signaling pathway,” whereas the pathway involving sIL-6R is known as the “trans-signaling pathway.”81,82,83 Both pathways initiate similar downstream signaling cascades via gp130,84 but the trans-signaling pathway is considered dominant, as IL-6 preferentially binds to sIL-6R. The resulting complex is capable of inducing IL-6 signaling in a variety of cells expressing gp130 via the circulatory system, whereas mIL-6R is expressed on a limited range of cell types, including macrophages, osteocytes, chondrocytes, monocytes, leukocytes, and hepatocytes.53,85,86
Additionally, a soluble form of gp130 (sgp130) is observed in the bloodstream. Sgp130 functions as an inhibitor of the IL-6-induced trans-signaling pathway by binding to IL-6-sIL-6R complexes with higher affinity than to IL-6-mIL-6R complexes.78,87 Upon binding, receptor homodimerization brings Janus kinases (JAKs), including JAK1, JAK2, and tyrosine kinase 2 (TYK2), into proximity, resulting in their activation. This activation phosphorylates tyrosine residues in the intracellular domains of gp130, triggering two main gp130-mediated signaling pathways: the signal transducer and activator of transcription (STAT) pathway, which facilitates the translocation of transcription factors such as STAT1, STAT3, and STAT5 into the nucleus to regulate target gene expression, and the ERK pathway, which is activated by the phosphorylation of tyrosine 759.77,80,82,85,88 Both signaling pathways are tightly regulated by suppressors of cytokine signaling (SOCS), which are downstream target genes of STAT3. Specifically, SOCS-1 and SOCS-3 are transcribed in response to JAK/STAT activation. SOCS-1 inhibits JAK/STAT signaling by binding to JAKs, while SOCS-3 associates with tyrosine 759 to terminate ERK signaling.78,82,88
Classic-signaling and trans-signaling pathways involving IL-6 could play opposing roles in OA pathogenesis. The classic-signaling pathway is typically associated with protective effects, such as increasing proteoglycan aggrecan synthesis in chondrocytes and promoting the production of tissue inhibitors of metalloproteinases, which counteract the effects of MMPs.86,88 In contrast, the trans-signaling pathway is thought to drive catabolic events in human OA chondrocytes, as evidenced by the observed inhibition of proteoglycan synthesis and the upregulation of cartilage-degrading enzymes.78,89 However, recent studies suggest that both pathways can induce opposite effects under different conditions,86,90 although the underlying mechanisms for this discrepancy remain unclear. The balance between the classic and trans-signaling pathways in chondrocytes is possibly influenced by mIL-6R expression, which can be upregulated by IL-1β or downregulated by transforming growth factor-β (TGF-β). Disruption of this balance may partially explain the shift between anabolic and catabolic events mediated by IL-6.86
Overall, IL-6 upregulates the expression of MMPs (MMP-1, MMP-3, and MMP-13) and ADAMTS family members (ADAMTS-4 and ADAMTS-5) in chondrocytes via the JAK/STAT and ERK signaling pathways.76,90 Furthermore, IL-6 stimulates the expression of RANKL via JAK/STAT signaling in synovial fibroblasts, disrupting the homeostasis between bone formation and resorption orchestrated by osteoblasts and osteoclasts, thus facilitating osteoclastogenesis and contributing to bone turnover. This leads to deleterious morphological changes in subchondral bone.78,82,86 IL-6 released by synovial fibroblasts acts synergistically with TNF-α to increase vascular endothelial growth factor production, which elevates oxygen tension, induces interstitial edema, and increases tissue pressure by increasing vascular permeability. These effects result in chondrocyte apoptosis and tissue damage.78,82,90 Furthermore, SOCS-3, which is induced by IL-6, inhibits chondrogenesis by reducing the levels of insulin-like growth factor-1 (IGF-1), an essential mediator of ECM, collagen II, and proteoglycan synthesis, as well as the differentiation of chondrogenic progenitor cells. Moreover, SOCS-3 also dampens IL-6 signaling, highlighting the paradoxical role of IL-6 in OA progression.86,88
Role of chondrocyte organelles in osteoarthritis
Chondrocytes, the only cellular component of cartilage, play crucial roles in maintaining the structural integrity and function of articular cartilage. The preservation of normal cartilage morphology relies on a dynamic balance of chondrocyte function, wherein the health and activity of chondrocyte organelles are essential. Disruptions in organelle homeostasis during OA pathology can lead to detrimental changes in chondrocyte differentiation and even apoptosis, which further exacerbate cartilage degeneration. A deeper understanding of the roles of chondrocyte organelles in OA progression offers valuable insights for identifying novel therapeutic targets and strategies aimed at preserving chondrocyte functionality.
Mitochondria
Mitochondria play a central role in cellular metabolism by producing the majority of adenosine triphosphate (ATP), which is essential for various physiological processes across nearly all human tissues. Mitochondrial dysfunction has been closely linked to several age-related diseases, including OA. Recent studies suggest that an imbalance between mitochondrial biogenesis and the clearance of damaged mitochondria contributes to OA onset and progression in chondrocytes.91,92
Mitochondria are composed of four main structural components: the central matrix, the inner mitochondrial membrane (IMM), the outer mitochondrial membrane (OMM), and the intermembrane space.93 The electron transport chain (ETC), located within the IMM, plays a pivotal role in ATP generation and maintaining the mitochondrial membrane potential (ΔΨm).93,94 The ETC, composed of complexes I-IV, ubiquinone, and cytochrome c, facilitates electron transfer, coupled with proton (H+) pumping across the membrane by complexes I, III, and IV. This proton gradient provides the electrochemical energy required for ATP synthase (Complex V) to convert ADP into ATP.93,95 However, this process generates byproducts: electrons from complexes I and II prematurely react with molecular oxygen (O2), potentially forming superoxide anions (O2−). These anions are detoxified by superoxide dismutase into hydrogen peroxide (H2O2), which, in the presence of metal ions such as Fe2+ and Cu2+, can generate highly reactive hydroxyl radicals (·OH). Moreover, superoxide anions can be translocated to the mitochondrial matrix through voltage-dependent anion channels (VDACs), leading to further ·OH formation and impacting the ΔΨm (Fig. 2).92,93,95 These reactive oxygen species (ROS) are implicated in the pathogenesis of OA, highlighting the delicate balance between energy production and oxidative stress. Although mitochondria are crucial for energy production, some studies suggest that chondrocytes predominantly rely on glycolysis for ATP synthesis because of their low-oxygen environment and lack of innervation, with only ~25% of ATP generated through mitochondrial oxidative phosphorylation.92,95 Nonetheless, a decrease in ATP production is observed in OA chondrocytes, which is attributed to reduced oxidative phosphorylation and a decrease in the number of mitochondria. These findings suggest that mitochondria are pivotal in OA progression despite the increased glycolytic activity in these cells.92

Role of mitochondria in the progression of OA. In healthy mitochondria, the ETC chain in the IMM provides sufficient ATP production to maintain chondrocyte homeostasis, and complexes I-III of the ETC chain have the capacity for ROS generation. Under normal circumstances, PINK1 is transported by TOM through the OMM and IMM, where it is retained in the central matrix with the assistance of the ΔΨm. In the matrix, PINK1’s MTS and TM domains are cleaved by MPP and PARL, leaving an unstable PINK1 remnant that is subsequently degraded via the ubiquitin–proteasome system. Consequently, Parkin remains in an inactive state, preventing mitophagy from occurring. Mitochondrial dysfunction, resulting from mtDNA damage, inflammasome activation, or ROS attack, leads to a depolarized ΔΨm, which prevents PINK1 from remaining in the matrix. The PINK1 accumulated on the OMM undergoes transphosphorylation, which activates Parkin to ubiquitinate OMM proteins, which bind to the phagophore via LC3-II localized on the phagophore membrane directly or through SQSTM1, which possesses an LC3-interacting region to bind to the phagophore indirectly. The phagophore subsequently matures into an autophagosome, which fuses with the lysosome to degrade damaged mitochondria. Appropriate mitophagy helps eliminate excessive ROS and maintain chondrocyte homeostasis, but excessive mitophagy can lead to chondrocyte apoptosis due to an insufficient energy supply. ETC electron transport chain, IMM inner mitochondrial membrane, ATP adenosine triphosphate, ROS reactive oxygen species, PINK1 PTEN-induced putative kinase 1, TOM translocases of the outer membrane, OMM outer mitochondrial membrane, OMMP outer mitochondrial membrane proteins, ΔΨm mitochondrial membrane potential, MPP mitochondrial processing peptidase, PARL presenilin-associated rhomboid-like protease, mtDNA mitochondrial DNA, LC3-II autophagic adapter protein light chain 3-II, SQSTM1 p62/Sequestosome 1
Mitochondrial biogenesis, the process by which mitochondria divide to generate new organelles, is regulated primarily by peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α).96 PGC-1α activates downstream transcription factors, including the nuclear respiratory factors NRF1 and NRF2, which promote mitochondrial transcription factor A (TFAM) expression. This, in turn, initiates mitochondrial DNA (mtDNA) replication and the production of new mitochondria.96,97 AMP-activated protein kinase (AMPK) modulates PGC-1α activity either through direct phosphorylation or by activating Silent Information Regulator 1 (SIRT1), a mitochondrial histone deacetylase that also influences PGC-1α activity.97,98 Studies have shown reduced mitochondrial biogenesis and an accumulation of dysfunctional mitochondria in OA chondrocytes. This imbalance leads to excessive ROS production, damages mitochondrial DNA and impairs ATP generation, contributing to chondrocyte apoptosis and cartilage degradation.92,99
In recent years, mitophagy has emerged as a potential therapeutic target for OA and other bone diseases. Mitophagy is the selective removal of damaged or dysfunctional mitochondria to protect cells from apoptosis.100 This process can occur via two main pathways: the Parkin RBR E3 ubiquitin-protein ligase (PRKN)-dependent pathway and the PRKN-independent pathway, with the former being the most well studied.91,93,100,101 PINK1 is a serine/threonine (Ser/Thr) kinase comprising an N-terminal mitochondrial targeting sequence (MTS), followed by an α-helical membrane (TM) segment and a Ser/Thr kinase domain, whereas parkin is an E3-ubiquitin ligase with an N-terminus termed the UBL domain.93,102 Under normal physiological circumstances, PINK1 is imported into mitochondria, where translocases of the outer membrane (TOM) carry the MTS of PINK1 through the OMM and IMM of mitochondria, allowing the MTS to be exposed to the central matrix with the help of the ΔΨm.93 The MTS is then cleaved by mitochondrial processing peptidase in the central matrix, and the TM segment is cleaved by presenilin-associated rhomboid-like protease (PARL) in the IMM. This process results in an unstable PINK1 remnant, which is released from the mitochondria to the cytosol for rapid degradation by the ubiquitin protease system.93,102 Moreover, the UBL domain of parkin inhibits its own E3-ubiquitin ligase activity at the N-terminus, resulting in inhibited mitophagy in normal cells.93 However, under pathological conditions, the mitochondria may depolarize due to the negative effects of proinflammatory mediators, mtDNA damage, or reactive oxygen species (ROS) attack, as experimentally confirmed in human OA chondrocytes.95,103 These events inhibit the translocation of the MTS of PINK1 into the central matrix, leading to the cessation of PINK1 degradation.93,102 Instead, accumulated PINK1 binds with TOM on the OMM surface as an integral molecule that undergoes transautophosphorylation to form complexes concomitant with high Parkin kinase activity, which recruits cytosolic Parkin to malfunctioning mitochondria.102,104
Once recruited, Parkin is activated by PINK1 phosphorylation at Ser65 in its UBL domain, which relieves its inhibitory effect on E3 ligase activity, allowing Parkin to phosphorylate ubiquitin (ph-Ub) and poly-Ub chains that bind to a plethora of OMM proteins, including mitofusin 1 (MFN1), MFN2, mitochondrial Rho-GTPase 1 (Miro1), and VDAC1. The ubiquitinated OMM proteins are capable of binding with phagophores via the autophagic adapter protein light chain 3-II (LC3-II) localized on the phagophore membrane directly or through p62/Sequestosome 1 (SQSTM1), which contains an LC3 interaction region that binds to phagophores indirectly.91,102 The phagophores subsequently mature into autophagosomes, which fuse with lysosomes to degrade damaged mitochondria (Fig. 2).105,106
Lysosome
Lysosomes, single-membrane organelles, are essential for degrading cellular waste and dysfunctional organelles through various hydrolytic enzymes within an acidic lumen and are maintained by the multisubunit V-ATPase complex.107,108 Autophagy occurs in three main forms, classified by substrate delivery to lysosomes: microautophagy, macroautophagy, and chaperone-mediated autophagy.107,109 Microautophagy involves the direct engulfment of small cytosolic particles into lysosomes for degradation, whereas macroautophagy is initiated by the formation of phagophores, which capture larger cytosolic material or damaged organelles. Phagophores mature into autophagosomes, which fuse with lysosomes to degrade their cargo.107,109,110 CMA is a selective autophagic process that recognizes specific protein motifs, facilitating their translocation to lysosomes for degradation via the heat shock cognate protein of 70 kDa (HSC70).111,112
Lysosomal cathepsins, a family of ~12 proteases, play a pivotal role in breaking down macromolecules by cleaving peptide bonds within the lysosomal matrix.107 Notably, studies indicate that chondrocytes internalize hydroxyapatite, a primary crystal contributing to cartilage calcification in OA.113,114 Excessive hydroxyapatite is transferred to lysosomes for dissolution through acidification,115,116 leading to a compromised acidic milieu within the lysosome that dramatically affects soluble lysosomal hydrolases, including acid sphingomyelinase (ASM), which modulates the balance of lysosomal lipid composition (particularly sphingomyelin), cathepsins responsible for dissolved hydroxyapatite matrix degradation, and lysosome-associated membrane proteins (LAMPs), which are critical for maintaining lysosomal morphology. This results in increased oxidative stress, accumulated dysfunctional mitochondria, and lysosomal membrane permeabilization (LMP).107,117
LMP triggers the release of cathepsins, particularly cathepsins B and D, into the cytosol, where they cleave the proapoptotic protein BID into its active form, t-BID,118,119 which translocates and inserts into the OMM, where it facilitates the recruitment of BAX, another cytosolic proapoptotic protein.120 BAX, upon binding to t-BID, oligomerizes to form pores in the OMM.121,122 The mitochondrial outer membrane permeabilization (MOMP) eventuates the release of cytochrome C, a protein that originally mediates electron transfer between complexes III and IV of the ETC and detoxifies ROS in the intermembrane space. However, once released into the cytosol, cytochrome C initiates chondrocyte apoptosis and exacerbates catabolic processes in OA by activating caspase 3 (Fig. 3).117

Mechanisms of lysosome-induced chondrocyte apoptosis. a Histological stratification of cartilage microstructure. b RUNX2-driven hypertrophic transition of superficial chondrocytes with type X collagen secretion as a hypertrophy biomarker. c Hydroxyapatites, derived from both chondrocyte uptake and hypertrophic chondrocyte synthesis, are transported to lysosomes for processing. This process may lead to LMP, which causes the release of cathepsins into the cytosol. The cathepsins cleave the proapoptotic protein BID into its active form, t-BID, which inserts into the OMM and recruits BAX. d The t-BID-BAX complex forms pores in the OMM, resulting in the leakage of cytochrome c from the IMS into the cytosol. Cytochrome c then activates caspase-3, triggering chondrocyte apoptosis. Abbreviations: I superficial layer, II transition layer, III deep layer, TNAP tissue-nonspecific alkaline phosphatase, MV matrix vesicle, HA hydroxyapatite, LMP lysosomal membrane permeabilization, OMM outer mitochondrial membrane, IMS intermembrane space
Targeting hydroxyapatite removal may offer a potential therapeutic strategy for mitigating LMP-induced chondrocyte apoptosis in OA. Understanding the mechanisms underlying hydroxyapatite formation is imperative. In healthy cartilage, chondrocytes in the superficial and middle layers remain uncalcified, while mineralizing hypertrophic chondrocytes, characterized by high expression of Runt-related transcription factor 2 (RUNX2) and type X collagen, are localized in the deep cartilage layers.123 RUNX2 regulates chondrocyte differentiation into mineralizing hypertrophic cells, with type X collagen serving as a hypertrophy marker.123,124,125 Pathological calcification occurs when uncalcified chondrocytes in the superficial layer aberrantly differentiate into mineralizing hypertrophic cells.126
The enzyme ectonucleotide pyrophosphatase/phosphodiesterase family member 1, located on hypertrophic chondrocyte membranes, converts ATP into inorganic pyrophosphate,127,128 which is hydrolyzed by tissue-nonspecific alkaline phosphatase into inorganic phosphate (Pi).124,126,127 The sodium-dependent phosphate transporters PiT-1 and PiT-2 transport excess Pi into matrix vesicles (MVs) secreted by hypertrophic chondrocytes.124,129 Within MVs, Pi interacts with calcium (Ca²⁺) internalized via Annexin V to form an amorphous calcium phosphate precursor (ACP).130 Upon interaction with collagen fibrils, ACP is converted into hydroxyapatite and released into the cytosol.123 Additionally, mitochondria are integral in maintaining Ca²⁺ and Pi homeostasis.127,131 Dysfunctional mitochondria contribute to the formation of ACPs, which are then delivered to lysosomes via mitophagy.123 However, conflicting clinical data suggest that mitophagy may play a protective role in maintaining chondrocyte survival.93,100,101 Further research is needed to elucidate the precise mechanisms linking mitophagy and cartilage calcification.
Endoplasmic reticulum
The ER is the largest membrane-bound organelle adjacent to the outer membrane of the eukaryotic cell nucleus. It is the major site of protein synthesis, lipid biogenesis, detoxification, and intracellular Ca²⁺ storage.132 In the early stages of OA, environmental insults or increased protein synthesis—stemming from the attempts of chondrocytes to repair damaged cartilage—often lead to the accumulation of misfolded proteins within the ER, resulting in ER stress that negatively affects cellular activity and viability (Fig. 4).133

Illustration of the ER stress response and its implications in OA. The diagram depicts the activation of the UPR due to the accumulation of misfolded proteins within the ER. Key ER stress sensors, including PERK, IRE1α, and ATF6α, are activated upon dissociation from the chaperone BiP. PERK phosphorylates eIF2α to attenuate protein synthesis, whereas IRE1α splices XBP1 mRNA to generate the active transcription factor XBP1s, which promotes the expression of chaperones and proteins involved in ER expansion. Upon cleavage, ATF6α translocates to the nucleus to activate UPR target genes that aid in protein folding and ER homeostasis. The figure also highlights the role of hypoxia, mutated collagen II, and AGEs in promoting ER stress. Prolonged ER stress can lead to the activation of proapoptotic pathways, including the CHOP pathway, and the production of matrix-degrading enzymes such as MMPs and ADAMTSs, contributing to cartilage degradation in OA. UPR unfolded protein response, PERK protein kinase-RNA-like endoplasmic reticulum kinase, IRE1α inositol-requiring enzyme 1 alpha, ATF6α activating transcription factor 6α, BiP glucose-regulated protein 78, eIF2α eukaryotic translation initiation factor 2A, ATF4 activating transcription factor 4, CHOP C/EBP homologous protein, XBP1 X-box binding protein 1, XBP1s spliced X-box binding protein 1, TRAF2 TNFR-associated factor 2, AGEs advanced glycation end products
Mild ER stress, characterized by limited accumulation of misfolded proteins, can be managed through the unfolded protein response (UPR). This protective mechanism aims to restore ER homeostasis by eliminating excess misfolded proteins via two key mechanisms: ER-associated degradation (ERAD) and autophagy (ER-phagy). ER-phagy, akin to mitophagy, involves the sequestration of dysfunctional organelles into autophagosomes, which subsequently fuse with lysosomes for degradation.133,134
In contrast, ERAD activation is mediated by three transmembrane ER proteins—protein kinase-RNA-like ER kinase (PERK), inositol-requiring enzyme 1 alpha (IRE1α), and activating transcription factor 6α (ATF6α)—which function as stress sensors.133,134,135 Under normal conditions, BiP (also known as glucose-regulated protein 78, GRP78) binds these sensors to keep them inactive in the absence of misfolded proteins. However, when misfolded proteins accumulate in the ER lumen, BiP dissociates from the sensors to bind the exposed hydrophobic regions of the misfolded proteins with higher affinity.135 The compensatory upregulation of BiP is consistently observed in both the cartilage and synovial fluid of OA patients, reflecting an adaptive effort to restore ER homeostasis under persistent stress conditions.136,137,138
BiP dissociation from PERK triggers PERK oligomerization and autophosphorylation, leading to the phosphorylation of eukaryotic translation initiation factor 2A (eIF2α). This inhibits cap-dependent translation to reduce the protein load in the ER.133,134 Phosphorylated eIF2α also promotes the translation of cap-independent mRNAs, including ATF4, which upregulates UPR target genes encoding chaperones to refold misfolded proteins. Additionally, ATF4 selectively activates C/EBP homologous protein (CHOP), a proapoptotic factor that triggers apoptosis if stress remains unresolved.133,135 Notably, CHOP expression is markedly upregulated in both the cartilage and synovial fluid of OA patients, indicating exacerbated CHOP-induced chondrocyte apoptosis in OA pathogenesis.136,137,138 SIRT1 has been shown to inhibit CHOP-induced apoptosis, making it a promising therapeutic target for mitigating ER stress.139
Following BiP dissociation, IRE1α dimerizes and undergoes autophosphorylation, acquiring endonuclease activity that splices X-box binding protein 1 (XBP1) mRNA, converting it into the functional transcription factor XBP1s. XBP1s upregulates protein chaperones, disulfide isomerases, and components of the protein translocation machinery, contributing to ER expansion. It also enhances the expression of ERAD components, supporting the recovery of ER homeostasis.133,134,135
Similarly, BiP dissociation from ATF6α enables its incorporation into a vesicle that migrates to the Golgi apparatus, where ATF6α is cleaved by site-1 (S1P) and site-2 proteases (S2P). The resulting active fragment translocates to the nucleus to activate UPR target genes involved in protein folding, secretion, ERAD, and ER expansion.134,135 Notably, ATF6α upregulation in OA chondrocytes augments XBP1s expression, as observed in OA cartilage biopsies, mediating adaptive responses that attenuate ER stress-induced chondrocyte apoptosis and ECM degradation.136,140 However, if ER stress persists, the UPR shifts toward a proapoptotic cascade through both CHOP-dependent and CHOP-independent pathways, culminating in chondrocyte apoptosis.133,134
Pathological ER stress has been implicated in OA progression. Factors such as hypoxia, mutated collagen II aggregates, and the accumulation of advanced glycation end products—an unavoidable byproduct of cellular senescence—contribute to ER stress in OA chondrocytes.133,134,138 Notably, ER stress upregulates tribbles homolog 3 (TRB3), a CHOP downstream proapoptotic protein that impairs the chondrocyte response to IGF-1, reducing ECM, collagen II, and proteoglycan production.138
Additionally, some studies suggest that IRE1α can activate the NF-κB, JNK, and MAPK signaling pathways through TRAF2, promoting the production of MMPs and ADAMTSs, which drive catabolic events in OA.134,138 Consistent with these findings, the expression of UPR sensors (ATF6α, PERK, and IRE1α) is markedly increased in OA cartilage compared with normal tissue, which is correlated with the degree of cartilage degradation.138,141,142 Thus, excessive ER stress plays a critical role in OA progression, highlighting the need for therapeutic strategies targeting ER stress pathways.
Organelle dysfunction and inflammation in osteoarthritis
Emerging evidence implicates organelle dysfunction as a critical driver of inflammatory cascades in OA, with pattern recognition receptors (PRRs) upregulated in OA chondrocytes playing a central mechanistic role. These PRRs, encompassing four principal classes—NOD-like receptors, Toll-like receptors, retinoic acid-inducible gene-I-like receptors, and C-type lectin receptors—are activated by both exogenous ligands and endogenous damage-associated molecular patterns (DAMPs), including mtDNA, cardiolipin, ROS, and metabolites (e.g., ATP).143,144,145
Mitochondria, widely recognized as evolutionary remnants of ancestral Alphaproteobacteria (the precursors of modern gram-negative bacteria), exhibit molecular similarities to bacterial components.144,145,146 Under physiological conditions, the integrity of the mitochondrial double membrane prevents the release of these DAMPs, thereby preventing inflammatory activation. However, in OA, pathological insults such as proinflammatory cytokines (e.g., IL-1, IL-6, and TNF-α) or ROS induce mitochondrial dysfunction. This triggers BAX/BAK-mediated MOMP, followed by osmotic pressure elevation within the mitochondrial matrix that displaces the IMM into the cytosol, ultimately leading to IMM rupture and the release of DAMPs (e.g., mtDNA and ATP) into the extracellular space—a mechanism corroborated by the elevated levels of DAMPs and ATP detected in OA synovial fluid.95,144
Cytosolic mtDNA induces inflammatory responses through interactions with specific intracellular receptors. Specifically, mtDNA activates the NLRP3 inflammasome, a multiprotein complex containing caspase-1, thereby promoting the maturation of IL-1β.144,145 Additionally, mtDNA engages with TLR9 expressed on the ER, activating the downstream MAPK and NF-κB signaling pathways.145 Furthermore, cytosolic mtDNA, either alone or in complex with curvature-associated proteins such as TFAM or high mobility group box 1 (HMGB1), activates the cGAS-STING pathway, which orchestrates NF-κB signaling and exacerbates inflammatory responses in OA.144,146
While the causal relationship between proinflammatory cytokines and LMP remains ambiguous, evidence confirms that under OA conditions, the combined pathological effects of hydroxyapatite crystal deposition and ROS-enriched microenvironments directly induce LMP.112,113 This lysosomal destabilization enables the leakage of multiple cysteine cathepsins (B, L, V, S, X, and Z), which play both specialized and overlapping functional roles in NLRP3 inflammasome activation. Although the exact mechanisms remain unclear, potential mechanisms may involve cysteine cathepsins, K⁺ efflux, oxidative stress, and Ca²⁺/Na⁺ influx.142
ER stress induced by proinflammatory cytokines, oxidative stress, and hypoxia in chondrocytes drives inflammatory responses through the UPR, which is regulated by three primary signaling pathways: the PERK, IRE1α, and ATF6α pathways.133,147,148 Specifically, PERK activation leads to eIF2α phosphorylation, which reduces IκB synthesis and consequently activates NF-κB signaling.149 IRE1α activation triggers TRAF2-mediated IκB degradation, further activating NF-κB while also engaging in JNK signaling. ATF6α similarly promotes IκB degradation to increase NF-κB activity.150 Notably, cytoplasmic leakage of BiP directly activates NF-κB, orchestrating a multifaceted inflammatory response in OA.150
Clinical manifestations of osteoarthritis
OA is characterized by structural joint degeneration, including osteophyte formation, cartilage defects, narrowed joint space, and subchondral bone remodeling. OA pain, one of the most prominent symptoms encouraging patients to seek medical intervention, often persists irrespective of these morphological alterations. Despite advances in OA treatment, current therapies for managing OA pain are largely palliative and unsatisfactory.151 None of the first-line agents, such as NSAIDs, opioids, hyaluronic acid hydrogels, or surgical procedures, have consistently demonstrated effective therapeutic outcomes.41 Over 75% of OA patients report the ongoing need for symptomatic relief.151
Morphological changes, including cartilage degradation, osteophyte formation, and periarticular tissue damage, have historically been regarded as the primary sources of pain in OA.41 However, imaging studies have revealed a relationship between the radiographic severity of OA and the intensity of pain experienced, suggesting that individual variations in pain perception may be influenced by genetic, environmental (e.g., obesity), psychological, and neurological factors. Given the poorly understood mechanisms underlying OA pain, the following section aims to systematically discuss the genesis and transmission of OA pain in the knee joint, offering insights into novel pain management strategies.
