
Introduction
India’s livestock sector is experiencing colossal growth; consequently, livestock resources have to be harnessed fully in order to achieve the goals of nutritional security and sustainability. However, paucity of reliable and economically viable diagnostics and vaccines cause the rise in morbidity of chronic diseases like Johne`s disease (JD) in animals. MAP is believed to infect animals at a young age and then remain latent for several years before symptoms appear. During this time, animals continue to shed MAP in varying amounts and patterns over a long period. In chronic cases, the long asymptomatic stage is followed by rapid progression to clinical disease which occurs after a period of anergy. An assay that detects the active phase of infection during the subclinical stage, when faecal shedding is low, is ideal. Such a test is urgently needed to help control the spread of infection. Confirmation of JD by culture, the gold standard, takes about 3–4 months. Available immuno-diagnostics mainly use M. avium group protein antigens, which have low specificity1,2. Although molecular methods are more sensitive and specific, they require specialized laboratories. To address this, researchers have developed new diagnostic methods that do not need complex lab equipment and can be used for rapid, point-of-care testing. Presently, different types of isothermal amplification assays suitable for general diagnostic laboratories are the promising fields for the development of rapid and point of care or pen side diagnostics. A wide variety of isothermal amplification techniques have been reported for amplification of DNA or RNA in the past two decades, including transcription-based amplification system (TAS)3, self-sustained sequence replication reaction (3SR)4, nucleic acid sequence based amplification (NASBA)5, strand displacement amplification (SDA)6, rolling circle replication (RCR)7, loop mediated isothermal amplification (LAMP)8, helicase dependent amplification (HDA)9 and single primer isothermal amplification (SPIA)10, cross-priming amplification (CPA)11 and polymerase spiral reaction (PSR)12,13,14. Among these assaysTAS, 3SR, NASBA, SDA, HDA and SPIA require multiple enzymes (three or more) and rigorous optimization. Only a few of these isothermal amplification methods (e.g., RCR, LAMP, CPA and PSR) can be efficiently performed at a constant temperature using one enzyme. However, RCR method can only amplify circular DNA while initial denaturation step is required for the satisfactory result of LAMP. CPA and PSR are novel and unique in context of isothermal nucleic acid amplification which require one enzyme and constant temperature and combines the advantage of both, simplicity of PCR and sensitivity, rapid and cost effectiveness of LAMP. Thus, this research programme is designed to develop and optimize the novel PSR assay for rapid visual detection of MAP DNA from faecal samples.
Materials and methods
Ethical permission
The study was approved with approval no. IAEC/18/25 by Institutional Animal Ethics Committee (IAEC), Uttar Pradesh Pandit Deen Dayal Upadhyaya Pashu Chikitsa Vigyan Vishwavidyalaya Evam Go-Anusandhan Sansthan (DUVASU), Mathura, Uttar Pradesh, India, with registration no. 386/PO/ReBi-S/ReRcBi-L/2001/CPCSEA.
Bacterial strains, clinical samples and genomic DNA extraction
MAP strain GJ-2215 used as positive control for development and optimization of the assay was obtained from Amity Centre for Mycobacterial Disease Research Laboratory, Amity Institute of Microbial Technology, Amity University Rajasthan, Jaipur, Rajasthan, India. A total ten Mycobacterium species/isolates, procured from Microbial Type Culture Collection and Gene Bank (MTCC) at Institute of Microbial Technology (IMTECH), Chandigarh, and M. bovis BCG vaccine strain along other pathogens commonly found in faecal samples and in cattle-shed environment available in department were used to determine the specificity of the developed assay (Table 1). Lyophilized cultures were revived as per instructions provided by MTCC using MB-7H9 broth media and inoculated on specific media in bio-safety cabinet. Inoculated slants and medium tubes were incubated as per the optimum temperature requirement of the different isolates (Supplementary file).
Clinical samples and processing
Random sampling procedure was employed to obtain reasonably valid and precise prevalence of MAP. Approximately, 5–10 gram of faecal samples were collected from sheep, goats, cattle and buffaloes per-rectally or collected from the centre of the freshly voided faeces, avoiding those contaminated with dust and/or urine. The samples were processed using modified centrifugation method16. Briefly, 2 to 5 g of faeces was finely grounded in sterilized pestle and mortar with sterile 1× PBS to prepare 5% suspension in 50 ml tubes. Faecal suspension was then vortex for 2 min, and allowed to stand undisturbed for 30 min. The upper layer was discarded and the middle layer was collected in sterile tubes for genomic DNA extraction.
Template DNA preparation
DNA extraction from faecal samples
The DNA isolation was carried out using the method described by Stabel et al.17 with some modifications. Processed sample suspensions were vortex for 5 s and allowed to settle for 2 min and vortex again for 5 s. Samples were then centrifuged at 200 rpm for 30 s and the supernatants from each sample were transferred to sterile 15 ml tube and diluted to 1:100 in 1x Tris- EDTA (TE) buffer. Then, 1.0 ml of diluted samples were transferred into sterile 1.5 ml eppendorf tubes and again centrifuged at 12,000 rpm for 2 min. Supernatant was discarded and the pellet was washed 2 times with 1 ml of 1x TE buffer. Pellet was re-suspended in 200 µl of 1x TE buffer, subsequently incubated in a heating water bath (Grant Instruments India Pvt. Ltd., India) at 100 °C for 10 min and then cooled for 5 min. This step was repeated for three times. Samples were stored at -20 °C till use in amplification.
DNA extraction from standard Mycobacterium isolates
The standard Mycobacterium isolates 0.5 McFarland equivalent suspension (HiMedia, India) in 1x PBS was boiled in a micro-centrifuge tube for 5 min and then snap-chilled on ice. Subsequently, the material was centrifuged at 10,000 g for 5 min, and the supernatant was used as a template for amplification.
Designing of MAP-PSR primers
PSR primers specific for MAP detection were designed as previously described13. The IS900 putative transposase (p43) gene was used for primer design. Full-length sequences from NCBI were aligned using ClustalW in MEGA-7.0 to identify conserved regions. The forward and reverse primers for the target 159 bp sequence of IS900 gene of MAP were analyzed to determine the best-fit score and possible presence of self–complementary or hairpin- loop structures using the online Oligo Analyzer Tool in the IDT database (http://eu.idtdna.com/calc/analyzer). Further, a stuffer oligonucleotide sequence of exogenous origin described by Gupta et al.13 was incorporated at the 5′ end of the primer set. The primers suggested were also checked in-silico for specificity using BLAST-N program. Primers used for PSR assay showing primer code, primer sequence and position within the IS900 gene (the stuffer sequence is presented in lower case and in italics) are shown in Table 2. The same PSR primers were also used in PCR amplification. The primers were custom synthesized in Imperial Life Sciences (P) Limited, Gurgaon, Haryana, India.