Osteoarthritis pain genesis
The action potentials generated by peripheral stimuli at the knee joint are transmitted predominantly through a dense network of sensory afferent neurons, which can be classified into large-diameter myelinated Aβ fibers, medium-diameter myelinated Aδ fibers, and small-diameter unmyelinated C fibers on the basis of differences in conduction velocity and stimulus thresholds.152 Nociceptive pain signals are transmitted primarily by Aδ and C fibers, both of which are pseudounipolar neurons typically referred to as nociceptors.153,154 Owing to their larger diameters, Aδ-fibers conduct pain signals more rapidly and are mainly responsible for transmitting sharp pain, whereas C-fibers convey more diffuse, burning pain.155,156
The cell bodies of nociceptors are clustered in the dorsal root ganglion (DRG) adjacent to the spinal cord, where each nociceptor gives rise to two axons: one extending toward peripheral tissues—including the capsule, ligaments, meniscus, periosteum, subchondral bone, and synovium of the knee joint—and the other projecting toward the dorsal horn of the spinal cord.154,155,156,157,158,159 Nociceptor nerve endings in the periphery express transient receptor potential (TRP) channels, including TRPV1, TRPA1, TRPM3, and TRPM8.160,161 The activation of these channels by proinflammatory cytokines (IL-1 and TNF-α) and ROS transforms noxious stimuli into electrical signals by initiating Ca²⁺ influx.162,163,164,165 Following this transduction, voltage-gated sodium (Nav) channels, such as Nav1.1, Nav1.6, Nav1.7, Nav1.8, and Nav1.9, amplify these signals via Na+ influx, ultimately reaching the threshold for action potential generation.157,166,167
Nerve growth factor (NGF) also plays a vital role in peripheral pain generation. Both local and systemic administration of NGF induce robust, long-lasting mechanical and thermal hyperalgesia in rodents and human volunteers, underscoring its proalgesic properties.168 In OA joints, cytokines such as TNF-α and IL-1 stimulate NGF secretion by nonneural cells such as macrophages, mast cells, fibroblast-like synoviocytes, and keratinocytes. This leads to elevated NGF levels, potentially contributing to the aberrant growth of sensory neurons and sympathetic fibers in otherwise aneural cartilage.169,170 NGF exerts its effects through two receptors localized on peripheral nociceptors: high-affinity tyrosine receptor kinase A (TrkA) and low-affinity p75 neurotrophin receptor (p75NTR).169,171 NGF binding to TrkA activates three key intracellular signaling pathways: the mitogen-activated protein kinase (MAPK), phospholipase C-γ, and phosphoinositide 3-kinase pathways.168,172 These NGF–TrkA complexes, along with activated signaling intermediates, are transported via endosomes to DRG neuron cell bodies, where they promote sensory neuron sprouting and upregulate the expression of pain-related proteins such as TRPV1, Nav1.8, substance P (SP), and calcitonin gene-related peptide (CGRP).168,169,170,173 Additionally, p75NTR enhances the sensitivity of TrkA to NGF by dimerizing with it, as p75NTR itself lacks catalytic activity.168,173 This NGF–p75NTR complex can mediate homeostatic responses in sensory neurons by inducing either apoptosis through c-Jun N-terminal kinase signaling or survival through the NF-κB pathway.171,174,175
The other end of Aδ-fiber axons terminates in laminae I and V of the dorsal horn of the spinal cord,152,156,176 where they transmit pain signals by directly synapsing with second-order neurons, such as spinothalamic and spinoreticular cells.156,176 The cell bodies of spinothalamic neurons are located primarily in laminae I and V, whereas spinoreticular neurons are found predominantly in laminae VII and VIII.156,177,178 In contrast, C-fiber axons terminate in laminae II, where they synapse with interneurons before transmitting pain signals to second-order neurons.152,156 Additionally, C-fibers are divided into peptidergic and nonpeptidergic fibers on the basis of their ability to produce SP and CGRP. Notably, bones are innervated by peptidergic but not nonpeptidergic C-fibers.153,176
Upon activation by noxious stimuli, the terminal endings of both types of nociceptors release neurotransmitters—including glutamate (Glu), SP, and CGRP—into the synaptic cleft to act on receptors of second-order neurons.176,179 In presynaptic terminals, Glu is packaged into vesicles via vesicular glutamate transporter 2 (VGLUT2), which facilitates its release.180 Once released, Glu binds to postsynaptic glutamate receptors, including metabotropic (mGluRs) and ionotropic (iGluRs) receptors located on second-order neurons.
Both SP and CGRP are synthesized in nociceptors and stored in dense-core vesicles under unstimulated conditions.180,181,182 Following neuronal depolarization, SP and CGRP are transported by fast axonal transport to the terminal endings of nociceptors. SP is released into the synaptic cleft to transmit noxious signals by binding with neurokinin 1 G-protein-coupled receptors (NK1Rs) on second-order neurons,182,183 whereas CGRP binds to a unique G protein-coupled receptor composed of three subunits: calcitonin-like receptor (CLR), receptor activity-modifying protein 1 (RAMP1), and receptor component protein (RCP).184
After receiving nociceptive input from nociceptors, the axons of spinothalamic and spinoreticular cells—referred to as the spinothalamic and spinoreticular tracts—transmit these nociceptive signals upward to the brain.176,185 Both tracts crossover at the anterior white commissure of the spinal cord. The spinothalamic tract then diverges into two subdivisions: the anterior spinothalamic tract, which projects to the ventral posterior lateral nucleus and is thought to mediate pain discrimination, and the lateral spinoreticular tract. In contrast, spinoreticular tracts terminate in the reticular formation of the medulla and pons, contributing to the affective and motivational components of pain (Fig. 5).156

Overview of pain transmission in the knee joint. a Synaptic interface between primary afferent nociceptors and second-order neurons in the spinal dorsal horn. b Connectivity patterns and regional distribution of nociceptors and second-order neurons in the spinal cord. c Neuronal pain signaling pathway. d Magnified peripheral nociceptor terminals showing Nav and TRP channel cooperation with NGF to initiate pain signaling during noxious chemical and mechanical stimulation. e The distribution of nociceptors at the periphery. The cell bodies of nociceptors cluster in the DRG, where each nociceptor extends two axons: one toward the periphery and the other to the dorsal horn of the spinal cord. At the periphery, nociceptor terminals transduce mechanical or chemical stimuli, including proinflammatory cytokines, inflammatory mediators such as those released from dysfunctional organelles, and mitochondrial ROS (mtROS), into pain signals via Ca²⁺/Na+ influx through TRP and Nav channels. Under inflammatory conditions, NGF secreted by Møs, MCs, and FLSs synergizes with cytokines and mtROS to increase TRP/Nav sensitization and SP expression, amplifying nociceptive signaling. Pain signals are transmitted to second-order spinal neurons through synaptic neurotransmitter release and then relayed to the brain for perceptual processing. AP action potential, SP substance P, ER endoplasmic reticulum, mtROS mitochondrial ROS, Mø macrophage, MC mast cell, FLS fibroblast-like synoviocyte, TRP transient receptor potential channel, Nav voltage-gated sodium channel
Organelle dysfunction and osteoarthritis pain
Organelle dysfunction within the OA microenvironment contributes substantially to pain sensitization. Mitochondrial dysfunction, triggered by ROS and proinflammatory cytokines, leads to excessive ROS production and BAX/BAK-mediated mitochondrial outer membrane permeabilization, resulting in the release of mtDNA and ATP. These DAMPs activate PRRs, thereby driving inflammatory responses. Similarly, lysosomal dysfunction caused by hydroxyapatite crystal deposition induces LMP and the leakage of cysteine cathepsins, which activate the NLRP3 inflammasome. ER stress due to the accumulation of misfolded proteins activates the UPR, which in turn triggers inflammatory cascades. Collectively, these organelle-derived signals sensitize TRP and Nav channels, amplifying nociceptive signaling in OA.
In addition to promoting inflammation, organelle dysfunction exacerbates OA pain by sensitizing TRP family members—particularly TRPV1, the most extensively studied in this context. TRPV1 is expressed on chondrocytes, intracellular organelles (mitochondria and the ER), and peripheral nociceptors.186 Under OA conditions, TRPV1 hyperactivation induces pathological Ca²⁺ influx, causing mitochondrial Ca²⁺ overload in both chondrocytes and nociceptors. This reduces the ΔΨm, leading to depolarization and creating a self-amplifying cycle in which ROS production and inflammation further sensitize TRP channels, ultimately potentiating OA pain signaling.186,187 Additionally, mitochondrial Ca²⁺ mobilization promotes the release of neurotransmitters from presynaptic nociceptors in the DRG, further amplifying nociceptive signal transduction in OA pain.186 Furthermore, TRPV1 hyperactivation directly mediates calcium efflux from the ER through its intrinsic ion channel, with the liberated Ca²⁺ being shuttled into mitochondria, where excessive uptake induces mitochondrial dysfunction. Moreover, aberrant Ca²⁺ regulation simultaneously triggers ER stress in both chondrocytes and peripheral nociceptors, activating the UPR pathway to amplify OA pain.188,189,190
TRPA1, which is localized to lysosomes in nociceptors, facilitates lysosomal Ca²⁺ release upon activation, enhancing neurotransmitter exocytosis and thus intensifying pain signaling.191,192,193 Additionally, nociceptor mitochondria absorb ~40% of the Ca²⁺ influx mediated by TRPM3 activation, potentially contributing to mitochondrial dysfunction and OA pain.194 However, the mechanistic links between TRPM3/TRPM8 activation and organelle dysregulation remain poorly understood and require further validation.
In the pain pathway of OA, excessive enhancement of OA pain signals leads to overactivation of neurotransmitter receptors, which causes dysfunction of organelles in second-order neurons. For example, disruption of Ca²⁺ homeostasis in nociceptors can result in increased release of neurotransmitters such as glutamate.186,191,192,193 The excessive release of glutamate causes overactivation of NMDA and AMPA receptors, leading to exaggerated Ca²⁺ influx that further results in mitochondrial depolarization and ER stress, potentially causing the death of second-order neurons.195,196,197 This may partially explain the association between OA and neurodegenerative diseases, as epidemiological evidence shows that individuals with OA have a 25% greater likelihood of being diagnosed with Parkinson’s disease (PD) or Alzheimer’s disease (AD).197
Potential drugs and strategies to treat osteoarthritis
Pharmacological treatment of osteoarthritis
Building on identified therapeutic targets for OA, pharmacological interventions can be structured around three pillars: anti-inflammation, analgesia, and the restoration of organelle homeostasis. Anti-inflammatory strategies aim to neutralize key cytokines (e.g., IL-1β, IL-6, and TNF-α) that drive cartilage degradation and synovial inflammation. Analgesic approaches target nociceptive signaling pathways to alleviate pain sensitization. Concurrently, therapies targeting mitochondrial dysfunction, ER stress, and lysosomal permeabilization in chondrocytes may disrupt the inflammation–organelle failure cycle. This tripartite framework offers a synergistic strategy to not only alleviate symptoms but also alter the OA disease trajectory.
Analgesics
Joint pain is the hallmark symptom of OA and a major contributor to reduced quality of life.198 The current management landscape includes physiotherapy, pharmacological treatment, and surgical interventions.158,199 Physiotherapy techniques such as heat or cold therapy enhance or restrict circulation to alleviate stiffness or reduce swelling. Lifestyle adjustments—such as weight loss and reduced joint loading—aim to decrease mechanical stress and associated pain.
Pharmacological agents commonly employed include NSAIDs, opioids, hyaluronic acid injections, corticosteroid injections, and growth factor injections such as platelet-rich plasma (PRP), which is derived from a patient’s blood and injected back into the affected joints to reduce pain. Despite their widespread use, these treatments fail to address the underlying causes of pain, and each treatment has limitations or adverse effects. For example, NSAIDs provide partial relief by inhibiting cyclooxygenase-1 (COX-1) and COX-2, which generate PGE2 but are ineffective in severe pain management and can lead to renal damage and gastrointestinal issues.200 Opioids offer modest benefits in chronic pain relief, but their use is limited by risks such as respiratory depression and addiction.201
Hyaluronic acid and corticosteroid injections require frequent administration because of their rapid in vivo metabolism,35,202 whereas the application of growth factors remains nonstandardized and requires further research to validate their efficacy.35 In end-stage OA, where pain becomes refractory to pharmacological treatments, surgical options, including total joint replacement, are often considered. While joint replacement has shown positive results in clinical settings, challenges such as joint infections, prosthesis dislocation, aseptic loosening, and the need for frequent revisions remain significant obstacles.203 Therefore, novel analgesic drugs with superior efficacy and fewer side effects are urgently needed to address OA-related pain.
Drawing upon the mechanisms underlying the etiology of OA pain delineated in the preceding section, the strategic targeting of TRP and Nav channels, which play pivotal roles in the peripheral detection and transmission of noxious stimuli, alongside NGF, which regulates their expression, and the neurotransmitters implicated in the transmission of pain signals to second-order neurons, has emerged as a promising therapeutic strategy. These approaches hold significant potential for the development of innovative analgesics tailored to alleviate pain associated with OA.204
TRP family-based analgesics
TRP channels are key players in pain perception, as they transduce noxious stimuli into pain signals at the joint. These channels, along with the regulation of NGF, represent promising targets for therapeutic intervention in OA pain.
TRPV1-based analgesics
The TRPV1 channel is the most extensively studied TRP channel. Several small molecules that act as TRPV1 antagonists have been identified and are expected to form the basis for a new class of pain-relieving drugs. Notable compounds include AMG517, MK2295, and AZD1386.205,206 However, systemic administration of these antagonists in preclinical studies has been associated with adverse effects, such as hyperthermia or burn-like injuries, due to the loss of the sensation of noxious heat.206,207 The mechanism underlying these side effects is believed to involve proton activation of TRPV1 (at least in rat models),206 where antagonists blocking proton activation lead to hyperthermia, whereas compounds enhancing proton sensitivity can cause hypothermia.208 In response, efforts have been made to develop TRPV1 antagonists that retain analgesic effects while avoiding interference with proton activation. The second generation of these antagonists has shown promising results in clinical trials. For example, SB-705489 (GlaxoSmithKline) reduced dental pain in rats and has completed phase 2 trials for dental pain, followed by a phase 2 rectal pain trial with no hyperthermia reported.205,206 NEO6860 has demonstrated an analgesic effect without altering body temperature in knee OA patients,162,209 and XEN-D0501, although causing a slight increase in body temperature in healthy individuals, has been approved by the FDA for treating erythromelalgia.208,210 These findings suggest that TRPV1 antagonists could offer new insights into pain management for OA. However, caution is needed, as some TRPV1 antagonists may exhibit varying effects across species (e.g., JYL1421 causes hypothermia in rats but hyperthermia in dogs and cynomolgus monkeys),206,208 underscoring the need for further research to clarify their therapeutic potential in OA pain relief.
Interestingly, preclinical studies have shown that TRPV1 agonists may also exert analgesic effects, a phenomenon that contrasts with the actions of TRPV1 antagonists. Topical or site-specific capsaicin injections, which are potent TRPV1 agonists derived from chili peppers, alleviate OA pain.205,211 This paradox can be explained by prolonged or repeated exposure to capsaicin causing a large influx of calcium ions into nociceptors, activating calcineurin, a protein phosphatase that dephosphorylates TRPV1. This results in the desensitization of TRPV1-expressing nociceptors.212,213 However, the clinical application of capsaicin is limited due to its initial pain-inducing effects at the administration site, rapid metabolism (with a half-life of 1.6 h), and side effects such as hypothermia, pruritus, erythema, and papules.205,213
To overcome these limitations, efforts have focused on developing long-lasting, tolerable TRPV1 agonists that avoid the adverse effects of capsaicin. Resiniferatoxin (RTX), an ultrapotent capsaicin analog, can reversibly desensitize nociceptors for several months with a single low-dose injection and produces similar effects upon second administration. RTX may also provide permanent analgesia through irreversible ablation of nociceptors and is currently undergoing preclinical testing.205,214,215 CNTX-4975, a synthetic form of trans-capsaicin, considerably reduced joint pain in OA patients after a single 1.0 mg intra-articular injection, with effects lasting up to 24 weeks. In a phase 2 multicenter, double-blind trial, treatment-emergent adverse events were comparable to those of the placebo.212 Zucapsaicin, the cis-isomer of capsaicin, was approved in 2010 as a topical analgesic for severe knee pain in OA patients and is associated with fewer side effects than traditional capsaicin.216 Other TRPV1 agonists, including NE19550, MRD-652, and CPIPC, have shown promising analgesic effects in animal models,205,213,216 but their clinical efficacy in human OA remains to be fully established.
TRPA1-based analgesics
The TRPA1 channel is commonly characterized as a nonselective cation channel that detects noxious cold, mechanical stimuli, and irritant chemicals.217,218 While TRPA1 activation by noxious cold in healthy tissues is debatable, its role in cold hypersensitivity following tissue injury is well supported by multiple studies.161 In various OA rat models induced by complete Freund adjuvant (CFA), carrageenan, MIA, and monosodium urate, direct activation of TRPA1 by ROS, IL-1β, and TNF-α induces both mechanical and cold hypersensitivity. Notably, genetic deletion or pharmacological blockade of TRPA1 channels reverses allodynia without inducing the thermosensory or thermoregulatory disturbances typically observed with TRPV1 antagonism.161,218 These findings highlight the potential of TRPA1 antagonists as novel analgesics for OA pain management. Several promising TRPA1 antagonists, including HC-030031, AP-18, and A-967079, have been shown to reduce mechanical and cold hypersensitivity in animal models of CFA-induced inflammatory pain.217,219 However, a recent study reported conflicting results, in which systemic administration of HC-030031 did not alleviate ongoing pain in MIA-induced OA rats.220 This discrepancy may arise from the significant difference in TRPA1 expression between species, as TRPA1 is expressed at a much higher percentage (~80%) in human DRG neurons than in mouse DRG neurons, suggesting that the analgesic potency of TRPA1 antagonists may be underestimated in preclinical studies.217,219 Further clinical research is needed to elucidate the therapeutic potential of TRPA1 antagonists in OA pain management.
However, some TRPA1 agonists function similarly to TRPV1 agonists, desensitizing TRPA1-expressing neurons to produce analgesia. For example, systemic administration of atractylodin (ATR), a TRPA1 agonist, leads to sustained activation of TRPA1 (>1 h), which mitigates nocifensive behaviors induced by another TRPA1 agonist, allyl isothiocyanate (AITC).221 Additionally, intravenous injection of Ms 9a-1 markedly enhances TRPA1 responsiveness to agonists, thereby exerting an antinociceptive effect on CFA-induced inflammatory pain in rats.222 The mechanisms underlying the pro- or antinociceptive effects of certain TRPA1 agonists remain unclear. One hypothesis is that while pronociceptive TRPA1 agonists reversibly modulate TRPA1, antinociceptive agonists may irreversibly bind to cysteine residues near the cation-passing gate of TRPA1, thereby preventing channel reopening.219 Despite the analgesic potency of TRPA1 agonists, many challenges remain unresolved. For example, the administration of TRPA1 agonists may cause initial pain that is intolerable to patients, and TRPA1 agonists often suffer from issues such as poor chemical properties, low selectivity, or weak efficacy. Additional studies may provide further prospects for the use of TRPA1 agonists in OA pain treatment.
TRPM8-based analgesics
TRPM8 is recognized as a sensor for both innocuous cool (15–25 °C) and noxious cold (<15 °C) temperatures and is activated by compounds that elicit cool sensations, such as menthol, icilin, and eucalyptol.223,224 In contrast to the well-defined mechanisms of TRPV1 and TRPA1 channels in nociception, TRPM8 appears to play a bidirectional role in the pain pathway but exhibits a distinct anti- or pronociceptive pattern in specific pain conditions.223 For example, in the CFA model of inflammatory pain, mechanical and heat pain are substantially reduced following menthol injection, whereas pretreatment with TRPM8 antagonists or genetic deletion of TRPM8 abolishes this analgesic effect.223,225 Inflammatory conditions, including low pH and “inflammatory soup,” inhibit TRPM8 activity, and the mRNA expression of TRPM8 is reduced in human OA chondrocytes.226 These findings suggest that activation of TRPM8 may attenuate mechanical and heat allodynia in patients with inflammatory pain.223 Additionally, given that most nociceptors express TRPV1, the lack of coexpression of TRPM8 with TRPV1 suggests that TRPM8 does not interfere with the perception of mechanical and heat stimuli but blocks the transmission of evoked action potentials to second-order neurons.223,227
Conversely, TRPM8 activation can lead to cold-induced pain following injury and exacerbate cold hyperalgesia in patients with neuropathies.223 The application of high concentrations (30–40% wt/vol) of menthol has been shown to cause cold pain, cold allodynia, and cold hyperalgesia in human trials.228,229 These data suggest that patients with mechanical or heat pain as their primary symptom may be better treated with TRPM8 agonists at a low concentration, whereas individuals with cold-hyperalgesia may respond better to TRPM8 antagonists. Therefore, TRPM8 agonists are likely to be preferable therapeutic options for OA pain.
However, the use of TRPM8 agonists in OA pain treatment has rarely been reported. In a study involving 20 OA patients, the topical application of 3.5% menthol gel to OA knee joints resulted in no significant improvements in pain behavior compared with placebo.230 Additionally, coculture of menthol with OA chondrocytes increased the expression of MMPs and proinflammatory mediators in vitro.226 These findings suggest that further studies are needed to confirm the potential analgesic effects of TRPM8 agonists in treating OA pain.
TRPM3-based analgesics
TRPM3 is also a nonselective cation-permeable channel that is predominantly expressed on nociceptors, where it mediates nocifensive responses to the endogenous neurosteroid pregnenolone sulfate (PS) and noxious heat.207,231,232 Intraplantar injection of PS in mice elicits pain behaviors that are absent in TRPM3-deficient mice, suggesting that TRPM3 activation is critical for initiating pain sensation.207,232 Several studies have demonstrated that TRPM3 expression in DRG neurons increases under inflammatory conditions and that administration of TRPM3 antagonists in rodents abolishes TRPM3-mediated pain without affecting core temperature. Additionally, these antagonists reduce the responsiveness of TRPV1 and TRPA1 channels to their respective agonists, thereby enhancing the analgesic effects of TRPM3 blockade.232,233 Although TRPM3 antagonism may attenuate heat sensation, the perception of noxious heat remains intact as long as the heat sensor, TRPV1, remains functional,218 underscoring the potential of TRPM3 antagonists as promising analgesics for OA pain treatment.
However, research on TRPM3 antagonists as analgesics is still in its infancy. Plant-derived TRPM3 antagonists, such as isosakuranetin, ononetin, and primidone, suffer from suboptimal pharmacokinetic properties and lack target selectivity, hindering their therapeutic applicability in humans.207 Further research is needed to address these challenges, potentially by encapsulating these antagonists in appropriate delivery systems. Unlike other TRP channels, which are typically tetramers with a central cation-permeable pore,234 recent findings suggest that TRPM3 comprises an alternative pore. This alternative pore, synergistically activated with the central pore, intensifies pain sensation.218,231 This novel mechanism has sparked interest in targeting the alternative pore of TRPM3, and the concurrent blockade of both pores may enhance analgesia, offering a new avenue for OA pain management.
Voltage-gated sodium channel-based analgesics
Blockade of Nav channels represents another promising strategy for alleviating OA pain, as these channels contribute to pain hypersensitivity in OA patients by initiating action potential firing and transmitting pain signals through Na+ influx in nociceptors.235 Current Nav blockers used for pain management, such as local anesthetics (e.g., lidocaine), class I antiarrhythmics (e.g., mexiletine), antiepileptics (e.g., carbamazepine), and antidepressants (e.g., TCAs), have demonstrated significant efficacy in pain relief but also cause severe adverse effects, such as dizziness, sedation, convulsions, and cardiotoxicity, as these drugs nonselectively act on multiple Nav subtypes related to motor and neurological functions,236,237 highlighting the need for selective Nav blockers to minimize off-target effects.
Nav subtypes, including Nav1.1 through Nav1.9, are expressed in various tissues and play pivotal roles in multiple physiological functions, necessitating careful consideration of potential side effects before specific subtypes are targeted.237 Blockade of Nav1.1, Nav1.2, and Nav1.3 channels, which are expressed primarily in the central nervous system (CNS), poses a risk of altered mental status or seizures.236 In contrast, the inhibition of Nav1.4 and Nav1.5 has been associated with muscular and cardiac channelopathies, respectively.238 Nav1.6 is expressed mainly in motor neurons, and blockade of this isoform leads to paresthesia and weakness.239 Nav1.9 is expressed in nociceptors and myenteric neurons in the gastrointestinal tract. Research on Nav1.9-specific blockers has been limited due to the rarity of clinical cases involving Nav1.9 mutations as well as challenges in developing Nav1.9-expressing cell models, which could explain the lack of promising Nav1.9-specific blockers despite clear evidence of Nav1.9 participation in pain signaling.236,240
The potential analgesic effects of Nav channel blockers are often outweighed by their severe side effects. In contrast, specific inhibitors of Nav1.7 and Nav1.8 have shown the most promising results in pain treatment, with manageable adverse effects.236 Blockade of Nav1.7 results in manageable anosmia, whereas inhibition of Nav1.8 is associated with a risk of visual disturbances.241,242 Studies in OA mouse models have revealed upregulated expression of Nav1.7 and Nav1.8 in nociceptors innervating the knee joint, which is correlated with the pain hypersensitivity observed in these models.243,244 Several Nav1.7- and Nav1.8-specific blockers have been shown to attenuate secondary allodynia and improve motor function in murine OA models. For example, PF-04856264, a selective Nav1.7 blocker, substantially alleviates OA pain and upregulates type II collagen levels following intra-articular injection in a murine meniscectomy model, with similar results observed in MIA-induced OA rat models through oral administration.245 ProTx II, a tarantula toxin that selectively blocks Nav1.7, demonstrated no analgesic efficacy following intravenous or intrathecal injection but was found to upregulate the expression of type II collagen and aggrecan while reducing MMP13 and ADAMTS5 levels.245,246,247 A recently designed peptide, PTx2-3127, which is based on ProTx II, has shown efficacy in chronic and thermal pain in rat models via intrathecal administration, positioning it as a prospective novel analgesic.248
However, intra-articular injection of A-803467, a potent and selective Nav1.8 blocker, considerably reduced hindlimb disability and secondary allodynia in MIA-induced OA model rats.247,249 While specific Nav blockers have shown promising analgesic effects in animal models of skeletal degenerative diseases, the pharmacological and toxicological profiles of these blockers in humans remain largely unknown, necessitating further studies before their clinical application.
NGF-based analgesics
NGF promotes the expression of the TRPV1 channel, Nav1.8 channel, CGRP, and SP, thereby enhancing pain sensation in OA patients. The inhibition of NGF signaling has emerged as a promising strategy for pain relief, with several clinical trials demonstrating that NGF antagonists outperform NSAIDs in terms of efficacy while avoiding severe side effects. These findings suggest that NGF is a prime target for the development of novel analgesics for OA pain management.168,169,173,250
Several strategies have been employed to inhibit NGF signaling, including (i) agents that capture free NGF to prevent its interaction with its receptor TrkA, (ii) agents that directly occupy the NGF-binding site, and (iii) agents that block TrkA receptors.168,172 NGF-capturing agents, such as humanized monoclonal antibodies, have shown remarkable specificity and potency in neutralizing NGF activity by directly binding to NGF.168,169,172 Notable examples include tanezumab (RN624), fasinumab (REGN475), and fulranumab, all of which have proven effective in alleviating OA pain. For example, tanezumab demonstrated superior analgesic effects to NSAIDs in treating hip or knee OA, although it was associated with a higher incidence of adverse events in a phase III trial conducted by Pfizer and Eli Lilly in 2019.251 Similarly, fasinumab and fulranumab considerably reduce pain and improve physical function in OA patients, with adverse events comparable to those of placebo in two phase II, randomized, double-blind, placebo-controlled trials.252,253
Additionally, several molecules that occupy NGF-binding sites and disrupt the interaction between NGF and its receptors, including ALE0540, PD90780, Ro 08-2750, Y1036, and PQC 083, have been investigated.172,173 Notably, systemic or spinal intrathecal administration of ALE0540 has demonstrated antiallodynic efficacy in animal models of neuropathic pain and thermally induced inflammatory pain.254 However, the analgesic effects of other molecules in this class remain to be fully elucidated.