Optimization and visualization of MAP-PSR
Based on previous reports, different reaction set-ups were tried out in 25 µl reaction volumes having different component and incubated at 63–66 °C for 60–75 min time period in water bath to get the desired amplification. The reaction mixture and temperature-time at which first amplification occurred was further optimized for various reagents, temperature and time successively to get clear separation of DNA band exhibiting the ideal laddering pattern with clear difference between positive and negative controls. Each optimized reaction condition was considered as initial reaction setup for the subsequent optimization. The MAP-PSR assay was optimized by testing various components. Primer concentrations ranged from 10 to 60 pmol in 10 pmol intervals. dNTPs (Sigma-Aldrich, St. Louis, USA) were tested from 0.8 to 1.8 mM with 0.2 mM steps. MgSO₄ (NEB, Ipswich, MA) was varied between 8 and 14 mM in 2 mM intervals. Betaine (Sigma-Aldrich, St. Louis, USA) concentrations ranged from 0.5 to 1.2 M in 0.1 M intervals. Bst DNA polymeraseLg Frag (NEB, Ipswich, MA) was used from 2 to 15 U with 2 U steps. Temperatures from 61 °C to 66 °C were tested in 1 °C intervals. Incubation times ranged from 30 to 90 min. A non-template control (NTC) was included in each run to detect contamination. The final amplified PSR products were visualized under visible light after the addition of 1 µl of 1:10 diluted SYBR Green-I (10,000× concentrated in DMSO; Invitrogen, USA). A positive reaction exhibited bright green color fluorescence which intensified on exposure of UV light, while a negative reaction remained orange. The amplified products were also resolved on 2.5% agarose gel electrophoresis in TAE buffer, stained with ethidium bromide (0.5 µg/ml) and photo was captured under UV light in a gel documentation system (Uvitech, Cambridge, UK).
Specificity of MAP-PSR
The specificity or exclusivity of the MAP-PSR assay was determined employing genomic DNA of 12 different Mycobacterium sps including the positive control MAP strain along with other pathogens commonly found in faecal samples and in cattle-shed environment (Table 1).
Analytical sensitivity of MAP-PSR
The analytical sensitivity was tested using ten-fold serial dilutions (10⁻¹ to 10⁻¹³) of MAP standard strain DNA at 6.1 ng/µl, measured with spectrophotometer (Eppendorf Biophotometer; Eppendorf India Ltd., Chennai, India). Results were compared with conventional PCR using the same MAP-PSR primers. The PCR reaction was carried out using 2x EmeraldAmpMax PCR master mix (TakaRa Bio Inc., Japan). A volume of 2.0 µl template DNA from each dilution was used with optimized reaction components and thermal cycling conditions. Amplification was performed using a Veriti thermocycler (Applied Biosystems, USA) (Supplementary file).
Validation with clinical samples
A total of 100 cattle fecal samples—50 PCR-positive and 50 PCR-negative—were randomly selected from samples previously tested using the OIE-recommended PCR. These were used to validate the MAP-PSR assay. Validation was performed alongside MAP-PCR standardized using MAP-PSR primers, with each reaction containing 2.0 µL of extracted DNA in a 25 µL volume. To confirm the specificity of the PSR amplification, selected samples that tested positive in both assays were subjected to Sanger sequencing.
Results
Optimization and standard PSR reaction for MAP-PSR assay
The first sign of amplification was observed at 65ºC incubated for 75 min with reaction components contained 2.5 µl of 10x Thermopol Bst reaction buffer, 5.0 µl of 1 M Betaine, 3.5 µl of 10 mM dNTP’s, 2.5 µl of 100 mM MgSO4, 40 pmol each forward and reverse primers, 12 U Bst DNA polymeraseLg Frag, 2.0 µl of template genomic DNA and nuclease-free water to make up the volume to 25 µl (Supplementary file). Further, following optimization the assay exhibited best amplification result at temperature of 64ºC for a period of 60 min with the final reaction composition of 8 mM MgSO4, 1.4 mM dNTPs, 1 M betaine, 8U Bst DNA polymeraseLg Frag, 30 pmol of each primer (Figs. 1 and 2). The standardized reaction condition was checked for desired amplification by visualizing development of turbidity and change in color after addition of 1.0 µl of 1:10 dilution SYBR Green-I dye. The color of SYBR Green-I dye changed from orange to fluorescent green which intensifies on exposure of UV light in positive reaction while dye retained its original orange color in negative control. The white turbidity was observed in positive reaction but there was no turbidity in negative control. The clear laddering pattern of DNA bands of amplified products was observed in 2.5% agarose gel electrophoresis. Thereafter, the standard reaction MAP-PSR assay was carried out for determination of specificity and sensitivity of the assay followed by evaluation of developed MAP-PSR assay in parallel with PCR using clinical samples.

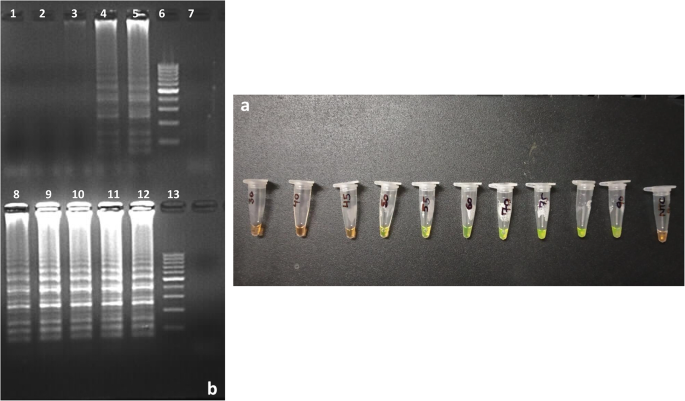
(a) Effect of incubation time on the PSR assays. Result of MAP-PSR amplification at different incubation time period showing positive amplification from Tube 4 to Tube 10 represented by change of SYBRGreen-1 to fluorescent green colour and no amplification in Tube 11 represented by no change in orange colour of dye (Tube 1: 30 min; Tube 2: 40 min M; Tube 3: 45 min; Tube 4: 50 min; Tube 5: 55 min; Tube 6: 60 min; Tube 7: 70 min; Tube 8: 75 min; Tube 9: 80 min; Tube 10: 90 min; Tube 11: NTC). (b) AGE (2.5%) showing the laddering pattern produced by DNA bands after amplification of IS900 gene of MAP in PSR assay at different incubation time. Lane 1: W1: 30 min; W2: 40 min; W3: 45 min; W4: 50 min; W5: 55 min; W6: 100 bp ladder; W7: NTC. Lane 2: W8: 60 min; W9: 70 min; W10: 75 min; W11: 80 min; W12: 90 min; W13: 100 bp ladder.

Optimized reaction PSR amplification result for MAP detection. (a) Visual detection of PSR amplification results based on development of turbidity in positive amplification and no turbidity development in negative samples. (b) Visual detection of PSR amplification results based on development fluorescent green colour in positive amplification and no colour change of SYBR Green-I dye in negative samples. (c) AGE (2.5%) showing the laddering pattern produced by DNA bands after amplification in PSR assay (W1: MAP standard isolate DNA as template, W2: 100 bp ladder, W3: NFW as template).
Specificity of MAP-PSR assay
The specificity reactions showed a positive result only for the MAP isolate DNA. No amplification was observed for the other 11 Mycobacterium species, common bacterial isolates, or the negative control, as confirmed by SYBR Green-I dye and 2.5% agarose gel electrophoresis.
Sensitivity determination of MAP-PSR assay in comparison with conventional PCR method
The sensitivity of the MAP-PSR assay was determined in terms of copy number and amount of DNA. For the determination of sensitivity of MAP-PSR assay, 6.1 ng/µl of MAP DNA (1.18 × 10⁶ copy number) was diluted 10-fold serially and compared with conventional PCR. PSR assay exhibited positive result up to 122 fg or ⁓23 copy number of the template. However, in the conventional PCR, the detection limit was found to be ten times lesser than PSR.
Evaluation of MAP-PSR assay with clinical samples
A total of 100 clinical samples were tested using the PSR assay, and 59 were found positive for MAP DNA. PCR testing of the same samples detected 50 positives, all of which were also positive by PSR. A randomly selected positive sample from both assays confirmed MAP specificity through sequence analysis, and the sequence was submitted to NCBI GenBank (accession number MZ923160).