The analgesic mechanisms of both NGF-capture agents and NGF-binding site antagonists rely on blocking NGF activity. However, this blockade may inadvertently disrupt homeostatic cellular responses mediated by NGF-p75NTR interactions, potentially complicating long-term use.173 Consequently, selective inhibitors of TrkA, which is a high-affinity NGF receptor, may offer greater clinical benefits. Nonetheless, TrkA is widely expressed in cholinergic neurons of the CNS, raising concerns about potential side effects in the CNS. To mitigate these risks, the use of TrkA inhibitors should ideally be restricted to peripheral applications.255
Most TrkA inhibitors target the ATP-binding site, a highly conserved motif shared among Trk family members, which may inadvertently inhibit other Trk receptors and exacerbate the CNS or other adverse effects.168,255,256 To address this, numerous small molecules with high selectivity for TrkA have been developed by targeting less conserved ligand-binding regions. An exemplary compound in this category is PF-06273340, a potent TrkA inhibitor with low cytotoxicity. PF-06273340 inhibits TrkA by 98% at a concentration of 1 μM and can be effluxed by P-glycoprotein (P-gp), a transporter that prevents its entry into the brain, thereby reducing the risk of side effects in the CNS.255,256 In preclinical models, PF-06273340 has shown efficacy in alleviating pain in an ultraviolet burn-induced hyperalgesia model of inflammatory pain, suggesting that it is a promising candidate for clinical application in OA pain management.256
Another promising TrkA inhibitor is AR786, which has good oral bioavailability and low CNS penetration. Chronic oral administration of AR786 effectively prevented pain in MIA-induced OA model rats.257 As research into TrkA-selective inhibitors continues to expand, these compounds hold considerable promise for addressing unmet medical needs in OA pain treatment.
Neurotransmitter-based analgesics
Neurotransmitters play a critical role in nociceptive transmission. Targeting their transport or receptor activity can reduce nociceptive signaling and offer analgesic benefits. SP is one such neurotransmitter involved in pain transmission, and its receptor, NK1R, has emerged as a potential therapeutic target. Despite the success of various NK1R antagonists in animal models, human trials have not demonstrated significant analgesic effects.183,258 This discrepancy may be due to species differences or the limited impact of blocking a single SP signaling pathway amid the broader network of neurotransmitters involved in nociception.258
In addition to SP, CGRP is another important neurotransmitter in nociception. This 37-amino acid peptide binds to an atypical receptor composed of CLR, RAMP1, and RCP, which is located on both nociceptors and second-order neurons.181 CGRP sensitizes nociceptors and transmits pain signals via its receptor, suggesting that blocking CGRP receptors could offer analgesic potential. While CGRP receptor antagonists are well established in migraine treatment, their role in OA pain remains less explored. However, two studies have shown that CGRP receptor antagonists considerably reverse nociceptor sensitization and pain-related behaviors in OA animal models. For example, the subcutaneous injection of BIBN4096BS in MIA-induced OA rats provided morphine-like analgesia within 4 h.259 Intra-arterial injection of CGRP 8-37 reversed joint nociceptor mechanosensitization in both MIA-induced and meniscectomy OA models.260 These studies confirmed that CGRP receptor antagonists block receptors on joint nociceptors to prevent sensitization. However, whether they also inhibit nociceptive transmission by acting on second-order neurons remains to be clarified.
Glutamate is another central neurotransmitter involved in nociceptive transmission. Pharmacological blockade of glutamate receptors, including iGluRs and mGluRs, has demonstrated analgesic effects in both preclinical and clinical studies.261,262 The iGluR family includes N-methyl-D-aspartate (NMDA) receptors, α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptors, kainate (KA) receptors, and δ (GluD) receptors. GluD receptors differ in that they require the intracellular ligand D-serine for activation rather than glutamate, and their role in nociception is still under investigation,263 so they are not covered here.
Excluding GluD, all iGluRs are ligand-gated ion channels permeable to Ca²⁺, Na+, and K+, causing depolarization of second-order neurons upon activation.264 NMDARs are heteromeric complexes typically containing the NR1 subunit and one or more NR2A-D subunits.265 While most subunits are ubiquitously expressed in the CNS, NR2B-containing NMDA receptors are more selectively distributed in the forebrain and superficial dorsal horn of the spinal cord.265,266 Traditional nonselective NMDA antagonists—such as ketamine, memantine, and MK-801—act on all NMDA subunits,267 but they can cause adverse effects such as neurotoxicity, motor impairment, learning impairment, and psychotomimetic symptoms at or near the antinociceptive dosage.268,269 To avoid these side effects, selective antagonists of NR2B-containing NMDA receptors represent a novel therapeutic option that might be devoid of the most prominent side effects of nonselective NMDA antagonists while retaining analgesic efficacy.
Ifenprodil is a potent and selective NR2B-containing NMDA receptor antagonist. Spinal administration of ifenprodil alleviates mechanical allodynia and neuropathic pain without cognitive or psychotomimetic side effects.265,266 Other newly developed NR2B-containing NMDA receptor antagonists, including CP101606, Ro256981, and CI1041, have shown antinociceptive effects without CNS side effects.266 However, the pharmacological properties of these drugs have never been tested in animal models of OA pain; more preclinical data will be necessary to assess their feasibility in relieving OA pain.
AMPA receptors, another iGluR subtype, are composed of GluR-A-D (also known as GluR1-4).270 GluR-B has an arginine residue at the pore-forming segment, which introduces additional positive charges into the pore that inhibit Ca²⁺ influx,271 increasing its antinociceptive properties and limiting its potential as a drug target. Unlike GluR-C and GluR-D, Ca²⁺-permeable AMPA receptors are predominantly composed of homomeric GluR-A subunits at second-order neurons of the spinal dorsal horn,270 and several animal studies have demonstrated that the activation of GluR-A-containing AMPA receptors leads to long-lasting nociceptive hypersensitivity,270,272 making GluR-A the most attractive target among other subunits in analgesia. However, selective antagonists for GluR-A remain elusive. Nonselective AMPA receptor antagonists, such as NBQX, have yielded mixed results in OA pain models. For example, in antigen-induced arthritis (AIA) rats, intra-articular injection of NBQX, an AMPA/KA receptor antagonist, reduced pain-related behaviors within 2 days,273 but it had no analgesic effect on posttraumatic OA rats.274 More data are needed to demonstrate the effectiveness of AMPA receptor antagonists in analgesia and the role of GluR-A antagonists in OA pain treatment.
Kainate receptors, the final members of the iGluR family, consist of five subunits, including GluK1-5 (formerly known as Glu5-7, KA1, and KA2).275 In contrast to the restricted localization of the NMDA receptor and AMPA receptor on postsynaptic neurons, the kainate receptor is distributed on both presynaptic and postsynaptic neurons at the spinal dorsal horn.275,276 Kainate receptors on presynaptic neurons (including nociceptors) mediate the release of glutamate, whereas those on postsynaptic neurons (including second-order neurons) contribute to nociceptive transmission by binding to glutamate.277 Among the kainate receptor subunits, GluK1 is predominantly expressed in nociceptors within the DRG, and it appears to play a crucial role in nociceptive transmission, as the kainate receptor exhibits an absolute requirement for GluK1, for which kainate receptor-mediated currents are not detected in GluK1 knockout DRG neurons.277,278 Recent studies have extensively explored the efficacy of several GluK1 antagonists in analgesia, and some have shown promising results in preclinical trials. MSVIII-19 (8,9-dideoxy-neodysiherbaine) is a potent GluK1 antagonist. An elegant experiment conducted by Chang-Shen Qiu and colleagues revealed that intrathecally administered MSVIII-19 reduced thermal hypersensitivity in animal models of inflammatory pain and both thermal hyperalgesia and mechanical hypersensitivity in a chronic construction injury model of neuropathic pain.275 LY467711 and LY525327 are two orally effective prodrugs that are metabolized into two selective GluK1 antagonists called LY458545 and LY457691 in vivo. Compared with morphine and ibuprofen, the oral administration of these two prodrugs has shown preferable analgesic efficacy in three classical pain models, including formalin-induced late-phase paw-licking pain, carrageenan-induced thermal hyperalgesia, and capsaicin-induced mechanical hyperalgesia.279
Finally, mGluRs, another critical family of glutamate receptors, also play crucial roles in the pain pathway. mGluRs can be divided into three groups on the basis of amino acid homology: group I (mGluR1 and mGluR5), group II (mGluR2 and mGluR3), and group III (mGluR4, mGluR6, mGluR7, and mGluR8).280 Like iGluRs, mGluRs are involved in transmitting pain signals, but the functions of different groups vary on the basis of their distributions. Group I mGluRs are expressed in both pre- and postsynaptic neurons and initiate action potential firing in second-order neurons when activated, thus contributing to pain signal transmission. In contrast, group II and III mGluRs are located predominantly at presynaptic terminals of nociceptors, where they act as autoreceptors to suppress nociceptor excitability upon activation.281,282 Given their distinct roles, targeting specific subtypes of mGluRs, such as blocking group I mGluRs or activating group II/III mGluRs, offers promising therapeutic strategies for OA pain relief. Numerous studies have demonstrated the analgesic potential of group I mGluR antagonists and group II/III mGluR agonists. The spinal administration of group I mGluRs such as LY367385 and AIDA has been shown to reverse thermal hyperalgesia in rats with knee inflammation without affecting basal perception.283 Activating group II mGluRs with the agonist APDC blocks nociceptor hyperexcitability without affecting basal membrane excitability.281 Other group II mGluR agonists (LY379268, LY354740, LY389795, and LCCG1) have demonstrated analgesic properties in many inflammatory pain models (capsaicin-treated and formalin-treated models),280 whereas group III mGluR agonists, such as ACPT-I and PHCCC, inhibit nociceptive behaviors in rats subjected to formalin tests and mechanical hyperalgesia in various models of inflammatory pain (carrageenan-treated and monoarthritic rats).282 These findings underscore the importance of mGluRs in nociceptive transmission and highlight their potential as therapeutic targets for OA pain treatment.
Anti-inflammatory effects
The devastating inflammation observed in the OA joint is closely linked to the massive release of proinflammatory cytokines, which contribute to hallmark clinical manifestations, such as swelling, synovitis, and inflammatory pain. Catabolic events elicited by these inflammatory cytokines further aggravate the inflammatory milieu, thereby exacerbating OA progression. Targeting key cytokines involved in OA progression—specifically IL-1, IL-6, and TNF-α—through pharmacological blockade may provide a foundation for the development of novel therapeutic strategies. This section systematically reviews potential therapeutic targets identified within the signaling pathways of IL-1, IL-6, and TNF-α, discusses corresponding therapeutic agents, and highlights the challenges involved in managing OA-related inflammation and preventing disease progression.
IL-1-targeting pharmacological agents
IL-1 exerts a variety of negative effects on the pathogenesis of OA, inducing both local and systemic inflammation. It upregulates MMPs and ADAMTS, inhibits the synthesis of the proteoglycans aggrecan and type II collagen, and disrupts the balance between osteoblasts and osteoclasts. These effects collectively lead to tissue degradation, bone erosion, and osteophyte formation. Given its centrality in OA pathology, IL-1 signaling represents a promising therapeutic target for mitigating disease progression. A deeper understanding of IL-1 signaling mechanisms is essential for identifying potential therapeutic interventions. On the basis of the involvement of IL-1 signaling in OA progression, therapeutic strategies targeting IL-1 can be categorized into three main approaches: (i) agents that antagonize IL-1RI, (ii) agents that neutralize circulating IL-1, and (iii) agents that inhibit the maturation of IL-1.
IL-1RI antagonists
IL-1Ra exerts anti-inflammatory effects by competitively antagonizing IL-1 at IL-1RI, thereby blocking downstream proinflammatory signaling without receptor activation. This mechanism suggests therapeutic potential for OA, a hypothesis supported by epidemiological evidence demonstrating an inverse correlation between serum IL-1Ra concentrations and the radiographic severity of knee OA.284,285 Despite its anti-inflammatory properties, the therapeutic application of IL-1Ra in OA remains limited by its short intra-articular half-life and suboptimal therapeutic efficacy. Current research focuses on bioengineering strategies to increase IL-1Ra pharmacokinetics. For example, Khaled et al. engineered a PLGA microsphere encapsulating IL-1Ra, demonstrating that intra-articular administration preserved the inherent cartilage-protective bioactivity of IL-1Ra while substantially enhancing disease-modifying efficacy in a rat model of ACLT-induced OA.286 Similarly, Shikhar et al. designed an IL-1Ra through cationic carrier-mediated modification, enabling electrostatic targeting to the negatively charged aggrecan-glycosaminoglycan matrix within cartilage. While unmodified IL-1Ra failed to attenuate IL-1-induced cartilage degradation over 16 days, the modified IL-1Ra considerably suppressed induced glycosaminoglycan loss and nitrite release concomitant with improvements in chondrocyte metabolism and viability.287
Despite promising preclinical efficacy, IL-1Ra protein-based therapies remain limited by the need for repeated administration. Gene therapy has emerged as a strategic alternative to circumvent this challenge, offering sustained IL-1Ra expression and novel therapeutic insights for OA management.288 Deng et al. developed a polyplex nanomicelle-based delivery system for mRNAs encoding IL-1Ra. Compared with the control, a single intra-articular injection of this construct in a rat model of temporomandibular joint OA alleviated pain and suppressed OA progression for 28 days, as evidenced by reduced pain-related behaviors and attenuated cartilage degradation.289 Alan et al. employed helper-dependent adenovirus (HDAd)-mediated intra-articular gene therapy to deliver the IL-1Ra-encoding gene, effectively preventing pathological progression in murine and equine OA models.290 Frisbie et al. designed an adenoviral vector expressing equine IL-1Ra DNA (Ad-EqIL-1Ra), which was administered via intra-articular injection in equine OA models. This intervention reduced joint pain-related lameness and pathological changes while exerting protective effects against proteoglycan loss. Notably, therapeutic efficacy was observed even when treatment was initiated 14 days post-OA induction, indicating potential utility in addressing established joint trauma.288
While these preclinical advancements in IL-1Ra delivery optimization underscore its therapeutic promise, clinical translation necessitates confronting inherent pharmacokinetic challenges. The translational gap between preclinical efficacy and clinical applicability becomes evident in human trials, where even mechanistically validated strategies face hurdles such as short half-lives and suboptimal biodistribution. For example, anakinra, a recombinant IL-1Ra, has been approved by the FDA for treating rheumatoid arthritis (RA).56,64,291 Clinical trials have demonstrated its safety and tolerability, with only rare cases of anaphylaxis. However, owing to its relatively short half-life of 4–6 h, daily subcutaneous injections are required to maintain therapeutic efficacy, which can lead to frequent injection-site reactions.49,50,64 AMG108, a humanized monoclonal antibody that targets IL-1RI, is still in clinical development for OA and RA treatment.56 Phase II trials have shown a favorable safety profile, with intravenous or subcutaneous administration, but the clinical efficacy has been limited, with only modest improvements observed in RA patients and no significant effects in OA patients to date.292,293
IL-1 neutralizing agents
Canakinumab is a humanized monoclonal antibody that selectively neutralizes IL-1β, with no effect on IL-1α-driven inflammation.56,291 With a long average half-life of 26.1 days,51 canakinumab is administered every 4–8 weeks via subcutaneous injection. It is approved for treating cryopyrin-associated periodic syndrome (CAPS) and systemic-onset juvenile idiopathic arthritis.49,56,64 Rilonacept, a dimeric fusion protein comprising the Fc region of human IgG1 and the extracellular IL-1 binding site of IL-1RI and IL-1RAcP, can neutralize circulating IL-1α, IL-1β, and IL-1Ra.51,56 The safety profile of rilonacept is still inconclusive due to limited clinical data, but it appears comparable to that of anakinra.50,56 It requires weekly subcutaneous injections and has been approved by the FDA for treating CAPS, recurrent pericarditis, and IL-1Ra deficiency (DIRA). However, indiscriminate neutralization of IL-1Ra may hinder the therapeutic efficacy of rilonacept.56,291 To optimize clinical outcomes, the modulation of rilonacept to preferentially neutralize IL-1α and IL-1β while preserving the affinity of IL-1Ra may be beneficial. Recent studies by Yusha Wang et al. revealed a de novo variant, p.Lys131Glu (K131E), in IL-1R1 that reduces its affinity for IL-1Ra. They further designed a novel IL-1-neutralizing agent, rilabnacept, upon the modulation of rilonacept with the K131E mutation, which showed promise in preclinical models of IL-1-driven diseases.291
In addition to the agents already in clinical use, several other neutralizing IL-1 agents are currently under investigation. These include bermekimab, a naturally occurring monoclonal antibody that neutralizes IL-1α; gevokizumab, a humanized monoclonal antibody that targets IL-1β; and lutikizumab, a dual-targeting antibody that blocks both IL-1α and IL-1β.56 These emerging therapies offer promising alternatives or adjuncts to existing treatments, potentially enhancing the ability to modulate the IL-1-mediated inflammatory pathways in OA. As clinical development advances, the efficacy and safety profiles of these agents will be key in determining their future role in managing IL-1-mediated diseases, including OA.
Caspase-1 inhibitors
VX-740 (also known as pralnacasan) is an orally active, reversible caspase-1 inhibitor developed in cooperation with Vertex Pharmaceuticals and Aventis.294 In murine OA models, high-dose VX-740 (50 mg/kg) substantially alleviated inflammation and reduced cartilage degradation and bone erosion indicators by 59% and 84%, respectively.295 The subsequent phase I clinical trials demonstrated favorable tolerability of VX-740 with 50% oral bioavailability. In phase II clinical trials involving 285 RA patients, VX-740 showed a trend toward symptom improvement but failed to reach statistical significance. Phase III trials for RA and OA treatment were discontinued because of the liver toxicity observed in long-term animal studies.50,294 VX-765 (belnacasan), another reversible caspase-1 inhibitor developed by Vertex Pharmaceuticals, has greater potency than VX-740.50 Phase I clinical trials have shown that VX-765 meets pharmacokinetic and pharmacodynamic objectives, while phase II trials for psoriasis were completed in 2005, although no further updates have been published.294 A recent study demonstrated that oral administration of VX-765 considerably alleviated inflammation and joint tissue damage in a murine OA model,296 suggesting its potential as a novel anti-inflammatory agent for OA treatment.
To summarize, currently FDA-approved anti-IL-1 drugs—such as anakinra, canakinumab, and rilonacept—are associated with common biologic-related side effects, including injection-site reactions, reversible neutropenia, and an increased risk of bacterial infections.49 Despite encouraging preclinical and early clinical findings, monotherapy with IL-1-targeting agents (such as anakinra, canakinumab, rilonacept, AMG 108, and VX-740) has not yielded substantial clinical improvements in RA or OA patients, although some trends toward relief have been noted.48 These findings suggest that exclusive inhibition of IL-1β may be insufficient for effective OA treatment. Combination therapies targeting IL-1β along with other anti-inflammatory agents may enhance the therapeutic efficacy of reducing OA-associated inflammation.
TNF-α-targeting pharmacological agents
As discussed earlier regarding TNF-α-mediated signaling in OA pathogenesis, therapeutic strategies targeting TNF-α can be categorized into three main approaches: (i) neutralizing both soluble and transmembrane sTNF-α and tmTNF-α; (ii) antagonizing TNFR-1 and TNFR-2 receptors; and (iii) inhibiting TACE, which reduces sTNF-α production and may partially prevent TNFR-1-expressing (including chondrocytes) apoptosis induced by the binding of sTNF-α and TNFR-1. However, since tmTNF-α also binds TNFR-1 with high affinity and TACE also cleaves TNFRs into soluble forms that act as natural TNF-α antagonists,297,298 TACE inhibition may yield minimal or even counterproductive effects. For example, marimastat, a TACE inhibitor, suppressed LPS-induced sTNF-α in mice but only slightly delayed LPS-induced lethality.298
Traditional TNF-α-targeting agents approved by the FDA include infliximab, etanercept, and adalimumab.68,298 Infliximab is a chimeric monoclonal antibody composed of a murine TNF-α-binding site fused with the Fc region of human IgG1, enabling it to bind specifically to TNF-α and block its interaction with TNFRs.68,299,300 When infliximab is administered intravenously with a half-life of 8–9.5 days, it is approved for treating Crohn’s disease (CD), ankylosing spondylitis (AS), psoriatic arthritis (PsA), psoriasis (PSO), and RA.68,301 However, its murine component may elicit human anti-chimeric antibodies (HACAs), reducing therapeutic efficacy—an effect that can be ameliorated with the coadministration of methotrexate.301 Adalimumab is a fully human IgG1 monoclonal antibody with a similar mechanism but lower immunogenicity.68,298 Nevertheless, long-term use may still lead to the development of anti-adalimumab antibodies.301 When administered subcutaneously with a half-life of 10–13 days, it is approved for PsA, PSO, AS, CD, juvenile idiopathic arthritis (JIA), uveitis, and RA.68
Etanercept is a fusion protein that combines the extracellular region of TNFR-2 and the Fc region of IgG1. It is administered subcutaneously with a half-life of 3–3.5 days and is approved for PsA, PSO, AS, and RA.68,299,301 All these agents contain an IgG1 Fc region, enabling them to induce lysis of tmTNF-α-producing cells (including chondrocytes) via complement-dependent cytotoxicity and antibody-dependent cell-mediated cytotoxicity, potentially exacerbating OA progression.302
This issue is addressed by certolizumab pegol, a recently approved TNF-α-targeting agent. It is a humanized monoclonal antibody lacking the Fc region and consists of a human IgG1 Fab fragment with a murine anti-TNF-α amino acid sequence covalently linked to a polyethylene glycol moiety.68,299 This PEGylation enhances pharmacokinetics, yielding a subcutaneous half-life of ~14 days. In CD, PSO, PsA, AS, and RA, certolizumab pegol avoids Fc-mediated cytotoxicity.68 However, similar to other TNF-α antagonists, it may still induce immunogenicity and antibody formation, limiting its long-term use. These agents also increase the risk of infections (particularly tuberculosis), malignancies, systemic lupus erythematosus-like syndromes, and demyelinating diseases.300 The latter two are thought to be associated with disrupted TNF-α-TNFR-2 signaling (discussed below).299
Recent studies underscore the role of TNFR-2 in TNF-α-associated autoimmunization. Approximately 30% of regulatory T (Treg) cells—key mediators in the suppression of immune overactivation—express TNFR-2 but not TNFR-1.303 The binding of TNF-α and TNFR-2 initiates expansion, prolonged survival, and increased phenotypic stability of Treg cells in humans, thus spurring research into the use of TNFR-2 agonists as potential therapies for autoimmune diseases.304 However, paradoxically, the proportion of Treg cells increases in most autoimmune diseases, including RA, following TNF-α inhibition therapy. This phenomenon may be explained by certain anti-TNF-α agents, such as adalimumab, which conversely increase the expression of tmTNF-α on monocytes, thereby promoting the interaction between tmTNF-α and TNFR-2.305 The change in the proportion of Treg cells caused by nonselective blockade of TNF-α-TNFR-2 signaling might partially account for the autoimmune-related adverse effects and infections observed with anti-TNF-α monoclonal antibody therapy.
However, the proinflammatory effects are predominantly mediated by TNFR-1, suggesting that selective antagonism of TNFR-1 may be a promising strategy for treating OA, as it preserves TNFR-2-mediated immunoregulation and potentially prevents the side effects typically associated with monoclonal antibody therapy.306 Several attempts have been made to inhibit TNFR-1, such as R1antTNF—a mutant form of human TNF-α that selectively blocks TNFR-1 by competing with TNF-α for the binding site—showing promise in OA inflammation treatment. However, its therapeutic effects are limited by its short half-life (~10 min) in murine plasma when it is administered intravenously. PEGylation of R1antTNF extended its half-life to 12 h without compromising bioactivity. Both prophylactic and therapeutic use of PEGylated R1antTNF showed comparable efficacy to etanercept in reducing the severity of arthritis in a murine CIA model without impairing the immune response to infections—an issue observed with etanercept.307
Atrosimab, a monoclonal antibody targeting TNFR-1, considerably reduces the development and severity of arthritis in murine models, showing effects comparable to those of infliximab, etanercept, and certolizumab pegol in both prophylactic and therapeutic settings.308 Other selective TNFR-1 antibodies (IZI-06.1, TNFR1-AlbudAb, Nb 70 and Nb 96), as well as small molecules (PMG, R1), have demonstrated the ability to prevent TNFR-1-induced apoptosis and cytokine production in certain cell types without compromising TNFR-2-mediated immune regulation, suggesting promising therapeutic strategies for OA.306 However, these agents still rely on a similar mechanism of blocking TNFR-1 by competing for the same TNF-α binding site, which may limit their therapeutic efficacy owing to the high affinity of both sTNF-α and tmTNF-α for TNFR-1.
Recent studies have also identified noncompetitive TNFR-1 inhibitors, such as DS41, DSA11, and Zafirlukast, which bind to distinct sites on TNFR-1 to stabilize its nonfunctional state, thereby blocking downstream signaling without interfering with TNFR-2 regulation.306 In conclusion, current pharmacological anti-TNF-α agents (infliximab, etanercept, adalimumab, and certolizumab pegol) have demonstrated substantial clinical efficacy in treating various forms of arthritis, particularly RA. With a deeper understanding of TNF-α signaling, selective TNFR-1 antagonists have been developed to minimize the side effects of traditional anti-TNF-α therapies while retaining their anti-inflammatory properties. These agents have shown promising therapeutic effects in preclinical arthritis models. However, their efficacy in OA treatment remains underexplored, and further research is needed to evaluate the potential of anti-TNF-α therapy in OA management.
IL-6-targeting pharmacological agents
The IL-6 pathway has emerged as a pivotal factor in the pathogenesis of various diseases, including OA. As such, targeting IL-6 represents a promising anti-inflammatory strategy for OA treatment. IL-6 has three key binding sites: site I, site II, and site III. Through site I, IL-6 binds to either mIL-6R or sIL-6R to form a membrane-bound or circulating complex, which subsequently interacts with dimerized gp130 via sites II and III, initiating classic or trans-signaling.309 As discussed in the previous section, this IL-6 pathway is a central focus of therapeutic intervention.
Therapeutic strategies targeting the IL-6 pathway can be summarized into several categories: (i) blockade of gp130, (ii) blockade of circulating IL-6, (iii) blockade of IL-6R, (iv) inhibition of ADAM10/ADAM17, (v) inhibition of sIL-6R transcription, and (vi) inhibition of intracellular participants such as JAKs and STAT3. While gp130 serves as a signaling receptor for multiple cytokines, its blockade could disrupt cytokine homeostasis. Animal studies have shown that gp130-deficient mice are nonviable,309,310 making gp130 inhibition an undesirable approach.
Conventional therapies primarily target circulating IL-6 or IL-6R via monoclonal antibodies. For example, tocilizumab is a humanized anti-IL-6R monoclonal antibody created by grafting the complementarity-determining regions of a mouse anti-human IL-6R antibody into human IgG1κ.311 It blocks both mIL-6R and sIL-6R by competing for site I on IL-6R. Tocilizumab has shown efficacy in protecting against joint destruction in RA treatment and is approved for treating RA, juvenile idiopathic arthritis, and other IL-6-related diseases.310
Other monoclonal antibodies targeting IL-6 (e.g., olokizumab, siltuximab, sirukumab, and clazakizumab) and IL-6R (e.g., sarilumab, satralizumab, and vobarilizumab) have also been shown to be effective in reducing joint inflammation and destruction.310,311 However, an increased risk of bacterial infections is commonly reported,83,310 likely due to the role of IL-6 in plasma-cell differentiation and antibody production.310,311 Further studies suggest that this risk is associated primarily with classic-signaling312,313,314, whereas trans-signaling drives catabolic activity in chondrocytes.315 For example, infections are the most common adverse events in tocilizumab-treated hand OA patients, whereas the trans-signaling-specific antagonist olamkicept has shown minimal immune impact and preserved bone repair in a murine femur osteotomy model.316,317 This distinction has fueled interest in trans-signaling inhibitors.