Discussion
The detection of MAP infections in livestock is crucial to control the spread among the animals and to humans. Culture of MAP is regarded as the gold standard for diagnosis of paraTB but it is cumbersome method, requires well-equipped laboratories and needs 8–12 weeks of time for MAP to grow (Angelidou et al., 2016). In addition to culture, several tests based on detection of cellular and humoral immune response are available but the clinical sensitivity and specificity are low (Vansnick et al., 2004; Mobious et al., 2008; Plain et al., 2014). Overcoming the sensitivity and specificity limitations, PCR assays is being looked as an alternative gold standard for detection of MAP in clinical samples in a time period of 90–120 min, but these assays also required highly sophisticated equipment and post amplification procedure for visualization of the products. However, increasing number of studies have reported that the PSR method provides a promising isothermal amplification assay that can be used for rapid detection of pathogens even in limited resource diagnostics facilities12,14,18. The present study has been successful in developing and validating PSR assay for rapid and sensitive detection MAP DNA within 60 min at 64 °C using a water bath and as per the literature available, this is the first report of optimization and evaluation of PSR for detection of MAP. The sensitivity was determined in terms of amount of DNA and copy number of template and it was found that the MAP-PSR sensitivity was 10-fold more than conventional PCR using the same set of primers and template concentration. Furthermore, the assay could specifically detect MAP without cross-reaction with other prevalent Mycobacterium species and other pathogens commonly found in faecal samples and in cattle-shed environment. The developed MAP-PSR assay when validated in 100 clinical faecal samples, was able to detect 9 more samples positive (n = 59) than conventional PCR (n = 50). Other isothermal assays have also been developed for detection of MAP. Among those, the MAP-LAMP developed by Trangoni et al.19 demonstrated the detection limit of 100 fg per reaction which was in accordance with those obtained with PCR. Hansen et al.20 developed a recombinase polymerase amplification (RPA) assay for rapid isothermal detection of MAP DNA with detection limit of 16 DNA molecules and 500 fg genomic DNA which corresponds to approximately 95 MAP genomes21. The sensitivity of RPA for detection of MAP was found to be 10-fold higher to real-time PCR. Although, Rolling circle amplification (RCA) is also an isothermal amplification technique that had a 10-fold higher sensitivity than conventional PCR22 but the use of three components, a recombinase, a single-stranded DNA binding protein (SSB), and a strand displacing polymerase to achieve denaturation of double stranded DNA makes the optimization a cumbersome task as compared to the much simpler format of PSR assay. Pathogen detection directly from clinical samples certainly is a challenge, and performance of developed a PSR assay in the study using a wide panel of field samples evidence that the assay may be a useful tool to diagnose animals suffering with Johne’s disease. Finally, the MAP-PSR developed in this study is overall more cost-effective (Supplementary file), due to its low equipment need, faster turnaround, and suitability for point-of-care testing that can detect MAP genomic DNA in faecal samples of ruminants with high sensitivity and specificity and can be applied by resource limited laboratory located at farm/district for screening of herd and subsequently culling of positive animals.
Data availability
Data is provided within the manuscript or supplementary information files.
References
-
Koo, H. C. et al. Analysis of the immune response to Mycobacterium avium subsp. Paratuberculosis in experimentally infected calves. Infect. Immun. 72 (12), 6870–6883 (2004).
-
Nielsen, S. S. Transitions in diagnostic tests used for detection of Mycobacterium avium subsp. Paratuberculosis infections in cattle. Vet. Microbiol. 132, 274–282 (2008).
-
Kwoh, D. Y. et al. Transcription-based amplification system and detection of amplified human immunodeficiency virus type 1 with a bead-based sandwich hybridization format. Proc. Natl. Acad. Sci. U.S.A. 86 (4), 1173–1177 (1989).
-
Fahy, E., Kwoh, D. Y. & Gingeras, T. R. Self-sustained sequence replication (3SR): an isothermal transcription-based amplification system alternative to PCR. PCR Meth Appl. 1, 25–33 (1991).