Olamkicept (also known as sgp130Fc), a humanized monoclonal antibody composed of two sgp130 proteins fused to the Fc region of human IgG, is designed to compete for sites II and III on IL-6 with membrane-bound gp130.309,318 While this configuration theoretically blocks both classic and trans-signaling by neutralizing the IL-6-sIL-6R and IL-6-mIL-6R complexes, animal studies have shown that olamkicept selectively inhibits trans-signaling unless it is administered at high doses,309,310,313,314 suggesting that sgp130 may bind to IL-6-sIL-6R with increased affinity. Olamkicept has demonstrated promise in preclinical arthritis models, with intra-articular injections considerably reducing joint inflammation and cartilage damage in RA mice, supporting the potential of selective IL-6 trans-signaling inhibition for OA.319
However, sgp130 is also involved in other cytokine pathways, such as the IL-11 pathway, raising concerns about disruption of cytokine homeostasis.318 A recent study addressed this issue by developing cs-130Fc, a monoclonal antibody that combines the binding region of sgp130 (domains D1–D3), a nanobody (VHH6) that targets the IL-6-sIL-6R complex, and a human IgG Fc region. Cs-130Fc was shown to double the potency of olamkicept in inhibiting IL-6 trans-signaling while reducing IL-11 trans-signal inhibition by eightfold,313,318 offering new insights into anti-inflammatory therapy for OA.
Targeting the mIL-6R-cleaving enzymes ADAM10 and ADAM17 or inhibiting the transcription of sIL-6R to prevent the formation of IL-6–sIL-6R complexes represents another alternative strategy for inhibiting trans-signaling. However, a recent study suggested that the downregulation of ADAM10 and ADAM17 promotes the differentiation of murine bone marrow cells and human blood cells into osteoclasts, which may exacerbate bone resorption in OA patients.320 In contrast, inhibiting sIL-6R transcription may be insufficient for blocking IL-6R trans-signaling, as only ~15% of sIL-6R production is derived from RNA transcription, and no available agents targeting this process have been reported.313
Although targeting IL-6 trans-signaling to inhibit inflammation while preserving host defense against bacterial infections is generally considered the most promising approach for anti-IL-6 therapy, a recent study revealed that IL-6R antibodies, the trans-signaling inhibitor sgp130, and the cleavage enzyme inhibitor TAPI2 did not reduce the expression of IL-6 or inflammatory mediators (NO, PGE2, and MMPs) in chondrocytes or increase chondrocyte viability in a late-stage human knee OA model. However, an intracellularly active gp130 inhibitor, SC144, substantially improved these conditions.315 These findings suggest that agents targeting extracellular IL-6 signaling may have limited therapeutic effectiveness once intracellular signaling is established in late-stage OA.83,315
Therefore, blocking intracellular signaling components, such as JAKs (especially JAK1, which predominantly activates IL-6) or STAT3, may be preferable options for advanced OA treatment.321 Several JAK and STAT3 inhibitors have been developed, including pan-JAK inhibitors (tofacitinib and baricitinib), JAK1-selective inhibitors (filgotinib), TYK2-selective inhibitors (deucravacitinib), and STAT3 inhibitors (OPB-31121, OPB-51602).310 However, the development of IL-6 intracellular inhibitors is associated with several challenges, including low cell membrane permeability,309 disruption of cytokine homeostasis due to the involvement of JAK and STATs in other cytokine pathways,313 and the nonselective blockade of both classic and trans-signaling (as both intracellular mechanisms are currently understood to be identical).309,310,312
Targeting global intracellular signaling could result in side effects associated with classic-signaling, such as increased susceptibility to bacterial infections. Recent studies have shown that the administration of STAT3 inhibitors is customarily accompanied by side effects of peripheral neuropathy, and only limited efficacy in improving cartilage lesions has been reported in OA mice.76,310 In conclusion, the selective inhibition of IL-6 trans-signaling remains the most promising therapeutic approach for the prevention or early treatment of OA until novel intracellular signaling discriminating targets or refined dosage forms of JAK and STAT3 inhibitors with reduced side effects are developed, which may provide new insights for advanced OA treatment (Table 1).
Dysfunctional organelle pharmacotherapy in osteoarthritis
Chondrocyte organelles function as a complex, compartmentalized network essential for normal cell operations. In OA, pathological changes in these organelles are closely linked to chondrocyte differentiation, apoptosis, and cartilage degeneration. Repairing dysfunctional organelles is a promising therapeutic strategy to restore chondrocyte function and support cartilage repair. Targeting organelle-related processes involved in OA progression may help reestablish the cellular balance and maintain cartilage integrity, suggesting viable strategies for future clinical OA treatment (Table 2).
Mitochondrial pharmacotherapy in osteoarthritis
Mitochondrial homeostasis is crucial for chondrocytes to generate sufficient cellular energy and maintain normal cellular function. However, studies have shown that mitochondrial biogenesis-related proteins, such as AMPK, SIRT1, PGC-1α, NRF1, NRF2, and TFAM, are downregulated in OA chondrocytes from both humans and mice.97,322 In addition, decreased expression of mitochondrial respiratory complexes I–IV and ATP synthase (Complex V) results in insufficient energy production.323 The buildup of dysfunctional mitochondria also leads to excessive ROS production, contributing to chondrocyte damage and apoptosis.101,324 Therefore, restoring mitochondrial biogenesis by upregulating biogenesis-related proteins or enhancing mitophagy through proteins such as PINK1, Parkin, TOM, and LC3 to remove damaged mitochondria is a promising therapeutic strategy for OA. Potential drugs targeting these pathways, categorized by their roles in mitochondrial biogenesis or mitophagy, are summarized in Table 3.
Lysosomal pharmacotherapy in osteoarthritis
Lysosomes play a vital role in degrading cellular waste and damaged organelles through hydrolytic enzymes, thereby supporting ECM homeostasis. Lysosomal destabilization, which triggers chondrocyte apoptosis, is implicated in the pathogenesis of OA. A study conducted by Tao et al. revealed that the phagocytosis of hydroxyapatite crystallites by chondrocytes can disrupt lysosomal integrity.113 Consequently, therapeutic agents capable of mitigating cartilage calcification may represent promising drugs for slowing OA progression by stabilizing lysosomes. Ideal candidates would inhibit chondrocyte hypertrophic differentiation or interfere with Ca²⁺ and Pi binding during cartilage calcification without causing severe side effects.
Over the past few years, several therapies aimed at reducing cartilage calcification have been explored at the cellular and animal levels, offering potential treatments for OA. Recent developments in this area are summarized in Table 3. These therapies help maintain the chondrocyte phenotype by downregulating the expression of calcification-promoting genes and proteins, such as RUNX2 and type X collagen, or by reducing ACP formation in MVs, which may prevent crystal-induced LMP. However, caution is warranted, as some of these therapies have not been tested in OA models, and no relevant animal experiments have been conducted.
Furthermore, the use of cathepsin inhibitors to preserve BID integrity, along with functional molecules that alleviate LMP—such as heat shock protein 70 (Hsp70), annexin A7, endosomal sorting complex required for transport protein III (ESCRT-III), LAMP-1, and LAMP-2—has been proposed as an alternative treatment to prevent lysosome-induced chondrocyte apoptosis.325,326 Current studies suggest that cathepsins B and D primarily cleave BID. Specific inhibitors have been developed for cathepsins B (such as CA-074 and CA-030) and D (e.g., pepstatin A), which are used to treat various diseases.327,328 However, the efficacy of these inhibitors in reducing lysosome-induced chondrocyte apoptosis remains unconfirmed, as no published studies have specifically addressed this issue. Notably, inhibitors of cathepsins K and L have been widely studied for OA treatment because of the high expression of these enzymes in osteoclasts, where they are responsible for collagen degradation in bone and cartilage.329,330 Additionally, Hsp70 is an upstream regulator of ASM involved in the normal metabolism of SM.331,332 Annexin A7 and ESCRT-III are components of the plasma membrane repair system that facilitate the resealing of damaged lysosomal membranes by aggregating at the injury site.326,332 Moreover, LAMP-1 and LAMP-2 constitute ~50% of lysosomal membrane proteins and are essential for maintaining lysosomal morphology.333 Thus, the exogenous administration of recombinant forms of these proteins or their agonists may help repair hydroxyapatite-induced LMP in chondrocytes. This approach is supported by a study showing that recombinant Hsp70 (rHsp70), combined with the Hsp70 inducer arimoclomol, corrected pathological lysosomal morphology in primary fibroblasts from patients with Niemann–Pick disease.334
Endoplasmic reticulum pharmacotherapy in osteoarthritis
The ER is a central organelle for the proper synthesis, maturation, and folding of proteins. Over one-third of proteins in eukaryotic cells are folded within the ER lumen.138 This substantial workload makes the ER highly susceptible to environmental stimuli, often leading to errors and the accumulation of misfolded proteins, which induce ER stress. The UPR signaling pathway, which is activated in response to ER stress, helps restore ER homeostasis to some extent. However, if stress becomes irreversible, it triggers apoptosis. Upon elaboration of ER stress-associated mechanisms, two therapeutic strategies are under investigation for pharmacological intervention: one focuses on reducing or eliminating the aggregation of misfolded proteins in the ER lumen, whereas the other targets the modulation of stress sensor activity (PERK, IRE1α, and ATF6α) or the enzymes involved in the UPR.335
PERK and IRE1α are known to initiate apoptotic processes in cells. Emerging studies have tested inhibitors of these sensors in various ER stress-related diseases, including cancer, neurodegenerative disorders, and obesity. GSK2656157, a PERK inhibitor, effectively reduces cancer growth but is discontinued because of its toxicity.336 ISRIB, a small molecule that inhibits eIF2α phosphorylation, thereby reducing CHOP expression, has been found to enhance cognitive memory in brain-injured mice without severe side effects.337 Inhibitors of IRE1α, such as 4µ8c, MKC3946, and STF-083010, reversed hepatic steatosis in animal models.335 While these agents may help alleviate ER stress during OA progression, their efficacy in OA models remains untested.
Another therapeutic strategy for treating ER stress involves reducing the aggregation of misfolded proteins. Certain small molecules and proteins can alleviate this aggregation in the ER, offering potential benefits for OA. Chemical chaperones are small molecular compounds that reduce the free movement of proteins, which helps prevent the aggregation of misfolded proteins. They also increase the free energy between misfolded and native proteins, promoting proper folding to restore a more stable state.338 However, chemical chaperones are nonselective and often require high concentrations to be effective, which may cause toxicity and limit their application in vivo.335,338,339 Nonetheless, two chemical chaperones, 4-phenylbutyric acid (4-PBA) and tauroursodeoxycholic acid (TUDCA), are FDA-approved for human use.339 Among these, 4-PBA reduced cartilage destruction in a murine meniscectomy model, whereas TUDCA decreased the expression of ER stress-related markers and increased type II collagen expression.340,341
Pharmacological chaperones (PCs) are another type of small molecule designed to selectively bind to misfolded proteins and bridge noncovalent interactions lost or loosened during misfolding, thereby promoting proper folding.342,343 Compared with chemical chaperones, PCs work at lower concentrations and are more selective, making them promising therapeutic agents for OA. Cellular molecular chaperones are proteins that prevent the aggregation of misfolded proteins by recognizing and binding to their hydrophobic regions.344 The most common group of molecular chaperones is heat shock proteins (Hsps), which are classified by their molecular weight into Hsp40s, Hsp60s, Hsp70s, Hsp90s, Hsp100s, and small Hsps. These proteins are also involved in macromolecular complex assembly, protein transport, degradation, and the refolding of misfolded proteins,345 making them promising candidates for treating ER stress. Some molecular chaperones tested in OA chondrocytes are summarized in Table 3.
Certain chemical compounds and natural products derived from plants, fruits, and bacteria significantly downregulate the expression of ER stress-induced apoptotic genes or proteins (such as BiP, IRE1α, PERK, eIF2α, ATF4, and CHOP), positioning them as potential candidates for OA treatment (Table 3). However, the exact mechanisms through which these compounds alleviate ER stress are not fully understood, and further research is needed to elucidate how they affect ER stress pathogenesis.
Organelle-targeted drug delivery systems
Pharmacological molecules that target chondrocyte organelles are often limited by membrane barriers, considerably reducing their therapeutic efficacy. To maximize the effectiveness of these molecules, additional pharmacological modifications are necessary to increase their absorption by chondrocytes and organelles, improve their cellular distribution, and minimize toxicity. Various cell-penetrating peptides and cell-penetrating poly(disulfide)s are commonly used for their ability to promote cellular uptake and facilitate efficient intracellular delivery of therapeutic cargos, including small molecules, plasmid DNA, siRNA, therapeutic proteins, viruses, imaging agents, and other nanomedicines.346 However, they are commonly modified on the surface of nanomedicines (nanoparticle, liposome, exosome, polymeric micelles) along with other functional ligands instead of being directly conjugated with pharmacological molecules due to a lack of cell/tissue specificity.347,348 These synthetic pharmacological formulations, referred to as drug delivery systems (DDSs), are designed to improve the physicochemical properties, pharmacokinetics, and organelle-targeting capabilities of pharmacological molecules. Proper DDS design can enhance the functionality of chondrocyte organelles, offering potential improvements in OA treatment.
Mitochondria-targeting
In OA, ROS and an inflammatory environment damage the normal functions of mtDNA, leading to an increased number of dysfunctional mitochondria. This dysfunction contributes to chondrocyte lysis and cartilage degradation, making the repair or elimination of dysfunctional mitochondria through mtDNA rehabilitation a promising approach for OA treatment. Current strategies to repair damaged mtDNA involve delivering healthy mtDNA into the matrices of dysfunctional mitochondria; introducing functional molecules to repair, degrade, or inhibit the replication of damaged mtDNA; or promoting the replication of healthy mtDNA in normal mitochondria to increase mitochondrial biogenesis.349 However, owing to the nonselective distribution of therapeutic molecules in vivo and the unique structure of mitochondria—with their hydrophobic, dense double membrane, stringent transport machinery, and negative potential (approximately −160 to −180 mV) across the IMM—delivering drugs to mitochondria remains challenging. Therefore, additional carriers are needed to overcome these barriers.350,351 Strategies targeting mitochondria are discussed below (Fig. 6).

Schematic representation of advanced therapeutic strategies for mitochondrial targeting in chondrocytes to address mitochondrial dysfunction in OA. The diagram highlights two principal strategies: the delivery of mtDNA, therapeutic proteins, and therapeutic genes to dysfunctional mitochondria to foster repair and the delivery of healthy mtDNA to healthy mitochondria to increase biogenesis. The figure emphasizes the application of liposomes and hybrid membrane-coated biomimetic nanoparticles as cutting-edge delivery systems for healthy mitochondria, leveraging the ΔΨm for binding. For dysfunctional mitochondria in OA, where the ΔΨm is compromised, potential-independent strategies such as SS-31 and gramicidin S derivatives are employed for targeting. The mitochondrial import machinery, including the TOM and TIM complexes, is crucial for the translocation of therapeutic agents into the mitochondrial matrix. TOM facilitates the initial import of nuclear-encoded proteins into the mitochondrial matrix, whereas TIM assists in their further translocation across the inner mitochondrial membrane, aided by mtHsp70, which utilizes ATP hydrolysis to drive the process. HMCB NPs hybrid membrane-coated biomimetic nanoparticles, LNPs lipophilic nanoparticles, TP therapeutic protein, TG therapeutic gene, CM chondrocyte membrane, MM mitochondrial membrane, CL cardiolipin, TOM translocase of the outer mitochondrial membrane, TIM translocase of the inner mitochondrial membrane, mtDNA mitochondrial DNA, ΔΨm mitochondrial membrane potential, mtHsp70 mitochondrial chaperone heat shock protein 70
Lipophilic cations
Mitochondria-targeting carriers have been extensively studied in recent years, and most of them share the common trait of possessing lipophilic and cationic properties that enable the selective targeting of normal mitochondria through electrostatic interactions.352 Lipophilic cations such as triphenylphosphonium (TPP)-based cations, rhodamine 123 (Rh123), cyanine cations, and cationic peptides are usually conjugated synergistically with other functional ligands on the surface of nanomedicines such as nanoparticles, liposomes, and vesicles, in which therapeutic molecules are encapsulated. This approach enhances tissue/cell specificity while increasing their permeability and bioavailability, regulating their distributions in vivo, and managing their release rates with minimal side effects.352,353,354 For example, Huang et al. engineered a PEG/TPP-modified nanoparticle loaded with a natural anti-inflammatory compound, rhein, for delivery to mitochondria in IL-1β-stimulated chondrocytes. These nanoparticles exhibited anti-inflammatory effects by inhibiting IL-1β, IL-6, TNF-α, and MMP-13; upregulated the expression of anabolic genes (Col2a1 and Acan); protected the ΔΨm; and inhibited chondrocyte apoptosis.355 Yu et al. developed the pH-sensitive and TPP-modified nanoparticle DOX-PLGA/CPT/PD on the basis of the acidic property of the tumor environment for the delivery of DOX to mitochondria in tumor cells. The DDS is composed of positively charged nanoparticles synthesized from poly-lactide-co-glycolic acid (PLGA) and a TPP-containing amphiphilic polymer, with a negatively charged PD shell on the outer layer. This formulation ensures electroneutrality at physiological pH, preventing rapid clearance and nonselective ionic interactions in vivo. In an acidic tumor environment, the PD shell detaches from the nanoparticle, releasing the TPP-modified nanoparticle through amino bond breakage, resulting in improved DOX uptake and increased mitochondrial DOX concentrations in tumor cells.350 Similarly, Yuan et al. developed a biodegradable silica nanoparticle conjugated with TPP and CPD on the surface to deliver macromolecules to the mitochondria. The results further demonstrated the successful delivery of the native protein FLBSA and an anti-MTCO2 antibody to the mitochondria in HeLa cells.356 These strategies can also be adapted to deliver functional molecules, such as the mitochondrial outer membrane protein MDI (CG3249) and C17orf80, which promote mtDNA replication in the normal mitochondria of chondrocytes to treat OA.357,358 However, developing pharmaceutical formulations tailored specifically for OA is essential to overcome challenges such as the rapid in vivo clearance of cationic ligand-modified nanomedicines and the limitations of lipophilic cations, which primarily localize to the mitochondrial surface rather than the matrix. Furthermore, nonselective ionic interactions between cationic ligands and anionic proteins in the bloodstream may induce unintended side effects.350,352
Potential-independent mitochondria-targeting strategies
The promising mitochondria-targeting properties of lipophilic cations have spurred significant interest in exploring their therapeutic potential in various mitochondria-related diseases. However, these strategies are unlikely to effectively target depolarized mitochondria with impaired ΔΨm in chondrocytes.353 Therefore, potential independent mitochondria-targeting approaches may offer more suitable alternatives for treating OA.
Szeto-Schiller 31 (SS-31), a short-chain peptide consisting of ~10 amino acids, targets the IMM independently of ΔΨm through interaction with cardiolipin, a unique phospholipid residing on the IMM.354,359 Owing to its small size, ease of synthesis, water solubility, favorable safety profile (with injection site reactions as the predominant side effect and no fatal events reported), and relatively long half-life demonstrated in animal studies (~2 h in rats, dogs, and monkeys),354 SS-31 is an ideal candidate for targeting dysfunctional mitochondria in chondrocytes.
Furthermore, derivatives of gramicidin S, which are traditionally known as antibiotics because of their ability to disrupt bacterial membranes, are also being explored as mitochondria-targeting carriers because of the similarities between bacterial and mitochondrial membranes. XJB-5-131, a derivative of gramicidin S, has been shown to be inserted into the IMM and deliver a ROS scavenger (4-AT) without dependence on the ΔΨm, suggesting its potential for targeting depolarized mitochondria in chondrocytes.354,360 However, no mitochondrion-targeted pharmaceutical formulations using SS-31 or gramicidin S derivatives have been established for OA treatment, although relevant insights can be gleaned from other studies.
For example, Zhang et al. developed a PEG-PLGA nanoparticle conjugated with SS-31 that encapsulated cyclosporine A (CsA@PLGA-PEG-SS31) for treating myocardial ischemia‒reperfusion. The results demonstrated enhanced mitochondria-targeting efficacy and desirable cardioprotective effects.359 Similarly, Kuang et al. designed SS-31-conjugated geranylgeranylacetone-loaded PLGA nanoparticles that exhibited specific accumulation in mitochondria and enhanced ΔΨm in hair cells.361
Recently, Li et al. reported the development of mitochondrion-targeted polyethylene glycol-fluorocarbon (PEGm-Fn) self-assembled micelles, which consist of a fluorinated core and a PEGylated shell. PEGm balances the strong hydrophobicity of Fn to form stable micelles, which disengage upon reaching the cell membrane due to its “stealth property.” Once internalized, Fn acts as a drug carrier and facilitates mitochondrial accumulation through interaction with cardiolipin on the IMM. These micelles exhibit electrically neutral properties, effective cellular uptake, endosomal escape, and successful mitochondrial targeting across various cell types.352 This strategy addresses both the challenges of cell membrane penetration and mitochondria targeting, positioning PEGm-Fn self-assembled micelles as a promising approach for mitochondrial targeting in chondrocytes.
Strategies for accessing the mitochondrial matrix
Strategies for accessing the mitochondrial matrix involve the intricate process of navigating the mitochondrial import machinery. While lipophilic cations and potential-independent mitochondria-targeting carriers are effective at delivering therapeutic cargo to mitochondria, their ability to reach the mitochondrial matrix, where therapeutic effects are exerted, is often limited. To overcome this limitation, modification of the therapeutic cargo is necessary.
Substances entering the mitochondria are tightly regulated by the mitochondrial import machinery, which includes various translocases, such as the TOM and translocase of the inner membrane (TIM) complexes.354 The TOM complex, an oligomeric structure equipped with multiple channels, is responsible for recognizing and translocating proteins across the OMM. In this process, TOM40, in collaboration with TOM20, which identifies protein presequences, catalyzes the initial import of proteins into the intermembrane space. TOM20, characterized by its ability to adopt multiple conformations and interaction modes, specifically recognizes the presequences of proteins destined for the mitochondrial matrix. Upon recognition, TOM20 facilitates the translocation of these proteins to the central receptor TOM22, thereby initiating their import into the intermembrane space.
Conversely, the translocation of proteins destined for the inner mitochondrial membrane is orchestrated by the TIM complex, which plays a crucial role in their passage across this barrier. The TIM23 complex, which serves as the channel core of the TIM machinery and is composed of TIM17, TIM21, and TIM50, is complemented by the motor protein PAM. This motor protein is encircled by the mitochondrial chaperone heat shock protein 70 (mtHsp70), which is instrumental in catalyzing ATP hydrolysis. This ATP hydrolysis powers the translocation of proteins into the mitochondrial matrix. The synergistic function of the TOM and TIM complexes is indispensable for the precise and efficient delivery of therapeutic agents to the mitochondrial matrix, ensuring the accurate transport of proteins to their designated locations within the mitochondria.354,362,363
Most mitochondrial-targeted proteins possess an MTS, which is essential for their transport to the matrix. Over half of the MTSs are localized at the N-terminus of proteins and often adopt an amphiphilic helical structure, with one positively charged surface and one hydrophobic surface—features believed to be necessary for matrix translocation.353,364 Conjugating MTSs with therapeutic genes or proteins in DDSs can facilitate their delivery to the mitochondrial matrix. However, proteins transported to the matrix by MTSs must typically be unfolded, which may compromise their therapeutic efficacy to some extent.354
Other mitochondrial-targeting strategies for osteoarthritis treatment
The size and complexity of DDSs often limit the efficiency of therapeutic cargo delivery to mitochondria.349,354 To address this issue, several innovative strategies have been proposed. Yamada et al. developed a mitochondrial fusogenic liposome (termed the MITO-Porter DDS) on the basis of the concept that mitochondria undergo fusion and fission to share biomolecules. This system directly delivers the encapsulated therapeutic cargo to mitochondria by fusing with the mitochondrial membrane, regardless of the physical properties of the cargo,349 making the MITO-Porter DDS a promising candidate for gene or protein delivery in OA treatment.
In recent years, biomimetic nanoparticles with hybrid membranes from cancer cells and mitochondria that encapsulate therapeutic molecules have been developed for cancer treatment by leveraging homotypic binding between cancer and mitochondrial membranes.365 Membrane-camouflaged nanoparticles combine the physicochemical benefits of synthetic materials with their biological features, enabling efficient mitochondrial delivery of therapeutic molecules by modulating the ΔΨm in target cells.366 A similar approach could be adapted for OA treatment. Deng et al. developed chondrocyte membrane-coated nanoparticles for the treatment of cartilage damage. They demonstrated that chondrocyte membranes exhibit inherent binding affinity to the ECM, promoting longer drug retention and facilitating homotypic binding between chondrocytes, which enhances nanoparticle internalization.367 These findings provide both theoretical and practical foundations for the development of biomimetic nanoparticles for chondrocyte mitochondrial targeting in OA treatment.
Lysosome targeting
The direct elimination of excess hydroxyapatite crystallites within dysfunctional lysosomes—either by restoring lysosomal acidity to enable degradability or by introducing crystal-degrading agents—is a critical component of OA treatment. Targeting palliative functional molecules that alleviate LMP in dysfunctional lysosomes further underscores the importance of lysosome-targeting strategies in OA therapy. One advantage of designing lysosome-targeting DDSs is that most naturally follow the endosome‒lysosome trafficking route,368 eliminating the need for additional lysosomal-targetable moieties. Furthermore, pH-sensitive DDSs that remain stable at physiological pH but release their encapsulated cargo in acidic environments, such as the lysosomal lumen (pH 4.5–5.0), are commonly utilized to increase lysosome targetability (Fig. 7).369,370

Schematic representation of dual therapeutic approaches for lysosomal dysfunction in OA, with a focus on the restoration of lysosomal acidity and the repair of lysosomal membranes. The restoration of lysosomal acidity is facilitated by the delivery of acidic nanoparticles designed to release their contents within the acidic lysosomal environment, thereby enhancing the degradation of hydroxyapatite crystallites. Concurrently, the delivery of functional molecules such as Hsp70 alleviates LMP and promotes lysosomal membrane repair. The repair mechanisms involve three distinct pathways: a ANXA7-mediated repair, where ANXA7 is recruited to the lysosomal membrane upon calcium influx. b ESCRT-mediated repair, involving the activation of ESCRT-III components and Ca²⁺-binding protein ALIX or ESCRT-I/II pathways in response to endolysosomal membrane damage, leading to the formation of a higher-order structure that closes the membrane breach by ATPase VPS4. c ASM-mediated repair, where ASM activity induces caveolae formation, followed by membrane closure and internalization of merged caveolae, thus restoring lysosomal membrane integrity. These strategies are crucial for mitigating lysosome-induced chondrocyte apoptosis and are highlighted as necessary for lysosome-targeting strategies in OA therapy. MV matrix vesicle, ASM acid sphingomyelinase, Hsp70 heat shock protein 70, LMP lysosomal membrane permeabilization, LAMPs lysosome-associated membrane proteins
With respect to hydroxyapatite crystallite elimination, no crystal-degrading agents other than hydrochloric acid, which directly degrade hydroxyapatite, have been reported to date.115,371 However, several studies have successfully restored lysosomal acidity. For example, Zeng et al. developed pH-sensitive polyester-coated nanoparticles (acNPs) encapsulating fluorinated diacid and tetrafluorosuccinic acid for nonalcoholic fatty liver disease treatment. The restoration of dysfunctional lysosomal acidification, improved mitochondrial function, and enhanced autophagy have been observed in HepG2 cells treated with acNPs.372 Zhang et al. designed two acidic nanoparticles based on PLGA and polylactic acid that effectively targeted macrophage lysosomes while rescuing lysosomal acidity.373 Therefore, restoring dysfunctional lysosomal acidification could be a viable strategy for eliminating cytosolic or lysosomal hydroxyapatite crystallites in chondrocytes.
Additionally, restoring LMP in chondrocytes could serve as a complementary therapeutic approach to mitigate lysosome-induced chondrocyte apoptosis, as such interventions alone may not address the underlying causes of LMP. While several studies have highlighted the protective roles of proteins such as Hsp70, LAMP-I, LAMP-II, Annexin A7, and ESCRT-III in maintaining lysosomal homeostasis,331,333,374 pharmacological agonists or recombinant proteins targeting these molecules for lysosome restoration remain paradoxically rare, except for a few studies investigating the use of Hsp70 or its agonist in lysosomal repair.375 Notably, exogenous administration of recombinant Hsp70 has been shown to regulate the activity of ASM on the lysosomes of primary fibroblasts, correcting the balance of lysosomal lipid composition and demonstrating the potential for alleviating LMP in chondrocytes.376 Further development of lysosome-targeting DDSs carrying Hsp70 or its agonists is warranted to increase lysosomal targeting, as nonselective upregulation of Hsp70 in the cytoplasm and membrane could disrupt normal physiological processes.377
Endoplasmic reticulum-targeting
Therapeutic agents for alleviating ER stress have been widely discussed in the previous section. To overcome limitations in their therapeutic efficacy, such as issues related to biocompatibility, tissue distribution, or physicochemical properties, sophisticated DDSs should be developed to specifically target the chondrocyte ER, where therapeutic agents often struggle to reach. Current ER-targeting carriers can be broadly categorized into small molecules and targeted peptides (Fig. 8).378

This diagram illustrates the sophisticated DDS designed to target the ER in chondrocytes, which is crucial for alleviating ER stress in OA. The left panel details the structure of ER-targeting scFv antibodies, which are conjugated with therapeutic genes to facilitate direct delivery to the ER, bypassing lysosomal degradation. The right panel outlines the targeting mechanism of ER-targeting ligands, which include sulfonyl ligands, chloride ligands, amphoteric ions, and nanoparticle ligands. These ligands interact with specific ER membrane proteins, such as the chloride pump, K+ channel, CCT, SRPR, and KDELR, to increase the ER-targeting ability of the DDS. The diagram highlights the potential of these ER-targeting strategies to restore chondrocyte ER homeostasis, thereby offering a promising approach for OA treatment. scFv single-chain fragment variable, CCT cytidylyltransferase, SPR signal recognition particle, SRPR signal recognition particle receptor, KDELR KDEL receptor
Small molecules, including sulfonyl ligands, chloride ligands, and amphoteric ionic ligands, are commonly employed as ER-targetable fluorogenic probes and are often conjugated with fluorophores or ER-targeting carriers when modified with DDSs.379 Sulfonyl ligands, particularly glibenclamide, and its derivatives are widely recognized for their ER-targeting capabilities, which are mediated by their interaction with ATP-sensitive K+ channels located on the ER membrane via the cyclohexyl sulfonylurea moiety.380 Chloride ligands also offer an effective ER-targeting strategy by selectively binding to the chloride pump on the ER membrane.379 Additionally, a recently discovered zwitterionic aggregation-induced emission luminogen (AIEgen), which incorporates a sulfonyl group, demonstrates ER targetability, primarily attributed to electrostatic interactions between the amphoteric ion and the positively charged phosphocholine cytidylyltransferase on the ER membrane surface. This ER targetability is not solely derived from the sulfonyl group, as replacing the sulfonate moiety with p-bromobenzene shifts the targetability from the ER to the mitochondria. This observation underscores the prerequisite role of amphoteric properties in mediating ER targeting.381,382
ER-targeting peptides, including KDEL, KKXX, RXR (where X represents any amino acid), Eriss, and pardaxin,379,383,384 have been explored for their potential in targeting the ER. However, pardaxin is excluded from the subsequent discussion because of its proapoptotic effects. Both ER-targeting small molecules and peptide-conjugated nanomedicines remain the subject of ongoing investigation for their potential to restore chondrocyte ER homeostasis in OA treatment. For example, Yen et al. developed curcumin-encapsulated PLGA nanoparticles modified with KDEL on their surface. These nanoparticles inhibit the calcium-dependent interaction between UPR chaperones and misfolded proteins, thereby facilitating their escape from the ER lumen.385 In another study, Wang et al. designed sulfonyl ligand-modified PLGA nanoparticles loaded with an ER stress-inducing agent to promote apoptosis in cancer cells for melanoma therapy.159 Similar strategies could be adapted for OA treatment by replacing the encapsulated agent with ER stress inhibitors to restore chondrocyte ER homeostasis.
Driven by rapid advancements in organelle-targeting strategies and biomedical technologies, novel approaches such as liposome fusion and monoclonal antibodies have emerged as promising solutions for ER targeting. Liposomes, as self-assembled phospholipid carriers, can be structurally modified to mimic the ER membrane, thereby increasing their ER-targeting potential. Alternatively, ER-targeting ligands can be incorporated onto the surface of liposomes to improve their targeting specificity.386,387 Pollock et al. developed an ER-targeting liposome with a phospholipid composition resembling that of a rough ER membrane that was purified from rat liver. The study demonstrated that the liposome utilizes scavenger and low-density lipoprotein receptor-mediated endocytosis, followed by caveolin- and microtubule-dependent trafficking to the ER. Once at the ER, the liposome fuses with the ER membrane, enabling continuous cargo release.388 Yuan et al. designed pardaxin-modified liposomes to induce apoptosis in cancer cells. Compared with nonmodified liposomes, these liposomes exhibited enhanced ER colocalization in cancer cells at 8 h,389 suggesting that similar liposome-based strategies—the replacement of pardaxin with nontoxic ER-targeting ligands—could be effective in restoring chondrocyte ER homeostasis for OA treatment.
In addition to restoring ER function, gene therapy holds promise for OA treatment by delivering genetic materials that promote articular protection to chondrocytes.390 However, conventional gene delivery methods typically follow the endosome‒lysosome route, leading to the degradation of gene payloads and diminished therapeutic efficacy.391 ER-targetable monoclonal antibodies offer a potential solution to this issue by circumventing lysosomal degradation while simultaneously delivering therapeutic genes directly to the ER, near the nucleus, where they are protected from degradation and can be efficiently delivered to the nucleus.392,393 These ER-targeting monoclonal antibodies are often designed via a single-chain fragment variable approach, in which the N-terminal single-chain fragment variable domain, which contains a target site-specific signal sequence, is conjugated with therapeutic genes, while the C-terminal ER-targeting signal KDEL is incorporated.379 However, the biosafety and efficacy of such ER-targeting monoclonal antibodies in restoring chondrocyte ER homeostasis require further validation.
Other osteoarthritis therapies
Emerging therapeutic innovations are reshaping OA management through novel biological strategies. This section explores cell-based immunotherapies such as mesenchymal stem cells (MSCs), which combine immunomodulatory effects with anti-inflammatory effects, and engineered extracellular vesicles (EVs) designed for targeted therapeutic delivery and biomarker discovery. These approaches highlight the diversification of OA interventions addressing both immune dysregulation and tissue repair.
Cell-based immunotherapy
Given that cytokines and chemokines in the OA microenvironment recruit activated neutrophils and monocytes into the knee joint, where the recruited immune cells secrete proinflammatory mediators, which in turn recruit additional immune cells (e.g., macrophages and effector T cells), creating a vicious cycle,394 immunotherapy has emerged as a promising therapeutic strategy for OA treatment. Current immunotherapeutic approaches for OA are categorized into two primary modalities: conventional and biologic therapies and cell-based therapies.395 Conventional and biologic therapies, which include anticytokine strategies, have been extensively delineated in the preceding sections. In contrast, cell-based therapies constitute a distinct therapeutic approach, with MSCs being the most prevalent modality for OA treatment.396 MSCs promote chondrogenesis through their capacity for chondrocyte differentiation, coupled with anti-inflammatory and immunomodulatory effects that synergistically suppress pathological inflammation and rebalance dysregulated immune responses,397 including promoting the transformation of proinflammatory M1 macrophages into anti-inflammatory M2 macrophages, inhibiting T-cell proliferation and activity, promoting the development of anti-inflammatory regulatory T (Treg) cells, and reducing B-cell proliferation and differentiation, thereby decreasing antibody production.395 Clinical trials have shown that intra-articular injection of MSCs into the knee joints of OA patients results in significant results in cartilage repair and enhanced chondrogenesis with recoverable mild to moderate side effects.397
Macrophages are highly plastic immune cells that serve as powerful producers of cytokines and chemokines in inflamed joints. Targeting macrophages as a therapeutic approach holds promise for the treatment of OA.398 A preclinical study demonstrated that intra-articular injection of genetically engineered macrophages locked in the M2 phenotype has anti-inflammatory and pro-regenerative effects in OA rats.399 Moreover, the ability of Treg cells, which have the capacity to mediate immune responses and suppress inflammation, is considered a promising therapeutic strategy for OA, as autoantibodies against type II collagen are detected in nearly 50% of OA patients.400 Sohn et al. designed nanoparticles loaded with antigens (e.g., collagen II) and rapamycin, which facilitate tolerogenic dendritic cell responses. Upon uptake of nanoparticles, dendritic cells differentiate into tolerogenic DCs and induce antigen-specific immunomodulatory responses (such as the induction of antigen-specific regulatory (Treg) cells), thereby modulating the immune response against type II collagen. Intra-articular injection of the nanoparticles inhibited cartilage degradation and relieved pain in OA model mice.401
Extracellular vesicles in osteoarthritis treatment
EVs have emerged as promising therapeutic candidates for OA, offering novel potential to address the persistent challenge of disease-modifying therapies in joint degeneration. EVs are generated primarily through endosomal pathways, wherein early endosomes undergo inward membrane budding to form numerous intraluminal vesicles. These late endosomes, known as multivesicular bodies, subsequently fuse with the plasma membrane, releasing their internal intraluminal vesicles into the extracellular space as EVs.402,403 Characterized by their lipid bilayer membrane structure, EVs differ fundamentally from conventional cell-based therapies in that they retain donor cell-derived surface markers, circumvent ethical concerns, and exhibit reduced risks of immune rejection and tumorigenesis.404 Their therapeutic utility is further enhanced by their inherent targeting capabilities and engineerable cargo-loading properties, positioning them as versatile platforms for OA intervention.
Conventional imaging techniques primarily detect OA at advanced stages, necessitating biomarkers for early diagnosis.405 EVs mediate intercellular crosstalk through the selective transfer of bioactive lipids, proteins, and RNA species, including mRNAs, microRNAs, and long noncoding RNAs, demonstrating clinically exploitable diagnostic potential via OA-specific molecular cargo.405,406 Recent studies have demonstrated s-associated biomarker alterations in OA pathophysiology. For example, plasma EV miR-193b-3p levels are considerably lower than those in healthy controls, whereas synovial fluid EV miR-182-5p levels progressively decrease with disease severity. Notably, the synovial fluid EV-derived lncRNA PCGEM1 was markedly elevated in late-stage OA patients compared with early-stage OA patients, and the serum EV circ_008365 was downregulated in OA patients.403,407,408 These stage-specific EV biomarker modifications may hold diagnostic potential for OA.
Preclinical studies have demonstrated that native EVs derived from MSCs, including bone marrow-derived MSCs (BMSCs), adipose-derived MSCs (AMSCs), synovial MSCs (SMSCs), and embryonic MSCs (EMSCs), attenuate cartilage degradation and suppress inflammatory responses, highlighting their therapeutic potential for OA treatment.407 For example, BMSCs were the first MSCs applied in OA therapeutics because of their clinical feasibility in isolation, proliferation, and cryopreservation.409 BMSC-derived EVs were found to suppress the expression of catabolic markers (ADAMTS5 and MMP-13) and proinflammatory cytokines (IL-1β, IL-6, and TNF-α) while increasing the expression of anabolic markers (COL II and aggrecan) in vitro and to mitigate cartilage degeneration and joint pathology in OA rat models.403,407 However, BMSC-based therapies have intrinsic limitations, including age-dependent decreases in stem cell quantity/differentiation capacity, invasive extraction procedures causing donor morbidity, and restricted bone marrow yields.410 In contrast to BMSCs, AMSCs exhibit superior tissue accessibility due to the ubiquitous distribution of adipose depots. AMSC-derived EVs (AMSC-EVs) have robust anti-OA effects through the downregulation of proinflammatory cytokines (IL-1β, TNF-α, and IL-6), the upregulation of anti-inflammatory IL-10, and the restoration of collagen II synthesis in OA chondrocytes.403,407 Intra-articular delivery of AMSC-EVs substantially attenuated OA progression in OA rats, reducing cartilage degeneration while preserving subchondral bone architecture.407 Similarly, EVs derived from SMSCs and EMSCs also exhibited significant anti-OA effects in both in vitro and in vivo models.407,411
EVs inherently encapsulate proteins, lipids, and nucleic acids, demonstrating exceptional biocompatibility and drug-loading capacity that positions them as potent therapeutic carriers.412 These vesicles deliver payloads through two complementary mechanisms: cellular internalization via clathrin-dependent or clathrin-independent endocytosis, phagocytosis, or micropinocytosis, and extracellular engagement through ligand‒receptor interactions that activate downstream signaling cascades to achieve therapeutic effects.413 However, naturally derived EVs exhibit limited targetability and inherent heterogeneity, even when they originate from an identical recipient cell type.414 To overcome these challenges, surface modification of EVs for targeted delivery, including genetic modification of donor cells to produce EVs with specific surface proteins or direct conjugation of targeting moieties to the EV surface, is employed to increase tissue-specific delivery precision, therapeutic bioavailability, and biosafety while minimizing immunogenicity and off-target toxicity.403,406 In OA treatment, EVs are commonly modified with chondrocyte affinity peptide (CAP) to enable precise chondrocyte targeting.415 Therapeutic cargo loading primarily utilizes two strategies: genetic modification of parent cells to overexpress native or engineered therapeutic molecules and exogenous methods such as passive incubation with drugs, electrostatic complexation using transfection reagents for nucleic acids, or physical techniques such as electroporation and sonication to transiently permeabilize EV membranes.415 Furthermore, hybrid EV-liposome systems have been developed to synergistically mitigate liposomal toxicity while overcoming the limited drug-loading capacity of natural EVs, offering a dual-functional platform for enhanced therapeutic delivery.415 Liang et al. developed chondrocyte-targeting EVs by transfecting dendritic cells with plasmids encoding a fusion protein combining CAP and the transmembrane protein lysosome-associated membrane glycoprotein 2b (Lamp2b), enabling EV surface display of CAP-Lamp2b. Subsequent electroporation transiently permeabilized EV membranes to encapsulate miR-140, a miRNA involved in cartilage development and homeostasis. Intra-articular administration of these bioengineered EVs in a rat OA model substantially alleviated OA progression.416 Xu et al. engineered EVs through surface modification with a fusion protein combining the MSC-binding peptide E7 and Lamp2b to achieve SMSC targeting, accompanied by electroporation-mediated encapsulation of kartogenin to induce SMSC differentiation into chondrocytes. This dual-functional approach demonstrated enhanced chondrogenesis both in vitro and in vivo.417
Additionally, the anionic lipid bilayer of native EVs impedes penetration through the negatively charged cartilage matrix, substantially limiting therapeutic efficacy in the deep zones of cartilage during OA treatment.415 Bajpayee et al. engineered charge-reversible EVs by anchoring arginine-rich cationic motifs (CPCs + 14Rs) to anionic EV surfaces via pH-responsive switches, shifting the zeta potential from −25.4 ± 1.1 mV to −2.5 ± 1.0 mV. In destabilized medial meniscus-induced OA mice, intra-articular administration of these modified EVs enhanced cartilage penetration into deep zones, effectively alleviating OA pathological progression.418
Conclusion and perspective
Current therapeutic strategies for symptomatic relief and pathological management of OA are insufficient. Traditional pharmacological treatments and surgical interventions are frequently associated with adverse side effects and often fail to provide meaningful pain relief or mitigate the catabolic processes driven by inflammation and dysfunction of cellular organelles. Consequently, a deeper understanding of the mechanisms underlying OA pain, coupled with the inflammation and destabilization of cellular organelles that occur during disease progression, offers a promising avenue for identifying novel therapeutic targets.
A key objective in OA pain management is to interrupt pain signal transmission before it reaches the brain and is perceived as pain. Pharmacological blockade of ion channels in peripheral nociceptors, which detect noxious stimuli, and inhibition of NGF, which sensitizes these ion channels, are promising approaches for attenuating OA pain. Additionally, targeting neurotransmitters involved in the synaptic transmission of nociceptive signals may further contribute to pain relief. Several agents targeting these pathways have demonstrated promising results in preclinical animal models. However, their clinical efficacy and feasibility for OA pain management require validation through rigorous clinical trials.
Furthermore, the inflammatory cytokines accompanying dysfunctional chondrocyte organelles exacerbate catabolic processes in the knee joint. Therapeutic agents targeting key components of the inflammatory signaling pathways involved in OA progression may surpass conventional anti-inflammatory drugs, offering greater specificity for OA treatment. In addition, dysfunctional chondrocyte organelles contribute to cartilage destruction through chondrocyte apoptosis, and pharmacological agents aimed at restoring organelle homeostasis in chondrocytes present an alternative approach for OA therapy. However, therapeutic intervention for dysfunctional organelles in OA treatment is still in its infancy, and their therapeutic efficacy requires further investigation.
Moreover, advancement of sophisticated DDSs for organelle-targeting therapeutics is essential to address the challenges associated with biological membrane permeability, a barrier that most of these agents struggle to overcome. Concurrently, these systems must enhance therapeutic efficacy, optimize in vivo distribution, and minimize toxicity. Given the dearth of studies on organelle-targeting DDSs in OA, methodologies derived from treatments for other pathological conditions may provide valuable frameworks for future research aimed at restoring the functionality of dysfunctional chondrocyte organelles.
The primary limitation of current studies on OA is that most of the therapeutic strategies described are based on targets identified through the exploration of OA-related mechanisms, offering a strong theoretical foundation but lacking experimental validation in OA models. This is particularly evident in treatments aimed at addressing imbalances in cellular organelles within osteoarthritic joints. For example, drugs designed to restore mitochondrial homeostasis have been evaluated predominantly in preclinical OA studies. However, the side effects of the majority of these drugs remain unknown, and their efficacy has been confirmed largely only in OA cellular models, with insufficient data on their in vivo therapeutic effects and optimal drug delivery formulations. Furthermore, the efficacy of pharmacological agents that target lysosomes and the ER has, in most cases, not been verified in OA models.
Given the challenges of delivering drugs to organelles within chondrocytes, the previous section detailed the design of organelle-targeted DDSs to enhance in vivo efficacy. However, these designs are primarily based on mechanistic considerations of organelle targeting and lack experimental validation specific to chondrocytes. To guide future OA treatment, a roadmap is proposed for the clinical translation of DDSs aimed at repairing OA-affected organelles. This involves refining in vivo drug formulations and conducting pharmacological and toxicological studies to elucidate drug behavior, including the half-life, efficacy, and side effects, in OA therapy.
To confirm that therapeutic agents target the intended sites, relevant biomarkers, such as organelle-related markers (e.g., ΔΨm, ATP, mtDNA, ROS, UPR chaperones, cathepsins, RUNX2, and type X collagen), should be monitored postadministration. Efficacy and toxicity data from animal models should be submitted to regulatory authorities (e.g., the FDA). Subsequently, phase 0 trials using microdoses with probes in healthy volunteers can leverage imaging to verify drug localization, followed by phase 1 trials to establish safe dosages for OA patients, monitor organelle biomarkers in synovial fluid, and evaluate overall therapeutic effects on OA.
References
-
Yao, Q. et al. Osteoarthritis: pathogenic signaling pathways and therapeutic targets. Signal Transduct. Target. Ther. 8, 56 (2023).
-
Martel-Pelletier, J. et al. Osteoarthritis. Nat. Rev. Dis. Prim. 2, 16072 (2016).
-
Castro-Domínguez, F. et al. Unmet needs in the osteoarthritis chronic moderate to severe pain management in Spain: a real word data study. Rheumatol. Ther. 8, 1113–1127 (2021).
-
Tang, S. A. et al. Osteoarthritis. Nat. Rev. Dis. Prim. 11, 10 (2025).
-
Jayakumar, P. et al. Comparison of an artificial intelligence-enabled patient decision aid vs educational material on decision quality, shared decision-making, patient experience, and functional outcomes in adults with knee osteoarthritis: a randomized clinical trial. JAMA Netw. Open. 4, e2037107 (2021).
-
Li, J. W., Ma, Y. S. & Xiao, L. K. Postoperative pain management in total knee arthroplasty. Orthop. Surg. 11, 755–761 (2019).
-
Courties, A. et al. Osteoarthritis year in review 2024: epidemiology and therapy. Osteoarthr. Cartil. 32, 1397–1404 (2024).
-
Gummlich, L. & Marshall, L. Osteoarthritis. Nat. Rev. Dis. Prim. 11, 11 (2025).
-
Allen, K. D., Thoma, L. M. & Golightly, Y. M. Epidemiology of osteoarthritis. Osteoarthr. Cartil. 30, 184–195 (2022).
-
Minnig, M. C. C., Golightly, Y. M. & Nelson, A. E. Epidemiology of osteoarthritis: literature update 2022-2023. Curr. Opin. Rheumatol. 36, 108–112 (2024).
-
Reyes, C. et al. Socio-economic status and the risk of developing hand, hip or knee osteoarthritis: a region-wide ecological study. Osteoarthr. Cartil. 23, 1323–1329 (2015).
-
Giorgino, R. et al. Knee osteoarthritis: epidemiology, pathogenesis, and mesenchymal stem cells: what else is new? An update. Int. J. Mol. Sci. 24, 7 (2023).
-
Li, H., Kong, W., Liang, Y. & Sun, H. Burden of osteoarthritis in China, 1990-2019: findings from the Global Burden of Disease Study 2019. Clin. Rheumatol. 43, 1189–1197 (2024).
-
Bortoluzzi, A., Furini, F. & Scirè, C. A. Osteoarthritis and its management—epidemiology, nutritional aspects and environmental factors. Autoimmun. Rev. 17, 1097–1104 (2018).
-
Pishgar, F. et al. Association between race and radiographic, symptomatic, and clinical hand osteoarthritis: a propensity score-matched study using osteoarthritis initiative data. Arthritis Rheumatol. 74, 453–461 (2022).
-
Callahan, L. F., Cleveland, R. J., Allen, K. D. & Golightly, Y. Racial/ethnic, socioeconomic, and geographic disparities in the epidemiology of knee and hip osteoarthritis. Rheum. Dis. Clin. North Am. 47, 1–20 (2021).
-
Steinmetz, J. D. et al. Global, regional, and national burden of osteoarthritis, 1990–2020 and projections to 2050: a systematic analysis for the Global Burden of Disease Study 2021. Lancet Rheumatol. 5, e508–e522 (2023).
-
Poulsen, E. et al. Knee osteoarthritis risk is increased 4-6 fold after knee injury—a systematic review and meta-analysis. Br. J. Sports Med. 53, 1454–1463 (2019).
-
Canetti, E. F. D. et al. Risk factors for development of lower limb osteoarthritis in physically demanding occupations: a systematic review and meta-analysis. Appl. Ergon. 86, 103097 (2020).
-
Jones, R. K. et al. A new approach to prevention of knee osteoarthritis: reducing medial load in the contralateral knee. J. Rheumatol. 40, 309–315 (2013).
-
Roos, E. M. & Arden, N. K. Strategies for the prevention of knee osteoarthritis. Nat. Rev. Rheumatol. 12, 92–101 (2016).
-
Takeda, H., Nakagawa, T., Nakamura, K. & Engebretsen, L. Prevention and management of knee osteoarthritis and knee cartilage injury in sports. Br. J. Sports Med. 45, 304–309 (2011).
-
de Melo, L. R. S. et al. National osteoarthritis strategy brief report: prevention of osteoarthritis. Aust. J. Gen. Pract. 49, 272–275 (2020).
-
Lee, R. & Kean, W. F. Obesity and knee osteoarthritis. Inflammopharmacology 20, 53–58 (2012).
-
Wluka, A. E., Lombard, C. B. & Cicuttini, F. M. Tackling obesity in knee osteoarthritis. Nat. Rev. Rheumatol. 9, 225–235 (2013).
-
Wing, R. R. & Phelan, S. Long-term weight loss maintenance. Am. J. Clin. Nutr. 82, 222 s–225 s (2005).
-
Thorogood, A. et al. Isolated aerobic exercise and weight loss: a systematic review and meta-analysis of randomized controlled trials. Am. J. Med. 124, 747–755 (2011).
-
Heinegård, D. & Saxne, T. The role of the cartilage matrix in osteoarthritis. Nat. Rev. Rheumatol. 7, 50–56 (2011).
-
Hootman, J. M. & Albohm, M. J. Anterior cruciate ligament injury prevention and primary prevention of knee osteoarthritis. J. Athl. Train. 47, 589–590 (2012).
-
Whittaker, J. L., Runhaar, J., Bierma-Zeinstra, S. & Roos, E. M. A lifespan approach to osteoarthritis prevention. Osteoarthr. Cartil. 29, 1638–1653 (2021).
-
Lien-Iversen, T. et al. Does surgery reduce knee osteoarthritis, meniscal injury and subsequent complications compared with non-surgery after ACL rupture with at least 10 years follow-up? A systematic review and meta-analysis. Br. J. Sports Med. 54, 592–598 (2020).
-
Madry, H. et al. Early osteoarthritis of the knee. Knee Surg. Sports Traumatol. Arthrosc. 24, 1753–1762 (2016).
-
Driban, J. B. et al. Defining and evaluating a novel outcome measure representing end-stage knee osteoarthritis: data from the Osteoarthritis Initiative. Clin. Rheumatol. 35, 2523–2530 (2016).
-
Mahmoudian, A. et al. Early-stage symptomatic osteoarthritis of the knee—time for action. Nat. Rev. Rheumatol. 17, 621–632 (2021).
-
Katz, J. N., Arant, K. R. & Loeser, R. F. Diagnosis and treatment of hip and knee osteoarthritis: a review. JAMA 325, 568–578 (2021).
-
Abramoff, B. & Caldera, F. E. Osteoarthritis: pathology, diagnosis, and treatment options. Med. Clin. North. Am. 104, 293–311 (2020).
-
Xiao, S. & Chen, L. The emerging landscape of nanotheranostic-based diagnosis and therapy for osteoarthritis. J. Control. Release 328, 817–833 (2020).
-
Park, E. H. & Fritz, J. The role of imaging in osteoarthritis. Best. Pract. Res. Clin. Rheumatol. 37, 101866 (2023).
-
Bijlsma, J. W., Berenbaum, F. & Lafeber, F. P. Osteoarthritis: an update with relevance for clinical practice. Lancet 377, 2115–2126 (2011).
-
Wang, S. et al. Recent advances in photoacoustic imaging for deep-tissue biomedical applications. Theranostics 6, 2394–2413 (2016).
-
Drevet, S. et al. New imaging tools for mouse models of osteoarthritis. Geroscience 44, 639–650 (2022).
-
van den Bosch, M. H. J., Blom, A. B. & van der Kraan, P. M. Inflammation in osteoarthritis: our view on its presence and involvement in disease development over the years. Osteoarthr. Cartil. 32, 355–364 (2024).
-
Kloppenburg, M. Inflammation is a relevant treatment target in osteoarthritis. Lancet 402, 1725–1726 (2023).
-
Kapoor, M. et al. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat. Rev. Rheumatol. 7, 33–42 (2011).
-
Chow, Y. Y. & Chin, K. Y. The role of inflammation in the pathogenesis of osteoarthritis. Mediat. Inflamm. 2020, 8293921 (2020).
-
Zhou, F. et al. Kinsenoside attenuates osteoarthritis by repolarizing macrophages through inactivating NF-κB/MAPK signaling and protecting chondrocytes. Acta Pharm. Sin. B. 9, 973–985 (2019).
-
Chiu, J. W. et al. IL-1α processing, signaling and its role in cancer progression. Cells 10, 92 (2021).
-
Gabay, C., Lamacchia, C. & Palmer, G. IL-1 pathways in inflammation and human diseases. Nat. Rev. Rheumatol. 6, 232–241 (2010).
-
Kahlenberg, J. M. Anti-inflammatory panacea? The expanding therapeutics of interleukin-1 blockade. Curr. Opin. Rheumatol. 28, 197–203 (2016).
-
Braddock, M. & Quinn, A. Targeting IL-1 in inflammatory disease: new opportunities for therapeutic intervention. Nat. Rev. Drug Discov. 3, 330–339 (2004).
-
Mistry, A., Savic, S. & van der Hilst, J. C. H. Interleukin-1 blockade: an update on emerging indications. BioDrugs 31, 207–221 (2017).
-
Li, Z. et al. p38MAPK signaling pathway in osteoarthritis: pathological and therapeutic aspects. J. Inflamm. Res. 15, 723–734 (2022).
-
Molnar, V. et al. Cytokines and chemokines involved in osteoarthritis pathogenesis. Int. J. Mol. Sci. 22, 9208 (2021).
-
van den Bosch, M. H. J., van Lent, P. & van der Kraan, P. M. Identifying effector molecules, cells, and cytokines of innate immunity in OA. Osteoarthr. Cartil. 28, 532–543 (2020).
-
Jenei-Lanzl, Z., Meurer, A. & Zaucke, F. Interleukin-1β signaling in osteoarthritis—chondrocytes in focus. Cell. Signal. 53, 212–223 (2019).
-
Abbate, A. et al. Interleukin-1 and the inflammasome as therapeutic targets in cardiovascular disease. Circ. Res. 126, 1260–1280 (2020).
-
Arend, W. P., Palmer, G. & Gabay, C. IL-1, IL-18, and IL-33 families of cytokines. Immunol. Rev. 223, 20–38 (2008).
-
Chowdhury, T. T., Bader, D. L. & Lee, D. A. Dynamic compression counteracts IL-1 beta-induced release of nitric oxide and PGE2 by superficial zone chondrocytes cultured in agarose constructs. Osteoarthr. Cartil. 11, 688–696 (2003).
-
Loeser, R. F. et al. Deletion of JNK enhances senescence in joint tissues and increases the severity of age-related osteoarthritis in mice. Arthritis Rheumatol. 72, 1679–1688 (2020).
-
Ge, H. -x et al. JNK pathway in osteoarthritis: pathological and therapeutic aspects. J. Recept. Signal Transd. 37, 431–436 (2017).
-
Catheline, S. E. et al. IKKβ-NF-κB signaling in adult chondrocytes promotes the onset of age-related osteoarthritis in mice. Sci. Signal. 14, eabf3535 (2021).
-
Qiu, B., Xu, X., Yi, P. & Hao, Y. Curcumin reinforces MSC-derived exosomes in attenuating osteoarthritis via modulating the miR-124/NF-kB and miR-143/ROCK1/TLR9 signalling pathways. J. Cell. Mol. Med. 24, 10855–10865 (2020).
-
Rigoglou, S. & Papavassiliou, A. G. The NF-κB signalling pathway in osteoarthritis. Int. J. Biochem. Cell Biol. 45, 2580–2584 (2013).
-
Jesus, A. A. & Goldbach-Mansky, R. IL-1 blockade in autoinflammatory syndromes. Annu. Rev. Med. 65, 223–244 (2014).
-
Wojdasiewicz, P., Poniatowski, ŁA. & Szukiewicz, D. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of osteoarthritis. Mediat. Inflamm. 2014, 561459 (2014).
-
van Loo, G. & Bertrand, M. J. M. Death by TNF: a road to inflammation. Nat. Rev. Immunol. 23, 289–303 (2023).
-
Zelová, H. & Hošek, J. TNF-α signalling and inflammation: interactions between old acquaintances. Inflamm. Res. 62, 641–651 (2013).
-
Jang, D. I. et al. The role of tumor necrosis factor alpha (TNF-α) in autoimmune disease and current TNF-α inhibitors in therapeutics. Int. J. Mol. Sci. 22, 2719 (2021).
-
Kitaura, H. et al. Role of the interaction of tumor necrosis factor-α and tumor necrosis factor receptors 1 and 2 in bone-related cells. Int. J. Mol. Sci. 23, 1481 (2022).
-
Souza, R. F., Caetano, M. A. F., Magalhães, H. I. R. & Castelucci, P. Study of tumor necrosis factor receptor in the inflammatory bowel disease. World J. Gastroenterol. 29, 2733–2746 (2023).
-
Moelants, E. A., Mortier, A., Van Damme, J. & Proost, P. Regulation of TNF-α with a focus on rheumatoid arthritis. Immunol. Cell Biol. 91, 393–401 (2013).
-
Motta, F., Barone, E., Sica, A. & Selmi, C. Inflammaging and osteoarthritis. Clin. Rev. Allergy Immunol. 64, 222–238 (2023).
-
Suto, T. et al. TNFR2 is critical for TNF-induced rheumatoid arthritis fibroblast-like synoviocyte inflammation. Rheumatology 61, 4535–4546 (2022).
-
Xiaoshi, J. et al. SETD7 mediates the vascular invasion in articular cartilage and chondrocytes apoptosis in osteoarthriis. FASEB J. 35, e21283 (2021).
-
Honorati, M. C., Cattini, L. & Facchini, A. IL-17, IL-1beta and TNF-alpha stimulate VEGF production by dedifferentiated chondrocytes. Osteoarthr. Cartil. 12, 683–691 (2004).
-
Latourte, A. et al. Systemic inhibition of IL-6/Stat3 signalling protects against experimental osteoarthritis. Ann. Rheum. Dis. 76, 748–755 (2017).
-
Liao, Y. et al. Interleukin-6 signaling mediates cartilage degradation and pain in posttraumatic osteoarthritis in a sex-specific manner. Sci. Signal. 15, eabn7082 (2022).
-
Kishimoto, T. & Kang, S. IL-6 Revisited: from rheumatoid arthritis to CAR T cell therapy and COVID-19. Annu. Rev. Immunol. 40, 323–348 (2022).
-
Del Giudice, M. & Gangestad, S. W. Rethinking IL-6 and CRP: Why they are more than inflammatory biomarkers, and why it matters. Brain Behav. Immun. 70, 61–75 (2018).
-
Pacifici, M. Osteoarthritis and chronic pain: interleukin-6 as a common denominator and therapeutic target. Sci. Signal. 15, eadd3702 (2022).
-
Wang, T. & He, C. Pro-inflammatory cytokines: the link between obesity and osteoarthritis. Cytokine Growth Factor Rev. 44, 38–50 (2018).
-
Tanaka, T., Narazaki, M. & Kishimoto, T. Interleukin (IL-6) immunotherapy. Cold Spring Harb. Perspect. Biol. 10, a028456 (2018).
-
Choy, E. H. et al. Translating IL-6 biology into effective treatments. Nat. Rev. Rheumatol. 16, 335–345 (2020).
-
Rose-John, S. Local and systemic effects of interleukin-6 (IL-6) in inflammation and cancer. FEBS Lett. 596, 557–566 (2022).
-
Jones, S. A. & Jenkins, B. J. Recent insights into targeting the IL-6 cytokine family in inflammatory diseases and cancer. Nat. Rev. Immunol. 18, 773–789 (2018).
-
Wiegertjes, R., van de Loo, F. A. J. & Blaney Davidson, E. N. A roadmap to target interleukin-6 in osteoarthritis. Rheumatology 59, 2681–2694 (2020).
-
Baran, P. et al. The balance of interleukin (IL)-6, IL-6·soluble IL-6 receptor (sIL-6R), and IL-6·sIL-6R·sgp130 complexes allows simultaneous classic and trans-signaling. J. Biol. Chem. 293, 6762–6775 (2018).
-
Mihara, M. et al. IL-6/IL-6 receptor system and its role in physiological and pathological conditions. Clin. Sci. 122, 143–159 (2012).
-
Unver, N. & McAllister, F. IL-6 family cytokines: key inflammatory mediators as biomarkers and potential therapeutic targets. Cytokine Growth Factor Rev. 41, 10–17 (2018).
-
Tsuchida, A. I. et al. Interleukin-6 is elevated in synovial fluid of patients with focal cartilage defects and stimulates cartilage matrix production in an in vitro regeneration model. Arthritis Res. Ther. 14, R262 (2012).
-
Liu, D. et al. Mitochondrial quality control in cartilage damage and osteoarthritis: new insights and potential therapeutic targets. Osteoarthr. Cartil. 30, 395–405 (2022).
-
He, Y. et al. The role of SIRT3-mediated mitochondrial homeostasis in osteoarthritis. Cell. Mol. Life Sci. 77, 3729–3743 (2020).
-
Wang, S. et al. The Role of Autophagy and Mitophagy in Bone Metabolic Disorders. Int. J. Biol. Sci. 16, 2675–2691 (2020).
-
Nunnari, J. & Suomalainen, A. Mitochondria: in sickness and in health. Cell 148, 1145–1159 (2012).
-
Blanco, F. J., Rego, I. & Ruiz-Romero, C. The role of mitochondria in osteoarthritis. Nat. Rev. Rheumatol. 7, 161–169 (2011).
-
Sun, J. et al. Sestrin2 overexpression attenuates osteoarthritis pain via induction of AMPK/PGC-1α-mediated mitochondrial biogenesis and suppression of neuroinflammation. Brain Behav. Immun. 102, 53–70 (2022).
-
Wang, Y. et al. Mitochondrial biogenesis is impaired in osteoarthritis chondrocytes but reversible via peroxisome proliferator-activated receptor γ coactivator 1α. Arthritis Rheumatol. 67, 2141–2153 (2015).
-
Wang, H. et al. PGC-1α in osteoarthritic chondrocytes: from mechanism to target of action. Front. Pharmacol. 14, 1169019 (2023).
-
He, Y., Makarczyk, M. J. & Lin, H. Role of mitochondria in mediating chondrocyte response to mechanical stimuli. Life Sci. 263, 118602 (2020).
-
Zeng, Z. et al. Mitophagy—a new target of bone disease. Biomolecules 12, 1420 (2022).
-
Sun, K. et al. Mitophagy in degenerative joint diseases. Autophagy 17, 2082–2092 (2021).
-
Wang, S. et al. The mitophagy pathway and its implications in human diseases. Signal Transduct. Target. Ther. 8, 304 (2023).
-
López-Armada, M. J. et al. Mitochondrial activity is modulated by TNFalpha and IL-1beta in normal human chondrocyte cells. Osteoarthr. Cartil. 14, 1011–1022 (2006).
-
Ashrafi, G. & Schwarz, T. L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 20, 31–42 (2013).
-
Pickles, S., Vigié, P. & Youle, R. J. Mitophagy and quality control mechanisms in mitochondrial. Maint. Curr. Biol. 28, R170–r185 (2018).
-
Poznyak, A. V. et al. Autophagy and mitophagy as essential components of atherosclerosis. Cells 10, 443 (2021).
-
Bonam, S. R., Wang, F. & Muller, S. Lysosomes as a therapeutic target. Nat. Rev. Drug Discov. 18, 923–948 (2019).
-
Yang, C. & Wang, X. Lysosome biogenesis: regulation and functions. J. Cell Biol. 220, e202102001 (2021).
-
Jaishy, B. & Abel, E. D. Lipids, lysosomes, and autophagy. J. Lipid Res. 57, 1619–1635 (2016).
-
Levine, B. & Kroemer, G. Autophagy in the pathogenesis of disease. Cell 132, 27–42 (2008).
-
Dong, S. et al. Chaperone-mediated autophagy sustains haematopoietic stem-cell function. Nature 591, 117–123 (2021).
-
Gros, F. & Muller, S. The role of lysosomes in metabolic and autoimmune diseases. Nat. Rev. Nephrol. 19, 366–383 (2023).
-
Ye, T. et al. Lysosomal destabilization: a missing link between pathological calcification and osteoarthritis. Bioact. Mater. 34, 37–50 (2024).
-
Jin, C. et al. NLRP3 inflammasome plays a critical role in the pathogenesis of hydroxyapatite-associated arthropathy. Proc. Natl. Acad. Sci. USA 108, 14867–14872 (2011).
-
Väänänen, H. K. & Laitala-Leinonen, T. Osteoclast lineage and function. Arch. Biochem. Biophys. 473, 132–138 (2008).
-
So, A. & Busso, N. Crystal-gazing into the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 7, 688–689 (2011).
-
Ansari, M. Y. et al. Lysosomal dysfunction in osteoarthritis and aged cartilage triggers apoptosis in chondrocytes through BAX mediated release of Cytochrome c. Osteoarthr. Cartil. 29, 100–112 (2021).
-
Yacoub, A. et al. Caspase-, cathepsin-, and PERK-dependent regulation of MDA-7/IL-24-induced cell killing in primary human glioma cells. Mol. Cancer Ther. 7, 297–313 (2008).
-
Hanzel, C. E., Almeira Gubiani, M. F. & Verstraeten, S. V. Endosomes and lysosomes are involved in early steps of Tl(III)-mediated apoptosis in rat pheochromocytoma (PC12) cells. Arch. Toxicol. 86, 1667–1680 (2012).
-
Esposti, M. D. The roles of Bid. Apoptosis 7, 433–440 (2002).
-
Rose, M. et al. Direct measurement of the affinity between tbid and bax in a mitochondria-like membrane. Int. J. Mol. Sci. 22, 8240 (2021).
-
Billen, L. P., Shamas-Din, A. & Andrews, D. W. Bid: a Bax-like BH3 protein. Oncogene 27(Suppl 1), S93–S104 (2008).
-
Bernabei, I., So, A., Busso, N. & Nasi, S. Cartilage calcification in osteoarthritis: mechanisms and clinical relevance. Nat. Rev. Rheumatol. 19, 10–27 (2023).
-
Taylor, A. M. Metabolic and endocrine diseases, cartilage calcification and arthritis. Curr. Opin. Rheumatol. 25, 198–203 (2013).
-
Nagata, K. et al. Runx2 and Runx3 differentially regulate articular chondrocytes during surgically induced osteoarthritis development. Nat. Commun. 13, 6187 (2022).
-
McCarthy, G. M. & Cheung, H. S. Point: hydroxyapatite crystal deposition is intimately involved in the pathogenesis and progression of human osteoarthritis. Curr. Rheumatol. Rep. 11, 141–147 (2009).
-
Yan, J. F. et al. Pathological calcification in osteoarthritis: an outcome or a disease initiator?. Biol. Rev. Camb. Philos. Soc. 95, 960–985 (2020).
-
Rosenthal, A. K. Articular cartilage vesicles and calcium crystal deposition diseases. Curr. Opin. Rheumatol. 28, 127–132 (2016).
-
Wang, X. et al. Stage-specific and location-specific cartilage calcification in osteoarthritis development. Ann. Rheum. Dis. 82, 393–402 (2023).
-
Bernabei, I. et al. CD11b deficiency favors cartilage calcification via increased matrix vesicles, apoptosis, and lysyl oxidase activity. Int. J. Mol. Sci. 24, 9776 (2023).
-
Naughton, D. P. Iron(III)-mediated intra-articular crystal deposition in arthritis: a therapeutic role for iron chelators. Med. Hypotheses 57, 120–122 (2001).
-
Schwarz, D. S. & Blower, M. D. The endoplasmic reticulum: structure, function and response to cellular signaling. Cell. Mol. Life Sci. 73, 79–94 (2016).
-
Lee, S. Y., Wong, P. F., Jamal, J. & Roebuck, M. M. Naturally-derived endoplasmic reticulum stress inhibitors for osteoarthritis?. Eur. J. Pharmacol. 922, 174903 (2022).
-
Rellmann, Y., Eidhof, E. & Dreier, R. Review: ER stress-induced cell death in osteoarthritic cartilage. Cell. Signal. 78, 109880 (2021).
-
Navid, F. & Colbert, R. A. Causes and consequences of endoplasmic reticulum stress in rheumatic disease. Nat. Rev. Rheumatol. 13, 25–40 (2017).
-
Lu, Y. et al. Endoplasmic reticulum stress-mediated apoptosis and autophagy in osteoarthritis: from molecular mechanisms to therapeutic applications. Cell Stress Chaperones 29, 805–830 (2024).
-
Liu, D. D. et al. Endoplasmic reticulum stress-related protein GRP78 and CHOP levels in synovial fluid correlate with disease progression of primary knee osteoarthritis: a cross-sectional study. J. Appl. Biomed. 22, 40–48 (2024).
-
Wen, Z. Q. et al. Insights into the underlying pathogenesis and therapeutic potential of endoplasmic reticulum stress in degenerative musculoskeletal diseases. Mil. Med. Res. 10, 54 (2023).
-
Yang, C. et al. DDIT3/CHOP promotes autophagy in chondrocytes via SIRT1-AKT pathway. Biochim. Biophys. Acta Mol. Cell Res. 1868, 119074 (2021).
-
Soh, L. J., Lee, S. Y., Roebuck, M. M. & Wong, P. F. Unravelling the interplay between ER stress, UPR and the cGAS-STING pathway: implications for osteoarthritis pathogenesis and treatment strategy. Life Sci. 357, 123112 (2024).
-
Hughes, A. et al. Endoplasmic reticulum stress and unfolded protein response in cartilage pathophysiology; contributing factors to apoptosis and osteoarthritis. Int. J. Mol. Sci. 18, 665 (2017).
-
Stahl-Meyer, J., Stahl-Meyer, K. & Jäättelä, M. Control of mitosis, inflammation, and cell motility by limited leakage of lysosomes. Curr. Opin. Cell Biol. 71, 29–37 (2021).
-
Herrero-Beaumont, G. et al. Targeting chronic innate inflammatory pathways, the main road to prevention of osteoarthritis progression. Biochem. Pharmacol. 165, 24–32 (2019).
-
Marchi, S. et al. Mitochondrial control of inflammation. Nat. Rev. Immunol. 23, 159–173 (2023).
-
Riley, J. S. & Tait, S. W. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 21, e49799 (2020).
-
Vringer, E. & Tait, S. W. G. Mitochondria and cell death-associated inflammation. Cell Death Differ. 30, 304–312 (2023).
-
Liu, Y. et al. Endoplasmic reticulum stress participates in the progress of senescence and apoptosis of osteoarthritis chondrocytes. Biochem. Biophys. Res. Commun. 491, 368–373 (2017).
-
Wang, Y. et al. Exosome modification to better alleviates endoplasmic reticulum stress induced chondrocyte apoptosis and osteoarthritis. Biochem. Pharmacol. 206, 115343 (2022).
-
Zhang, K. & Kaufman, R. J. From endoplasmic-reticulum stress to the inflammatory response. Nature 454, 455–462 (2008).
-
Hasnain, S. Z. et al. The interplay between endoplasmic reticulum stress and inflammation. Immunol. Cell Biol. 90, 260–270 (2012).
-
Pan, F. & Jones, G. Clinical perspective on pain and pain phenotypes in osteoarthritis. Curr. Rheumatol. Rep. 20, 79 (2018).
-
D’Mello, R. & Dickenson, A. H. Spinal cord mechanisms of pain. Br. J. Anaesth. 101, 8–16 (2008).
-
Miller, R. E. et al. The role of peripheral nociceptive neurons in the pathophysiology of osteoarthritis pain. Curr. Osteoporos. Rep. 13, 318–326 (2015).
-
Syx, D., Tran, P. B., Miller, R. E. & Malfait, A. M. Peripheral mechanisms contributing to osteoarthritis pain. Curr. Rheumatol. Rep. 20, 9 (2018).
-
O’Neill, T. W. & Felson, D. T. Mechanisms of osteoarthritis (OA) pain. Curr. Osteoporos. Rep. 16, 611–616 (2018).
-
Fong, A. & Schug, S. A. Pathophysiology of pain: a practical primer. Plast. Reconstr. Surg. 134, 8 s–14 s (2014).
-
Perrot, S. Osteoarthritis pain. Best. Pract. Res. Clin. Rheumatol. 29, 90–97 (2015).
-
Dieppe, P. A. & Lohmander, L. S. Pathogenesis and management of pain in osteoarthritis. Lancet 365, 965–973 (2005).
-
Wang, F. et al. Nanoparticle-mediated celastrol ER targeting delivery amplify immunogenic cell death in melanoma. J. Adv. Res. 24, 248 (2024).
-
Laing, R. J. & Dhaka, A. ThermoTRPs and Pain. Neuroscientist 22, 171–187 (2016).
-
Souza Monteiro de Araujo, D., Nassini, R., Geppetti, P. & De Logu, F. TRPA1 as a therapeutic target for nociceptive pain. Expert Opin. Ther. Targets 24, 997–1008 (2020).
-
Marwaha, L. et al. TRP channels: potential drug target for neuropathic pain. Inflammopharmacology 24, 305–317 (2016).
-
Gouin, O. et al. TRPV1 and TRPA1 in cutaneous neurogenic and chronic inflammation: pro-inflammatory response induced by their activation and their sensitization. Protein Cell 8, 644–661 (2017).
-
Aguilera-Lizarraga, J. et al. Pro-inflammatory mediators sensitise transient receptor potential melastatin 3 cation channel (TRPM3) function in mouse sensory neurons. Neuropharmacology 271, 110391 (2025).
-
Conaghan, P. G., Cook, A. D., Hamilton, J. A. & Tak, P. P. Therapeutic options for targeting inflammatory osteoarthritis pain. Nat. Rev. Rheumatol. 15, 355–363 (2019).
-
Goodwin, G. & McMahon, S. B. The physiological function of different voltage-gated sodium channels in pain. Nat. Rev. Neurosci. 22, 263–274 (2021).
-
Hameed, S. Na(v)1.7 and Na(v)1.8: Role in the pathophysiology of pain. Mol. Pain. 15, 1744806919858801 (2019).
-
Hefti, F. F. et al. Novel class of pain drugs based on antagonism of NGF. Trends Pharmacol. Sci. 27, 85–91 (2006).
-
Denk, F., Bennett, D. L. & McMahon, S. B. Nerve growth factor and pain mechanisms. Annu. Rev. Neurosci. 40, 307–325 (2017).
-
Yu, H. et al. Osteoarthritis pain. Int. J. Mol. Sci. 23, 4642 (2022).
-
Castellini, C. et al. The effect of interaction NGF/p75(NTR) in sperm cells: a rabbit model. Cells 11, 1035 (2022).
-
Oo, W. M. & Hunter, D. J. Nerve growth factor (NGF) inhibitors and related agents for chronic musculoskeletal pain: a comprehensive review. BioDrugs 35, 611–641 (2021).
-
Norman, B. H. & McDermott, J. S. Targeting the nerve growth factor (NGF) pathway in drug discovery. potential applications to new therapies for chronic pain. J. Med. Chem. 60, 66–88 (2017).
-
Follis, R. M. et al. Metabolic control of sensory neuron survival by the p75 neurotrophin receptor in Schwann cells. J. Neurosci. 41, 8710–8724 (2021).
-
Xu, J. et al. NGF-p75 signaling coordinates skeletal cell migration during bone repair. Sci. Adv. 8, eabl5716 (2022).
-
Havelin, J. & King, T. Mechanisms underlying bone and joint pain. Curr. Osteoporos. Rep. 16, 763–771 (2018).
-
Haber, L. H., Moore, B. D. & Willis, W. D. Electrophysiological response properties of spinoreticular neurons in the monkey. J. Clin. Neurol. 207, 75–84 (1982).
-
Willis, W. D., Kenshalo, D. R. Jr. & Leonard, R. B. The cells of origin of the primate spinothalamic tract. J. Clin. Neurol. 188, 543–573 (1979).
-
Dubin, A. E. & Patapoutian, A. Nociceptors: the sensors of the pain pathway. J. Clin. Investig. 120, 3760–3772 (2010).
-
Liu, X. J. & Salter, M. W. Glutamate receptor phosphorylation and trafficking in pain plasticity in spinal cord dorsal horn. Eur. J. Neurosci. 32, 278–289 (2010).
-
Russell, F. A. et al. Calcitonin gene-related peptide: physiology and pathophysiology. Physiol. Rev. 94, 1099–1142 (2014).
-
Zieglgänsberger, W. Substance P and pain chronicity. Cell Tissue Res. 375, 227–241 (2019).
-
Navratilova, E. & Porreca, F. Substance P and inflammatory pain: getting it wrong and right simultaneously. Neuron 101, 353–355 (2019).
-
Russo, A. F. Calcitonin gene-related peptide (CGRP): a new target for migraine. Annu. Rev. Pharmacol. Toxicol. 55, 533–552 (2015).
-
Thakur, M., Dickenson, A. H. & Baron, R. Osteoarthritis pain: nociceptive or neuropathic?. Nat. Rev. Rheumatol. 10, 374–380 (2014).
-
Juárez-Contreras, R. et al. TRPV1 channel: a noxious signal transducer that affects mitochondrial function. Int. J. Mol. Sci. 21, 8882 (2020).
-
Lv, Z. et al. TRPV1 alleviates osteoarthritis by inhibiting M1 macrophage polarization via Ca(2+)/CaMKII/Nrf2 signaling pathway. Cell Death Dis. 12, 504 (2021).
-
Vestuto, V. et al. New frontiers on ER stress modulation: are TRP channels the leading actors?. Int. J. Mol. Sci. 24, 185 (2022).
-
Tessier, N. et al. TRPV1 channels are new players in the reticulum-mitochondria Ca(2+) coupling in a rat cardiomyoblast cell line. Cells 12, 2322 (2023).
-
Sukumaran, P., Schaar, A., Sun, Y. & Singh, B. B. Functional role of TRP channels in modulating ER stress and Autophagy. Cell Calcium 60, 123–132 (2016).
-
Zhang, X., Hu, M., Yang, Y. & Xu, H. Organellar TRP channels. Nat. Struct. Mol. Biol. 25, 1009–1018 (2018).
-
Gu, M. & Xu, H. A painful TR(i)P to lysosomes. J. Cell Biol. 215, 309–312 (2016).
-
Shang, S. et al. Intracellular TRPA1 mediates Ca2+ release from lysosomes in dorsal root ganglion neurons. J. Cell Biol. 215, 369–381 (2016).
-
Chauhan, S. et al. Muscarinic acetylcholine type 1 receptor antagonism activates TRPM3 to augment mitochondrial function and drive axonal repair in adult sensory neurons. Mol. Metab. 92, 102083 (2025).
-
Mira, R. G. & Cerpa, W. Building a bridge between NMDAR-mediated excitotoxicity and mitochondrial dysfunction in chronic and acute diseases. Cell. Mol. Neurobiol. 41, 1413–1430 (2021).
-
Alberdi, E. et al. Amyloid beta oligomers induce Ca2+ dysregulation and neuronal death through activation of ionotropic glutamate receptors. Cell Calcium 47, 264–272 (2010).
-
Dong, Y., Kalueff, A. V. & Song, C. N-methyl-d-aspartate receptor-mediated calcium overload and endoplasmic reticulum stress are involved in interleukin-1beta-induced neuronal apoptosis in rat hippocampus. J. Neuroimmunol. 307, 7–13 (2017).
-
Wang, A., Leong, D. J., Cardoso, L. & Sun, H. B. Nutraceuticals and osteoarthritis pain. Pharmacol. Ther. 187, 167–179 (2018).
-
Marshall, L. Osteoarthritis. Nat. Rev. Dis. Prim. 2, 16073 (2016).
-
Enthoven, W. T. et al. Non-steroidal anti-inflammatory drugs for chronic low back pain. Cochrane Database Syst. Rev. 2, Cd012087 (2016).
-
Krebs, E. E. et al. Effect of opioid vs nonopioid medications on pain-related function in patients with chronic back pain or hip or knee osteoarthritis pain: the SPACE randomized clinical trial. JAMA 319, 872–882 (2018).
-
Chircov, C., Grumezescu, A. M. & Bejenaru, L. E. Hyaluronic acid-based scaffolds for tissue engineering. Rom. J. Morphol. Embryol. 59, 71–76 (2018).
-
Otto-Lambertz, C. et al. Periprosthetic infection in joint replacement. Dtsch. Arztebl. Int. 114, 347–353 (2017).
-
Malfait, A.-M. & Obeidat, A. M. Targeting osteoarthritis where it hurts: the osteochondral junction. Nat. Rev. Rheumatol. 21, 319–320 (2025).
-
Wang, Y. et al. Capsaicin has an anti-obesity effect through alterations in gut microbiota populations and short-chain fatty acid concentrations. Food Nutr. Res. 64, 19 (2020).
-
Iftinca, M., Defaye, M. & Altier, C. TRPV1-targeted drugs in development for human pain conditions. Drugs 81, 7–27 (2021).
-
Koivisto, A. P., Voets, T., Iadarola, M. J. & Szallasi, A. Targeting TRP channels for pain relief: a review of current evidence from bench to bedside. Curr. Opin. Pharmacol. 75, 102447 (2024).
-
Garami, A. et al. Hyperthermia induced by transient receptor potential vanilloid-1 (TRPV1) antagonists in human clinical trials: insights from mathematical modeling and meta-analysis. Pharmacol. Ther. 208, 107474 (2020).
-
Arsenault, P. et al. NEO6860, modality-selective TRPV1 antagonist: a randomized, controlled, proof-of-concept trial in patients with osteoarthritis knee pain. Pain. Rep. 3, e696 (2018).
-
Round, P., Priestley, A. & Robinson, J. An investigation of the safety and pharmacokinetics of the novel TRPV1 antagonist XEN-D0501 in healthy subjects. Br. J. Clin. Pharmacol. 72, 921–931 (2011).
-
Derry, S. et al. Topical capsaicin (high concentration) for chronic neuropathic pain in adults. Cochrane Database Syst. Rev. 1, Cd007393 (2017).
-
Stevens, R. M. et al. Randomized, double-blind, placebo-controlled trial of intraarticular trans-capsaicin for pain associated with osteoarthritis of the knee. Arthritis Rheumatol. 71, 1524–1533 (2019).
-
Dong, L. et al. Identification of a partial and selective TRPV1 agonist CPIPC for alleviation of inflammatory pain. Molecules 27, 5428 (2022).
-
Arora, V., Campbell, J. N. & Chung, M. K. Fight fire with fire: neurobiology of capsaicin-induced analgesia for chronic pain. Pharmacol. Ther. 220, 107743 (2021).
-
Szallasi, A. Resiniferatoxin: nature’s precision medicine to silence TRPV1-positive afferents. Int. J. Mol. Sci. 24, 15042 (2023).
-
Ann, J. et al. Discovery of nonpungent transient receptor potential vanilloid 1 (TRPV1) Agonist as strong topical analgesic. J. Med. Chem. 63, 418–424 (2020).
-
Hu, Z. et al. Transient receptor potential ankyrin 1 (TRPA1) modulators: recent update and future perspective. Eur. J. Med. Chem. 257, 115392 (2023).
-
Bamps, D., Vriens, J., de Hoon, J. & Voets, T. TRP channel cooperation for nociception: therapeutic opportunities. Annu. Rev. Pharmacol. Toxicol. 61, 655–677 (2021).
-
Andrade, E. L., Meotti, F. C. & Calixto, J. B. TRPA1 antagonists as potential analgesic drugs. Pharmacol. Ther. 133, 189–204 (2012).
-
Okun, A. et al. Afferent drive elicits ongoing pain in a model of advanced osteoarthritis. Pain 153, 924–933 (2012).
-
Kanda, H. et al. Atractylodin produces antinociceptive effect through a long-lasting TRPA1 channel activation. Int. J. Mol. Sci. 22, 3614 (2021).
-
Logashina, Y. A. et al. Analysis of structural determinants of peptide MS 9a-1 essential for potentiating of TRPA1 channel. Mar. Drugs 20, 465 (2022).
-
Weyer, A. D. & Lehto, S. G. Development of TRPM8 antagonists to treat chronic pain and migraine. Pharmaceuticals 10, 37 (2017).
-
Julius, D. TRP channels and pain. Annu. Rev. Cell. Dev. Biol. 29, 355–384 (2013).
-
Liu, B. et al. TRPM8 is the principal mediator of menthol-induced analgesia of acute and inflammatory pain. Pain 154, 2169–2177 (2013).
-
Halonen, L. et al. Human osteoarthritic chondrocytes express nineteen different TRP-genes-TRPA1 and TRPM8 as potential drug targets. Int. J. Mol. Sci. 24, 10057 (2023).
-
Dai, Y. TRPs and pain. Semin. Immunopathol. 38, 277–291 (2016).
-
Patel, R. et al. Anti-hyperalgesic effects of a novel TRPM8 agonist in neuropathic rats: a comparison with topical menthol. Pain 155, 2097–2107 (2014).
-
De Caro, C. et al. Antinociceptive effect of two novel transient receptor potential melastatin 8 antagonists in acute and chronic pain models in rat. Br. J. Pharmacol. 175, 1691–1706 (2018).
-
Topp, R., Brosky, J. A. Jr. & Pieschel, D. The effect of either topical menthol or a placebo on functioning and knee pain among patients with knee OA. J. Geriatr. Phys. Ther. 36, 92–99 (2013).
-
Liman, E. R. TRP channels: pain enters through the side door. Nat. Cell Biol. 10, 171–172 (2014).
-
Vriens, J. et al. TRPM3 is a nociceptor channel involved in the detection of noxious heat. Neuron 70, 482–494 (2011).
-
Mulier, M. et al. Upregulation of TRPM3 in nociceptors innervating inflamed tissue. Elife 9, e61103 (2020).
-
Vriens, J. et al. Opening of an alternative ion permeation pathway in a nociceptor TRP channel. Nat. Cell Biol. 10, 188–195 (2014).
-
Bennett, D. L. et al. The role of voltage-gated sodium channels in pain signaling. Physiol. Rev. 99, 1079–1151 (2019).
-
Kushnarev, M., Pirvulescu, I. P., Candido, K. D. & Knezevic, N. N. Neuropathic pain: preclinical and early clinical progress with voltage-gated sodium channel blockers. Expert Opin. Investig. Drugs 29, 259–271 (2020).
-
Lai, J., Porreca, F., Hunter, J. C. & Gold, M. S. Voltage-gated sodium channels and hyperalgesia. Annu. Rev. Pharmacol. Toxicol. 44, 371–397 (2004).
-
Loussouarn, G. et al. Physiological and pathophysiological insights of Nav1.4 and Nav1.5 comparison. Front. Pharmacol. 6, 314 (2015).
-
Drouillas, B. et al. Persistent Nav1.1 and Nav1.6 currents drive spinal locomotor functions through nonlinear dynamics. Cell Rep. 42, 113085 (2023).
-
Dib-Hajj, S. D., Black, J. A. & Waxman, S. G. NaV1.9: a sodium channel linked to human pain. Nat. Rev. Neurosci. 16, 511–519 (2015).
-
O’Brien, B. J. et al. Tetrodotoxin-resistant voltage-gated sodium channels Na(v)1.8 and Na(v)1.9 are expressed in the retina. J. Clin. Neurol. 508, 940–951 (2008).
-
MacDonald, D. I. et al. A central mechanism of analgesia in mice and humans lacking the sodium channel Na(V)1.7. Neuron 109, 1497–1512.e1496 (2021).
-
Reid, A. R., Côté, P. D. & McDougall, J. J. Long-term blockade of nociceptive Na(v)1.7 channels is analgesic in rat models of knee arthritis. Biomolecules 12, 1571 (2022).
-
Zhu, J. et al. Aberrant subchondral osteoblastic metabolism modifies Na(V)1.8 for osteoarthritis. Elife 9, e57656 (2020).
-
Fu, W. et al. Na(v)1.7 as a chondrocyte regulator and therapeutic target for osteoarthritis. Nature 625, 557–565 (2024).
-
Torres-Pérez, J. V. et al. The NA(v)1.7 blocker protoxin II reduces burn injury-induced spinal nociceptive processing. J. Mol. Med. (Berl.) 96, 75–84 (2018).
-
Rahman, W. & Dickenson, A. H. Osteoarthritis-dependent changes in antinociceptive action of Nav1.7 and Nav1.8 sodium channel blockers: an in vivo electrophysiological study in the rat. Neuroscience 295, 103–116 (2015).
-
Nguyen, P. T. et al. Computational design of peptides to target Na(V)1.7 channel with high potency and selectivity for the treatment of pain. Elife 11, e81727 (2022).
-
Schuelert, N. & McDougall, J. J. Involvement of Nav 1.8 sodium ion channels in the transduction of mechanical pain in a rodent model of osteoarthritis. Arthritis Res. Ther. 14, R5 (2012).
-
Collison, J. Anti-NGF therapy improves osteoarthritis pain. Nat. Rev. Rheumatol. 15, 450–450 (2019).
-
Katz, J. N. Tanezumab for painful osteoarthritis. JAMA 322, 30–32 (2019).
-
Sanga, P. et al. Long-term safety and efficacy of fulranumab in patients with moderate-to-severe osteoarthritis pain: a phase II randomized, double-blind, placebo-controlled extension study. Arthritis Rheumatol. 69, 763–773 (2017).
-
Dakin, P. et al. The efficacy, tolerability, and joint safety of fasinumab in osteoarthritis pain: a phase IIb/III double-blind, placebo-controlled, randomized clinical trial. Arthritis Rheumatol. 71, 1824–1834 (2019).
-
Owolabi, J. B. et al. Characterization of antiallodynic actions of ALE-0540, a novel nerve growth factor receptor antagonist, in the rat. J. Pharmacol. Exp. Ther. 289, 1271–1276 (1999).
-
Bagal, S. K. et al. Discovery of potent, selective, and peripherally restricted pan-Trk kinase inhibitors for the treatment of pain. J. Med. Chem. 61, 6779–6800 (2018).
-
Skerratt, S. E. et al. The discovery of a potent, selective, and peripherally restricted pan-Trk inhibitor (PF-06273340) for the treatment of pain. J. Med. Chem. 59, 10084–10099 (2016).
-
Nwosu, L. N., Mapp, P. I., Chapman, V. & Walsh, D. A. Blocking the tropomyosin receptor kinase A (TrkA) receptor inhibits pain behaviour in two rat models of osteoarthritis. Ann. Rheum. Dis. 75, 1246–1254 (2016).
-
Hill, R. NK1 (substance P) receptor antagonists-why are they not analgesic in humans?. Trends Pharmacol. Sci. 21, 244–246 (2000).
-
Hirsch, S. et al. The CGRP receptor antagonist BIBN4096BS peripherally alleviates inflammatory pain in rats. Pain 154, 700–707 (2013).
-
Bullock, C. M. et al. Peripheral calcitonin gene-related peptide receptor activation and mechanical sensitization of the joint in rat models of osteoarthritis pain. Arthritis Rheumatol. 66, 2188–2200 (2014).
-
Fundytus, M. E. Glutamate receptors and nociception: implications for the drug treatment of pain. CNS Drugs 15, 29–58 (2001).
-
Pud, D. et al. The NMDA receptor antagonist amantadine reduces surgical neuropathic pain in cancer patients: a double blind, randomized, placebo controlled trial. Pain 75, 349–354 (1998).
-
Chin, A. C. & Lau, A. Y. Structural biology and thermodynamics of GluD receptors. Neuropharmacology 191, 108542 (2021).
-
Kreutzwiser, D. & Tawfic, Q. A. Expanding role of NMDA receptor antagonists in the management of pain. CNS Drugs 33, 347–374 (2019).
-
Qu, X. X. et al. Role of the spinal cord NR2B-containing NMDA receptors in the development of neuropathic pain. Exp. Neurol. 215, 298–307 (2009).
-
Chizh, B. A., Headley, P. M. & Tzschentke, T. M. NMDA receptor antagonists as analgesics: focus on the NR2B subtype. Trends Pharmacol. Sci. 22, 636–642 (2001).
-
Shin, H. J., Na, H. S. & Do, S. H. Magnesium and Pain. Nutrients 12, 2184 (2020).
-
Chałupnik, P. & Szymańska, E. Kainate receptor antagonists: recent advances and therapeutic perspective. Int. J. Mol. Sci. 24, 1908 (2023).
-
Kohl, B. K. & Dannhardt, G. The NMDA receptor complex: a promising target for novel antiepileptic strategies. Curr. Med. Chem. 8, 1275–1289 (2001).
-
Hartmann, B. et al. The AMPA receptor subunits GluR-A and GluR-B reciprocally modulate spinal synaptic plasticity and inflammatory pain. Neuron 44, 637–650 (2004).
-
Isaac, J. T., Ashby, M. C. & McBain, C. J. The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron 54, 859–871 (2007).
-
Gangadharan, V. et al. Peripheral calcium-permeable AMPA receptors regulate chronic inflammatory pain in mice. J. Clin. Investig. 121, 1608–1623 (2011).
-
Bonnet, C. S. et al. AMPA/kainate glutamate receptors contribute to inflammation, degeneration and pain related behaviour in inflammatory stages of arthritis. Ann. Rheum. Dis. 74, 242–251 (2015).
-
Bonnet, C. S. et al. AMPA/kainate glutamate receptor antagonists prevent posttraumatic osteoarthritis. JCI Insight 5, e134055 (2020).
-
Qiu, C. S. et al. Antinociceptive effects of MSVIII-19, a functional antagonist of the GluK1 kainate receptor. Pain 152, 1052–1060 (2011).
-
Negrete-Díaz, J. V., Falcón-Moya, R. & Rodríguez-Moreno, A. Kainate receptors: from synaptic activity to disease. FEBS J. 289, 5074–5088 (2022).
-
Wu, L. J., Ko, S. W. & Zhuo, M. Kainate receptors and pain: from dorsal root ganglion to the anterior cingulate cortex. Curr. Pharm. Des. 13, 1597–1605 (2007).
-
Lerma, J. Kainate receptor physiology. Curr. Opin. Pharmacol. 6, 89–97 (2006).
-
Dominguez, E. et al. Two prodrugs of potent and selective GluR5 kainate receptor antagonists actives in three animal models of pain. J. Med. Chem. 48, 4200–4203 (2005).
-
Bleakman, D., Alt, A. & Nisenbaum, E. S. Glutamate receptors and pain. Semin. Cell Dev. Biol. 17, 592–604 (2006).
-
Davidson, S. et al. Group II mGluRs suppress hyperexcitability in mouse and human nociceptors. Pain 157, 2081–2088 (2016).
-
Goudet, C. et al. Group III metabotropic glutamate receptors inhibit hyperalgesia in animal models of inflammation and neuropathic pain. Pain 137, 112–124 (2008).
-
Zhang, L., Lu, Y., Chen, Y. & Westlund, K. N. Group I metabotropic glutamate receptor antagonists block secondary thermal hyperalgesia in rats with knee joint inflammation. J. Pharmacol. Exp. Ther. 300, 149–156 (2002).
-
Ma, C. A. et al. The association of plasma IL-1Ra and related cytokines with radiographic severity of early knee osteoarthritis. Osteoarthr. Cartil. Open. 2, 100046 (2020).
-
Attur, M. et al. Plasma levels of interleukin-1 receptor antagonist (IL1Ra) predict radiographic progression of symptomatic knee osteoarthritis. Osteoarthr. Cartil. 23, 1915–1924 (2015).
-
Elsaid, K. A., Ubhe, A., Shaman, Z. & D’Souza, G. Intra-articular interleukin-1 receptor antagonist (IL1-ra) microspheres for posttraumatic osteoarthritis: in vitro biological activity and in vivo disease modifying effect. J. Exp. Orthop. 3, 18 (2016).
-
Mehta, S. et al. Sustained intra-cartilage delivery of interleukin-1 receptor antagonist using cationic peptide and protein-based carriers. Osteoarthr. Cartil. 31, 780–792 (2023).
-
Frisbie, D. D. et al. Treatment of experimental equine osteoarthritis by in vivo delivery of the equine interleukin-1 receptor antagonist gene. Gene Ther. 9, 12–20 (2002).
-
Deng, J. et al. Anti-inflammatory therapy for temporomandibular joint osteoarthritis using mRNA medicine encoding interleukin-1 receptor antagonist. Pharmaceutics 14, 1785 (2022).
-
Nixon, A. J. et al. Disease-modifying osteoarthritis treatment with interleukin-1 receptor antagonist gene therapy in small and large animal models. Arthritis Rheumatol. 70, 1757–1768 (2018).
-
Wang, Y. et al. Identification of an IL-1 receptor mutation driving autoinflammation directs IL-1-targeted drug design. Immunity 56, 1485–1501.e1487 (2023).
-
Cardiel, M. H. et al. A phase 2 randomized, double-blind study of AMG 108, a fully human monoclonal antibody to IL-1R, in patients with rheumatoid arthritis. Arthritis Res. Ther. 12, R192 (2010).
-
Cohen, S. B. et al. A randomized, double-blind study of AMG 108 (a fully human monoclonal antibody to IL-1R1) in patients with osteoarthritis of the knee. Arthritis Res. Ther. 13, R125 (2011).
-
Modi, P., Shah, B. M. & Patel, S. Interleukin-1β converting enzyme (ICE): a comprehensive review on discovery and development of caspase-1 inhibitors. Eur. J. Med. Chem. 261, 115861 (2023).
-
Rudolphi, K. et al. Pralnacasan, an inhibitor of interleukin-1beta converting enzyme, reduces joint damage in two murine models of osteoarthritis. Osteoarthr. Cartil. 11, 738–746 (2003).
-
Huang, W., Tu, J., Qiao, A. & He, C. Effects of VX765 on osteoarthritis and chondrocyte inflammation in rats]. CJRRS 38, 74–81 (2024).
-
Dykstra-Aiello, C. et al. A wake-like state in vitro induced by transmembrane TNF/soluble TNF receptor reverse signaling. Brain Behav. Immun. 94, 245–258 (2021).
-
Palladino, M. A., Bahjat, F. R., Theodorakis, E. A. & Moldawer, L. L. Anti-TNF-alpha therapies: the next generation. Nat. Rev. Drug Discov. 2, 736–746 (2003).
-
Tracey, D. et al. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol. Ther. 117, 244–279 (2008).
-
Karampetsou, M. P., Liossis, S. N. & Sfikakis, P. P. TNF-α antagonists beyond approved indications: stories of success and prospects for the future. QJM 103, 917–928 (2010).
-
Erickson, A. R. & Mikuls, T. R. Switching anti-TNF-alpha agents: what is the evidence?. Curr. Rheumatol. Rep. 9, 416–420 (2007).
-
Horiuchi, T. et al. Transmembrane TNF-alpha: structure, function and interaction with anti-TNF agents. Rheumatology 49, 1215–1228 (2010).
-
Medler, J. & Wajant, H. Tumor necrosis factor receptor-2 (TNFR2): an overview of an emerging drug target. Expert Opin. Ther. Targets 23, 295–307 (2019).
-
Salomon, B. L. Insights into the biology and therapeutic implications of TNF and regulatory T cells. Nat. Rev. Rheumatol. 17, 487–504 (2021).
-
Zou, H. et al. Modulation of regulatory T cell activity by TNF receptor type II-targeting pharmacological agents. Front. Immunol. 9, 594 (2018).
-
Zhang, N., Wang, Z. & Zhao, Y. Selective inhibition of tumor necrosis factor receptor-1 (TNFR1) for the treatment of autoimmune diseases. Cytokine Growth Factor Rev. 55, 80–85 (2020).
-
Shibata, H. et al. The treatment of established murine collagen-induced arthritis with a TNFR1-selective antagonistic mutant TNF. Biomaterials 30, 6638–6647 (2009).
-
Richter, F. et al. The TNFR1 antagonist atrosimab is therapeutic in mouse models of acute and chronic inflammation. Front. Immunol. 12, 705485 (2021).
-
McElvaney, O. J., Curley, G. F., Rose-John, S. & McElvaney, N. G. Interleukin-6: obstacles to targeting a complex cytokine in critical illness. Lancet Respir. Med. 9, 643–654 (2021).
-
Kang, S., Tanaka, T., Narazaki, M. & Kishimoto, T. Targeting interleukin-6 signaling in clinic. Immunity 50, 1007–1023 (2019).
-
Yao, X. et al. Targeting interleukin-6 in inflammatory autoimmune diseases and cancers. Pharmacol. Ther. 141, 125–139 (2014).
-
Rose-John, S., Winthrop, K. & Calabrese, L. The role of IL-6 in host defence against infections: immunobiology and clinical implications. Nat. Rev. Rheumatol. 13, 399–409 (2017).
-
Rose-John, S. et al. Targeting IL-6 trans-signalling: past, present and future prospects. Nat. Rev. Immunol. 23, 666–681 (2023).
-
Rose-John, S. The soluble interleukin 6 receptor: advanced therapeutic options in inflammation. Clin. Pharmacol. Ther. 102, 591–598 (2017).
-
Eitner, A. et al. Importance of IL-6 trans-signaling and high autocrine IL-6 production in human osteoarthritic chondrocyte metabolism. Osteoarthr. Cartil. 32, 561–573 (2024).
-
Prystaz, K. et al. Distinct effects of IL-6 classic and trans-signaling in bone fracture healing. Am. J. Pathol. 188, 474–490 (2018).
-
Richette, P. et al. Efficacy of tocilizumab in patients with hand osteoarthritis: double blind, randomised, placebo-controlled, multicentre trial. Ann. Rheum. Dis. 80, 349–355 (2021).
-
Heise, D. et al. Selective inhibition of IL-6 trans-signaling by a miniaturized, optimized chimeric soluble gp130 inhibits T(H)17 cell expansion. Sci. Signal. 14, eabc3480 (2021).
-
Nowell, M. A. et al. Soluble IL-6 receptor governs IL-6 activity in experimental arthritis: blockade of arthritis severity by soluble glycoprotein 130. J. Immunol. 171, 3202–3209 (2003).
-
Babendreyer, A. et al. Downregulation of the metalloproteinases ADAM10 or ADAM17 promotes osteoclast differentiation. Cell Commun. Signal. 22, 322 (2024).
-
Schaper, F. & Rose-John, S. Interleukin-6: biology, signaling and strategies of blockade. Cytokine Growth Factor Rev. 26, 475–487 (2015).
-
Cheng, C. et al. miR-5581 contributes to osteoarthritis by targeting NRF1 to disturb the proliferation and functions of chondrocytes. Am. J. Pathol. 193, 1234–1247 (2023).
-
Fernández-Moreno, M., Rego-Pérez, I. & Blanco, F. J. Is osteoarthritis a mitochondrial disease? What is the evidence. Curr. Opin. Rheumatol. 34, 46–53 (2022).
-
Cao, H. et al. Advances in the study of mitophagy in osteoarthritis. J. Zhejiang Univ. Sci. B 25, 197–211 (2024).
-
Piao, S. & Amaravadi, R. K. Targeting the lysosome in cancer. Ann. N. Y. Acad. Sci. 1371, 45–54 (2016).
-
Skowyra, M. L., Schlesinger, P. H., Naismith, T. V. & Hanson, P. I. Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science 360, eaar5078 (2018).
-
Kozak, A. et al. A new cathepsin D targeting drug delivery system based on immunoliposomes functionalized with lipidated pepstatin A. Pharmaceutics 15, 2464 (2023).
-
Terzani, F. et al. Synthesis and biological evaluation of selective pepstatin based trifluoromethylated inhibitors of cathepsin D. Eur. J. Med. Chem. 267, 116178 (2024).
-
Connor, J. R. et al. Protective effects of a cathepsin K inhibitor, SB-553484, in the canine partial medial meniscectomy model of osteoarthritis. Osteoarthr. Cartil. 17, 1236–1243 (2009).
-
Suzuki, N., Takimoto, K. & Kawashima, N. Cathepsin K inhibitor regulates inflammation and bone destruction in experimentally induced rat periapical lesions. J. Endod. 41, 1474–1479 (2015).
-
Papadopoulos, C., Kravic, B. & Meyer, H. Repair or lysophagy: dealing with damaged lysosomes. J. Mol. Biol. 432, 231–239 (2020).
-
Qing, Y. et al. Targeting lysosomal HSP70 induces acid sphingomyelinase-mediated disturbance of lipid metabolism and leads to cell death in T cell malignancies. Clin. Transl. Med. 13, e1229 (2023).
-
Tanaka, T. et al. LAMP3 inhibits autophagy and contributes to cell death by lysosomal membrane permeabilization. Autophagy 18, 1629–1647 (2022).
-
Wang, F., Gómez-Sintes, R. & Boya, P. Lysosomal membrane permeabilization and cell death. Traffic 19, 918–931 (2018).
-
Marciniak, S. J., Chambers, J. E. & Ron, D. Pharmacological targeting of endoplasmic reticulum stress in disease. Nat. Rev. Drug Discov. 21, 115–140 (2022).
-
Bonsignore, G., Martinotti, S. & Ranzato, E. Endoplasmic reticulum stress and cancer: could unfolded protein response be a druggable target for cancer therapy?. Int. J. Mol. Sci. 24, 1566 (2023).
-
Ghemrawi, R. & Khair, M. Endoplasmic reticulum stress and unfolded protein response in neurodegenerative diseases. Int. J. Mol. Sci. 21, 6127 (2020).
-
Loo, T. W. & Clarke, D. M. Chemical and pharmacological chaperones as new therapeutic agents. Expert Rev. Mol. Med. 9, 1–18 (2007).
-
Engin, F. & Hotamisligil, G. S. Restoring endoplasmic reticulum function by chemical chaperones: an emerging therapeutic approach for metabolic diseases. Diab. Obes. Metab. 12(Suppl 2), 108–115 (2010).
-
Li, K. et al. DEPTOR prevents osteoarthritis development via interplay with TRC8 to reduce endoplasmic reticulum stress in chondrocytes. J. Bone Miner. Res. 36, 400–411 (2021).
-
Liu, C. et al. Tauroursodeoxycholic acid suppresses endoplasmic reticulum stress in the chondrocytes of patients with osteoarthritis. Int. J. Mol. Med. 36, 1081–1087 (2015).
-
Tran, M. L., Génisson, Y., Ballereau, S. & Dehoux, C. Second-generation pharmacological chaperones: beyond inhibitors. Molecules 25, 3145 (2020).
-
Grasso, D., Galderisi, S., Santucci, A. & Bernini, A. Pharmacological chaperones and protein conformational diseases: approaches of computational structural biology. Int. J. Mol. Sci. 24, 5819 (2023).
-
Chaudhuri, T. K. & Paul, S. Protein-misfolding diseases and chaperone-based therapeutic approaches. FEBS J. 273, 1331–1349 (2006).
-
Kim, Y. E. et al. Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 82, 323–355 (2013).
-
Ramsey, J. D. & Flynn, N. H. Cell-penetrating peptides transport therapeutics into cells. Pharmacol. Ther. 154, 78–86 (2015).
-
He, Y., Li, F. & Huang, Y. Smart cell-penetrating peptide-based techniques for intracellular delivery of therapeutic macromolecules. Adv. Protein Chem. Struct. Biol. 112, 183–220 (2018).
-
Foged, C. & Nielsen, H. M. Cell-penetrating peptides for drug delivery across membrane barriers. Expert Opin. Drug Deliv. 5, 105–117 (2008).
-
Yamada, Y. & Harashima, H. RNA delivery to mitochondria. Handb. Exp. Pharmacol. 284, 329–339 (2024).
-
Yu, H. et al. Tumor acidity activated triphenylphosphonium-based mitochondrial targeting nanocarriers for overcoming drug resistance of cancer therapy. Theranostics 9, 7033–7050 (2019).
-
Jean, S. R. et al. Peptide-mediated delivery of chemical probes and therapeutics to mitochondria. Acc. Chem. Res. 49, 1893–1902 (2016).
-
Liu, Y., Zhang, J., Tu, Y. & Zhu, L. Potential-independent intracellular drug delivery and mitochondrial targeting. ACS Nano 16, 1409–1420 (2022).
-
Yamada, Y. et al. Power of mitochondrial drug delivery systems to produce innovative nanomedicines. Adv. Drug Deliv. Rev. 154-155, 187–209 (2020).
-
Lu, P., Bruno, B. J., Rabenau, M. & Lim, C. S. Delivery of drugs and macromolecules to the mitochondria for cancer therapy. J. Control. Release 240, 38–51 (2016).
-
Huang, H. et al. Construction of mitochondrial-targeting nano-prodrug for enhanced Rhein delivery and treatment for osteoarthritis in vitro. Int. J. Pharm. 661, 124397 (2024).
-
Yuan, P. et al. Mitochondria-targeting, intracellular delivery of native proteins using biodegradable silica nanoparticles. Angew. Chem. Int. Ed. Engl. 58, 7657–7661 (2019).
-
Wu, H. et al. C17orf80 binds the mitochondrial genome to promote its replication. J. Cell Biol. 222, e202302037 (2023).
-
Zhang, Y., Chen, Y., Gucek, M. & Xu, H. The mitochondrial outer membrane protein MDI promotes local protein synthesis and mtDNA replication. EMBO J. 35, 1045–1057 (2016).
-
Zhang, C. X. et al. Mitochondria-targeted cyclosporin A delivery system to treat myocardial ischemia reperfusion injury of rats. J. Nanobiotechnol. 17, 18 (2019).
-
Liu, N. K. et al. Restoring mitochondrial cardiolipin homeostasis reduces cell death and promotes recovery after spinal cord injury. Cell Death Dis. 13, 1058 (2022).
-
Kuang, X. et al. SS-31 peptide enables mitochondrial targeting drug delivery: a promising therapeutic alteration to prevent hair cell damage from aminoglycosides. Drug Deliv. 24, 1750–1761 (2017).
-
Horten, P., Colina-Tenorio, L. & Rampelt, H. Biogenesis of mitochondrial metabolite carriers. Biomolecules 10, 1008 (2020).
-
Busch, J. D., Fielden, L. F., Pfanner, N. & Wiedemann, N. Mitochondrial protein transport: versatility of translocases and mechanisms. Mol. Cell. 83, 890–910 (2023).
-
Yoshinaga, N. & Numata, K. Rational designs at the forefront of mitochondria-targeted gene delivery: recent progress and future perspectives. ACS Biomater. Sci. Eng. 8, 348–359 (2022).
-
Zou, Y. et al. Cancer cell-mitochondria hybrid membrane coated Gboxin loaded nanomedicines for glioblastoma treatment. Nat. Commun. 14, 4557 (2023).
-
Huang, X. et al. Sequences flanking the transmembrane segments facilitate mitochondrial localization and membrane fusion by mitofusin. Proc. Natl. Acad. Sci. USA 114, E9863–e9872 (2017).
-
Deng, R. et al. Chondrocyte membrane-coated nanoparticles promote drug retention and halt cartilage damage in rat and canine osteoarthritis. Sci. Transl. Med. 16, eadh9751 (2024).
-
Uzhytchak, M. et al. Lysosomal nanotoxicity: impact of nanomedicines on lysosomal function. Adv. Drug Deliv. Rev. 197, 114828 (2023).
-
Hu, M., Chen, J., Liu, S. & Xu, H. The acid gate in the lysosome. Autophagy 19, 1368–1370 (2023).
-
Lin, Y. X. et al. pH-sensitive polymeric nanoparticles modulate autophagic effect via lysosome impairment. Small 12, 2921–2931 (2016).
-
Väänänen, H. K., Zhao, H., Mulari, M. & Halleen, J. M. The cell biology of osteoclast function. J. Cell Sci. 113(Pt 3), 377–381 (2000).
-
Zeng, J. et al. Restoration of lysosomal acidification rescues autophagy and metabolic dysfunction in non-alcoholic fatty liver disease. Nat. Commun. 14, 2573 (2023).
-
Zhang, X. et al. Use of acidic nanoparticles to rescue macrophage lysosomal dysfunction in atherosclerosis. Autophagy 19, 886–903 (2023).
-
Petersen, N. H. & Kirkegaard, T. HSP70 and lysosomal storage disorders: novel therapeutic opportunities. Biochem. Soc. Trans. 38, 1479–1483 (2010).
-
Kirkegaard, T. et al. Heat shock protein-based therapy as a potential candidate for treating the sphingolipidoses. Sci. Transl. Med. 8, 355ra118 (2016).
-
Kirkegaard, T. et al. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature 463, 549–553 (2010).
-
Cao, M., Luo, X., Wu, K. & He, X. Targeting lysosomes in human disease: from basic research to clinical applications. Signal Transduct. Target. Ther. 6, 379 (2021).
-
Qiao, L. et al. Research on endoplasmic reticulum-targeting fluorescent probes and endoplasmic reticulum stress-mediated nanoanticancer strategies: a review. Colloids Surf. B. 208, 112046 (2021).
-
Shi, Y. et al. Pharmaceutical strategies for endoplasmic reticulum-targeting and their prospects of application. J. Control. Release 329, 337–352 (2021).
-
Zhou, Y. et al. Endoplasmic reticulum-localized two-photon-absorbing boron dipyrromethenes as advanced photosensitizers for photodynamic therapy. J. Med. Chem. 61, 3952–3961 (2018).
-
Alam, P. et al. Red AIE-active fluorescent probes with tunable organelle-specific targeting. Adv. Funct. Mater. 30, 1909268 (2020).
-
Miao, Z. et al. Endoplasmic reticulum-targeting AIE photosensitizers to boost immunogenic cell death for immunotherapy of bladder carcinoma. ACS Appl. Mater. Interfaces 16, 245–260 (2024).
-
Matsuo, K. et al. Efficient generation of antigen-specific cellular immunity by vaccination with poly(gamma-glutamic acid) nanoparticles entrapping endoplasmic reticulum-targeted peptides. Biochem. Biophys. Res. Commun. 362, 1069–1072 (2007).
-
Ting, C. H. et al. The mechanisms by which pardaxin, a natural cationic antimicrobial peptide, targets the endoplasmic reticulum and induces c-FOS. Biomaterials 35, 3627–3640 (2014).
-
Yen, C. L. et al. Targeted delivery of curcumin rescues endoplasmic reticulum-retained mutant NOX2 protein and avoids leukocyte apoptosis. J. Immunol. 202, 3394–3403 (2019).
-
Zahednezhad, F. et al. Liposome and immune system interplay: challenges and potentials. J. Control. Release 305, 194–209 (2019).
-
Almeida, B., Nag, O. K., Rogers, K. E. & Delehanty, J. B. Recent progress in bioconjugation strategies for liposome-mediated drug delivery. Molecules 25, 5672 (2020).
-
Pollock, S. et al. Uptake and trafficking of liposomes to the endoplasmic reticulum. FASEB J. 24, 1866–1878 (2010).
-
Yuan, X. et al. Virus-like nonvirus cationic liposome for efficient gene delivery via endoplasmic reticulum pathway. ACS Cent. Sci. 6, 174–188 (2020).
-
Chen, H. et al. Urchin-like ceria nanoparticles for enhanced gene therapy of osteoarthritis. Sci. Adv. 9, eadf0988 (2023).
-
Qin, B. et al. Targeting DNA to the endoplasmic reticulum efficiently enhances gene delivery and therapy. Nanoscale 12, 18249–18262 (2020).
-
Reilly, M. J., Larsen, J. D. & Sullivan, M. O. Polyplexes traffic through caveolae to the Golgi and endoplasmic reticulum en route to the nucleus. Mol. Pharm. 9, 1280–1290 (2012).
-
Qi, L. Y. et al. Enhanced nuclear gene delivery via integrating and streamlining intracellular pathway. J. Control. Release 341, 511–523 (2022).
-
Klimak, M. et al. Immunoengineering the next generation of arthritis therapies. Acta Biomater. 133, 74–86 (2021).
-
Moulin, D. et al. The role of the immune system in osteoarthritis: mechanisms, challenges and future directions. Nat. Rev. Rheumatol. 21, 221–236 (2025).
-
Yu, H., Huang, Y. & Yang, L. Research progress in the use of mesenchymal stem cells and their derived exosomes in the treatment of osteoarthritis. Ageing Res. Rev. 80, 101684 (2022).
-
Alahdal, M. et al. Potential efficacy of dendritic cell immunomodulation in the treatment of osteoarthritis. Rheumatology 60, 507–517 (2021).
-
Dai, M. et al. Cartilage repair in degenerative osteoarthritis mediated by squid type II collagen via immunomodulating activation of M2 macrophages, inhibiting apoptosis and hypertrophy of chondrocytes. Biomaterials 180, 91–103 (2018).
-
Liang, C. et al. Engineered M2a macrophages for the treatment of osteoarthritis. Front. Immunol. 13, 1054938 (2022).
-
McHugh, J. T(reg) cell-inducing nanoparticles show promise for treating OA. Nat. Rev. Rheumatol. 19, 62 (2023).
-
Sohn, H. S. et al. Tolerogenic nanoparticles induce type II collagen-specific regulatory T cells and ameliorate osteoarthritis. Sci. Adv. 8, eabo5284 (2022).
-
Xian Bo, S. et al. The research progress of exosomes in osteoarthritis, with particular emphasis on the therapeutic effect. Front. Pharmacol. 13, 731756 (2022).
-
Zhou, Q., Cai, Y., Jiang, Y. & Lin, X. Exosomes in osteoarthritis and cartilage injury: advanced development and potential therapeutic strategies. Int. J. Biol. Sci. 16, 1811–1820 (2020).
-
Xia, Y., Zhang, J., Liu, G. & Wolfram, J. Immunogenicity of extracellular vesicles. Adv. Mater. 36, e2403199 (2024).
-
Wu, Y. et al. Exosomes rewire the cartilage microenvironment in osteoarthritis: from intercellular communication to therapeutic strategies. Int. J. Oral. Sci. 14, 40 (2022).
-
Wang, X. et al. Mechanism of osteoarthritis treatment by exosomes. Chin. Med. J. 138, 367–369 (2025).
-
Okuyan, H. M., Coşkun, A. & Begen, M. A. Current status, opportunities, and challenges of exosomes in diagnosis and treatment of osteoarthritis. Life Sci. 362, 123365 (2025).
-
Ni, Z. et al. Exosomes: roles and therapeutic potential in osteoarthritis. Bone Res. 8, 25 (2020).
-
Gupta, P. K., Das, A. K., Chullikana, A. & Majumdar, A. S. Mesenchymal stem cells for cartilage repair in osteoarthritis. Stem Cell Res. Ther. 3, 25 (2012).
-
Xu, Y. et al. USP26 combats age-related declines in self-renewal and multipotent differentiation of BMSC by maintaining mitochondrial homeostasis. Adv. Sci. 11, e2406428 (2024).
-
Asghar, S. et al. Exosomes in intercellular communication and implications for osteoarthritis. Rheumatology 59, 57–68 (2020).
-
Elsharkasy, O. M. et al. Extracellular vesicles as drug delivery systems: why and how?. Adv. Drug Deliv. Rev. 159, 332–343 (2020).
-
Wang, C. et al. Therapeutic potential of exosome-based personalized delivery platform in chronic inflammatory diseases. Asian J. Pharm. Sci. 18, 100772 (2023).
-
Carney, R. P. et al. Harnessing extracellular vesicle heterogeneity for diagnostic and therapeutic applications. Nat. Nanotechnol. 20, 14–25 (2025).
-
Selvadoss, A., Baby, H. M., Zhang, H. & Bajpayee, A. G. Harnessing exosomes for advanced osteoarthritis therapy. Nanoscale 16, 19174–19191 (2024).
-
Liang, Y. et al. Chondrocyte-targeted microRNA delivery by engineered exosomes toward a cell-free osteoarthritis therapy. ACS Appl. Mater. Interfaces 12, 36938–36947 (2020).
-
Xu, X. et al. Exosome-mediated delivery of kartogenin for chondrogenesis of synovial fluid-derived mesenchymal stem cells and cartilage regeneration. Biomaterials 269, 120539 (2021).
-
Zhang, C. et al. Charge-reversed exosomes for targeted gene delivery to cartilage for osteoarthritis treatment. Small Methods 8, e2301443 (2024).
-
Gavva, N. R. et al. Repeated administration of vanilloid receptor TRPV1 antagonists attenuates hyperthermia elicited by TRPV1 blockade. J. Pharmacol. Exp. Ther. 323, 128–137 (2007).
-
Eid, S. R. Therapeutic targeting of TRP channels-the TR(i)P to pain relief. Curr. Top. Med. Chem. 11, 2118–2130 (2011).
-
Mobasheri, A., Rannou, F., Ivanavicius, S. & Conaghan, P. G. Targeting the TRPV1 pain pathway in osteoarthritis of the knee. Expert Opin. Ther. Targets 28, 843–856 (2024).
-
Papakosta, M. et al. The chimeric approach reveals that differences in the TRPV1 pore domain determine species-specific sensitivity to block of heat activation. J. Biol. Chem. 286, 39663–39672 (2011).
-
Campbell, J. N. et al. Injectable capsaicin for the management of pain due to osteoarthritis. Molecules 26, 778 (2021).
-
Iadarola, M. J. et al. Long-term pain relief in canine osteoarthritis by a single intra-articular injection of resiniferatoxin, a potent TRPV1 agonist. Pain 159, 2105–2114 (2018).
-
Alsalem, M. et al. Anti-nociceptive and desensitizing effects of olvanil on capsaicin-induced thermal hyperalgesia in the rat. BMC Pharmacol. Toxicol. 17, 31 (2016).
-
Eid, S. R. et al. HC-030031, a TRPA1 selective antagonist, attenuates inflammatory- and neuropathy-induced mechanical hypersensitivity. Mol. Pain. 4, 48 (2008).
-
Petrus, M. et al. A role of TRPA1 in mechanical hyperalgesia is revealed by pharmacological inhibition. Mol. Pain. 3, 40 (2007).
-
Defalco, J. et al. Oxime derivatives related to AP18: agonists and antagonists of the TRPA1 receptor. Bioorg. Med. Chem. Lett. 20, 276–279 (2010).
-
Maleeva, E. E. et al. Potentiating TRPA1 by sea anemone peptide Ms 9a-1 reduces pain and inflammation in a model of osteoarthritis. Mar. Drugs 21, 617 (2023).
-
Lu, S. & Fang, C. Isosakuranetin inhibits subchondral osteoclastogenesis for attenuating osteoarthritis via suppressing NF-κB/CXCL2 axis. Int. Immunopharmacol. 143, 113321 (2024).
-
Wei, H., Chen, Z., Koivisto, A. & Pertovaara, A. Spinal mechanisms contributing to the development of pain hypersensitivity induced by sphingolipids in the rat. Pharmacol. Rep. 73, 672–679 (2021).
-
Krügel, U., Straub, I., Beckmann, H. & Schaefer, M. Primidone inhibits TRPM3 and attenuates thermal nociception in vivo. Pain 158, 856–867 (2017).
-
Eker, H. E., Cok, O. Y., Aribogan, A. & Arslan, G. The efficacy of intra-articular lidocaine administration in chronic knee pain due to osteoarthritis: a randomized, double-blind, controlled study. Anaesth. Crit. Care Pain. Med. 36, 109–114 (2017).
-
Romman, A., Salama-Hanna, J. & Dwivedi, S. Mexiletine usage in a chronic pain clinic: indications, tolerability, and side effects. Pain. Physician 21, E573–e579 (2018).
-
Fassoulaki, A., Patris, K., Sarantopoulos, C. & Hogan, Q. The analgesic effect of gabapentin and mexiletine after breast surgery for cancer. Anesth. Analg. 95, 985–991 (2002).
-
Pellock, J. M. Carbamazepine side effects in children and adults. Epilepsia 28(Suppl 3), S64–S70 (1987).
-
Vaid, R., Sohail, A. & Amir, N. Carbamazepine in osteoarthritis treatment: a novel approach targeting Nav1.7 channels. World J. Orthop. 15, 602–604 (2024).
-
Ferreira, G. E. et al. Efficacy and safety of antidepressants for the treatment of back pain and osteoarthritis: systematic review and meta-analysis. BMJ 372, m4825 (2021).
-
Deuis, J. R. et al. Analgesic effects of GpTx-1, PF-04856264 and CNV1014802 in a mouse model of NaV1.7-mediated pain. Toxins 8, 78 (2016).
-
Mueller, A. et al. Antiallodynic effects of the selective NaV1.7 inhibitor Pn3a in a mouse model of acute postsurgical pain: evidence for analgesic synergy with opioids and baclofen. Pain 160, 1766–1780 (2019).
-
Jarvis, M. F. et al. A-803467, a potent and selective Nav1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc. Natl. Acad. Sci. USA 104, 8520–8525 (2007).
-
DiMartino, S. J. et al. Efficacy and safety of fasinumab in an NSAID-controlled study in patients with pain due to osteoarthritis of the knee or hip. BMC Musculoskelet. Disord. 26, 192 (2025).
-
Kelly, K. M. et al. Safety and efficacy of fulranumab in osteoarthritis of the hip and knee: results from four early terminated phase III randomized studies. Curr. Med. Res. Opin. 35, 2117–2127 (2019).
-
Sheffield, K. S., Kennedy, A. E., Scott, J. A. & Ross, G. M. Characterizing nerve growth factor-p75(NTR) interactions and small molecule inhibition using surface plasmon resonance spectroscopy. Anal. Biochem. 493, 21–26 (2016).
-
Eibl, J. K., Chapelsky, S. A. & Ross, G. M. Multipotent neurotrophin antagonist targets brain-derived neurotrophic factor and nerve growth factor. J. Pharmacol. Exp. Ther. 332, 446–454 (2010).
-
Peltoniemi, M. A., Hagelberg, N. M., Olkkola, K. T. & Saari, T. I. Ketamine: a review of clinical pharmacokinetics and pharmacodynamics in anesthesia and pain therapy. Clin. Pharmacokinet. 55, 1059–1077 (2016).
-
Bell, R. F. Ketamine for chronic noncancer pain: concerns regarding toxicity. Curr. Opin. Support Pa. 6, 183–187 (2012).
-
Cheng, Q. et al. Memantine attenuates the development of osteoarthritis by blocking NMDA receptor mediated calcium overload and chondrocyte senescence. J. Orthop. Transl. 48, 204–216 (2024).
-
Huang, Z. et al. MK801 regulates the expression of key osteoarthritis factors in osteoarthritis synovial fibroblasts through complement C5. Res. Vet. Sci. 136, 377–384 (2021).
-
Egunlusi, A. O. & Joubert, J. NMDA receptor antagonists: emerging insights into molecular mechanisms and clinical applications in neurological disorders. Pharmaceuticals 17, 639 (2024).
-
Boyce, S. et al. Selective NMDA NR2B antagonists induce antinociception without motor dysfunction: correlation with restricted localisation of NR2B subunit in dorsal horn. Neuropharmacology 38, 611–623 (1999).
-
Lees, G. J. Pharmacology of AMPA/kainate receptor ligands and their therapeutic potential in neurological and psychiatric disorders. Drugs 59, 33–78 (2000).
-
Lane, N. & Felson, D. A promising treatment for osteoarthritis?. Ann. Intern. Med. 173, 580–581 (2020).
-
Lovell, D. J. et al. Long-term safety and efficacy of rilonacept in patients with systemic juvenile idiopathic arthritis. Arthritis Rheum. 65, 2486–2496 (2013).
-
Arnold, D. D., Yalamanoglu, A. & Boyman, O. Systematic review of safety and efficacy of IL-1-targeted biologics in treating immune-mediated disorders. Front. Immunol. 13, 888392 (2022).
-
Kloppenburg, M. et al. Phase IIa, placebo-controlled, randomised study of lutikizumab, an anti-interleukin-1α and anti-interleukin-1β dual variable domain immunoglobulin, in patients with erosive hand osteoarthritis. Ann. Rheum. Dis. 78, 413–420 (2019).
-
Fioravanti, A., Fabbroni, M., Cerase, A. & Galeazzi, M. Treatment of erosive osteoarthritis of the hands by intra-articular infliximab injections: a pilot study. Rheumatol. Int. 29, 961–965 (2009).
-
Zhang, Q. et al. Efficacy of infliximab in a rabbit model of osteoarthritis. Connect. Tissue Res. 53, 355–358 (2012).
-
Kloppenburg, M. et al. Etanercept in patients with inflammatory hand osteoarthritis (EHOA): a multicentre, randomised, double-blind, placebo-controlled trial. Ann. Rheum. Dis. 77, 1757–1764 (2018).
-
Chevalier, X. et al. Adalimumab in patients with hand osteoarthritis refractory to analgesics and NSAIDs: a randomised, multicentre, double-blind, placebo-controlled trial. Ann. Rheum. Dis. 74, 1697–1705 (2015).
-
Sarzi-Puttini, P. et al. Clinical, ultrasound, and predictability outcomes following certolizumab pegol treatment (with methotrexate) in patients with moderate-to-severe rheumatoid arthritis: 52-week results from the CZP-SPEED study. Adv. Ther. 35, 1153–1168 (2018).
-
Steeland, S. et al. Generation and characterization of small single domain antibodies inhibiting human tumor necrosis factor receptor 1. J. Biol. Chem. 290, 4022–4037 (2015).
-
Cao, Y. et al. Identification of a ligand for tumor necrosis factor receptor from Chinese herbs by combination of surface plasmon resonance biosensor and UPLC-MS. Anal. Bioanal. Chem. 408, 5359–5367 (2016).
-
Chen, S. et al. Discovery of novel ligands for TNF-α and TNF receptor-1 through structure-based virtual screening and biological assay. J. Chem. Inf. Model. 57, 1101–1111 (2017).
-
Lei, C. et al. Zafirlukast attenuates advanced glycation end-products (AGEs)-induced degradation of articular extracellular matrix (ECM). Int. Immunopharmacol. 68, 68–73 (2019).
-
Smolen, J. S. et al. Olokizumab versus placebo or adalimumab in rheumatoid arthritis. N. Engl. J. Med. 387, 715–726 (2022).
-
Leurs, A. et al. Remission of refractory systemic-onset juvenile idiopathic arthritis after treatment with siltuximab. J. Clin. Rheumatol. 25, 40–42 (2019).
-
Takeuchi, T. et al. Sirukumab for rheumatoid arthritis: the phase III SIRROUND-D study. Ann. Rheum. Dis. 76, 2001–2008 (2017).
-
Weinblatt, M. E. et al. The efficacy and safety of subcutaneous clazakizumab in patients with moderate-to-severe rheumatoid arthritis and an inadequate response to methotrexate: results from a multinational, phase IIb, randomized, double-blind, placebo/active-controlled, dose-ranging study. Arthritis Rheumatol. 67, 2591–2600 (2015).
-
Wells, A. F. et al. Immunogenicity of sarilumab monotherapy in patients with rheumatoid arthritis who were inadequate responders or intolerant to disease-modifying antirheumatic drugs. Rheumatol. Ther. 6, 339–352 (2019).
-
Fujita, R. et al. Urosepsis risk in neuromyelitis optica spectrum disorder patients administered satralizumab. Intern. Med. 62, 3317–3320 (2023).
-
Dörner, T. et al. FRI0239 results of a phase 2b study of vobarilizumab, an anti-interleukin-6 receptor nanobody, as monotherapy in patients with moderate to severe rheumatoid arthritis. Ann. Rheum. Dis. 76, 575 (2017).
-
Chiu, Y. S. et al. The JAK inhibitor Tofacitinib inhibits structural damage in osteoarthritis by modulating JAK1/TNF-alpha/IL-6 signaling through Mir-149-5p. Bone 151, 116024 (2021).
-
Wang, Q., Chen, Z. & Dai, S. M. Baricitinib is a potential treatment in inflammatory osteoarthritis: a proof of concept study. Rheumatology 61, e213–e215 (2022).
-
Lee, Y. H. & Song, G. G. Relative efficacy and safety of tofacitinib, baricitinib, upadacitinib, and filgotinib in comparison to adalimumab in patients with active rheumatoid arthritis. Z. Rheumatol. 79, 785–796 (2020).
-
Mease, P. J. et al. Efficacy and safety of selective TYK2 inhibitor, deucravacitinib, in a phase II trial in psoriatic arthritis. Ann. Rheum. Dis. 81, 815–822 (2022).
-
Papp, K. et al. Phase 2 trial of selective tyrosine kinase 2 inhibition in psoriasis. N. Engl. J. Med. 379, 1313–1321 (2018).
-
Yamaura, K. et al. Therapeutic potential of senolytic agent quercetin in osteoarthritis: a systematic review and meta-analysis of preclinical studies. Ageing Res. Rev. 90, 101989 (2023).
-
Kan, S. et al. FGF19 increases mitochondrial biogenesis and fusion in chondrocytes via the AMPKα-p38/MAPK pathway. Cell Commun. Signal. 21, 55 (2023).
-
Chen, D., Guo, J. & Li, L. Catalpol promotes mitochondrial biogenesis in chondrocytes. Arch. Physiol. Biochem. 128, 802–808 (2022).
-
D’Amico, D. et al. Urolithin A improves mitochondrial health, reduces cartilage degeneration, and alleviates pain in osteoarthritis. Aging Cell 21, e13662 (2022).
-
Xia, X. et al. Retuning mitochondrial apoptosis/mitophagy balance via SIRT3-energized and microenvironment-modulated hydrogel microspheres to impede osteoarthritis. Adv. Healthc. Mater. 12, e2302475 (2023).
-
Xin, R. et al. Mitochonic acid-5 inhibits reactive oxygen species production and improves human chondrocyte survival by upregulating SIRT3-mediated, parkin-dependent mitophagy. Front. Pharmacol. 13, 911716 (2022).
-
Liu, L. et al. The physiological metabolite α-ketoglutarate ameliorates osteoarthritis by regulating mitophagy and oxidative stress. Redox Biol. 62, 102663 (2023).
-
Mei, R. et al. 17β-estradiol induces mitophagy upregulation to protect chondrocytes via the SIRT1-mediated AMPK/mTOR signaling pathway. Front. Endocrinol. 11, 615250 (2020).
-
Yang, G. et al. Astaxanthin suppresses oxidative stress and calcification in vertebral cartilage endplate via activating Nrf-2/HO-1 signaling pathway. Int. Immunopharmacol. 119, 110159 (2023).
-
Chan, B. et al. Adseverin, an actin-binding protein, modulates hypertrophic chondrocyte differentiation and osteoarthritis progression. Sci. Adv. 9, eadf1130 (2023).
-
Zhang, Z. et al. Melatonin protects vertebral endplate chondrocytes against apoptosis and calcification via the Sirt1-autophagy pathway. J. Cell. Mol. Med. 23, 177–193 (2019).
-
Peng, R. et al. Klf10 is involved in extracellular matrix calcification of chondrocytes alleviating chondrocyte senescence. J. Transl. Med. 22, 52 (2024).
-
Elayyan, J. et al. Lef1 ablation alleviates cartilage mineralization following posttraumatic osteoarthritis induction. Proc. Natl. Acad. Sci. USA 119, e2116855119 (2022).
-
Ma, Y. et al. Anti-hypertrophic effect of synovium-derived stromal cells on costal chondrocytes promotes cartilage repairs. J. Orthop. Transl. 32, 59–68 (2022).
-
Grossin, L. et al. Induction of heat shock protein 70 (Hsp70) by proteasome inhibitor MG 132 protects articular chondrocytes from cellular death in vitro and in vivo. Biorheology 41, 521–534 (2004).
-
Grossin, L. et al. Gene transfer with HSP 70 in rat chondrocytes confers cytoprotection in vitro and during experimental osteoarthritis. FASEB J. 20, 65–75 (2006).
-
Siebelt, M. et al. Hsp90 inhibition protects against biomechanically induced osteoarthritis in rats. Arthritis Rheum. 65, 2102–2112 (2013).
-
Zhu, Z. et al. The natural product salicin alleviates osteoarthritis progression by binding to IRE1α and inhibiting endoplasmic reticulum stress through the IRE1α-IκBα-p65 signaling pathway. Exp. Mol. Med. 54, 1927–1939 (2022).
-
Xie, C. L. et al. Vitexin alleviates ER-stress-activated apoptosis and the related inflammation in chondrocytes and inhibits the degeneration of cartilage in rats. Food Funct. 9, 5740–5749 (2018).
-
Zhang, Z. et al. Safranal treatment induces Sirt1 expression and inhibits endoplasmic reticulum stress in mouse chondrocytes and alleviates osteoarthritis progression in a mouse model. J. Agric. Food Chem. 70, 9748–9759 (2022).
-
Tang, Q. et al. Trehalose ameliorates oxidative stress-mediated mitochondrial dysfunction and ER stress via selective autophagy stimulation and autophagic flux restoration in osteoarthritis development. Cell Death Dis. 8, e3081 (2017).
-
Liu, Z. et al. Dapagliflozin suppress endoplasmic reticulum stress mediated apoptosis of chondrocytes by activating Sirt1. Chem. Biol. Interact. 384, 110724 (2023).
Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (82293684), the CAMS Innovation Fund for Medical Sciences (CIFMS) (2021-I2M-1-026). We would like to thank Editage (www.editage.cn) for English language editing.
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, Y., Han, Y., Sun, Y. et al. Osteoarthritis: molecular pathogenesis and potential therapeutic options. Sig Transduct Target Ther 11, 81 (2026). https://doi.org/10.1038/s41392-025-02556-6
-
Received:
-
Accepted:
-
Published:
-
Version of record:
-
DOI: https://doi.org/10.1038/s41392-025-02556-6