-
Compton, J. Nucleic acid sequence-based amplification. Nature 350, 91–92 (1991).
-
Walker, G. T. et al. Strand displacement amplification-an isothermal, in vitro DNA amplification technique. Nucleic Acids Res. 20 (7), 1691–1696 (1992).
-
Baner, J., Nilsson, M., Mendel-Hartvig, M. & Landegren, U. Signal amplification of padlock probes by rolling circle replication. Nucleic Acids Res. 26 (22), 5073–5078 (1998).
-
Notomi, T. et al. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 28 (12), E63 (2000).
-
Vincent, M., Xu, Y. & Kong, H. Helicase-dependent isothermal DNA amplification. EMBO Rep. 5 (8), 795–800 (2004).
-
Kurn, N. et al. Novel isothermal, linear nucleic acid amplification systems for highly multiplexed applications. Clin. Chem. 51 (10), 1973 (2005).
-
Fang, R. et al. Cross-priming amplification for rapid detection of Mycobacterium tuberculosis in sputum specimens. J. Clin. Microbiol. 47 (3), 845–847 (2009).
-
Das, A. et al. Rapid visual isothermal nucleic acid-based detection assay of Brucella species by polymerase spiral reaction. J. Appl. Microbiol. 125 (3), 646–654 (2018).
-
Gupta, V. et al. Polymerase spiral reaction (PSR): a novel, visual isothermal amplification method for detection of canine parvovirus 2 genomic DNA. Arch. Virol. 162 (7), 1995–2001 (2017).
-
Malla, J. A. et al. Novel polymerase spiral reaction (PSR) for rapid visual detection of bovine herpesvirus 1 genomic DNA from aborted bovine fetus and semen. Gene 644, 107–112 (2018).
-
Jain, M. et al. Development of rELISA using novel markers for the diagnosis of paratuberculosis. J. Immunol. Methods 497, 113105. https://doi.org/10.1016/j.jim.2021.113105 (2021).
-
Ristow, P., Silva, M. G., de Souza, F. & Lilenbaum, W. Evaluation of Mycobacterium avium subsp. Paratuberculosis faecal culture protocols and media. Pesq Vet. Bras. 26 (1), 1–4 (2006).
-
Stabel, J. R., Bosworth, T. L., Kirkbride, T. A., Forde, R. L. & Whitlock, R. H. A simple, rapid, and effective method for the extraction of Mycobacterium paratuberculosis DNA from fecal samples for polymerase chain reaction. J. Vet. Diagn. Invest. 16, 22–30 (2004).
-
Woźniakowski, G. et al. Polymerase cross-linking spiral reaction (PCLSR) for detection of African swine fever virus (ASFV) in pigs and wild boars. Sci. Rep. 7, 42903 (2017).
-
Trangoni, M. et al. LAMP technology: rapid identification of Brucella and Mycobacterium avium subsp. Paratuberculosis. Braz J. Microbiol. 46, 619–626 (2015).
-
Hansen, S., Schafer, J., Fechner, K., Czerny, C. P. & Abd El-Wahed, A. Development of a recombinase polymerase amplification assay for rapid detection of the Mycobacterium avium subsp. Paratuberculosis. PLoS One. 11 (12), e0168733 (2016).
-
Fechner, K. et al. Distribution of Mycobacterium avium subsp. Paratuberculosis in a sub-clinical naturally infected German Fleckvieh bull. Transbound. Emerg. Dis. 64, 916–928 (2015).
-
Wang, X. R. et al. Rapid detection of Staphylococcus aureus by loop-mediated isothermal amplification. Appl. Biochem. Biotech. 175, 882–891 (2015).
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
The study was approved with approval no. IAEC/18/25 by Institutional Animal Ethics Committee (IAEC) of DUVASU, Mathura, Uttar Pradesh, India, registered with, Committee for Control and Supervision of Experiments on Animals (CCSEA), Government of India with registration no. 386/PO/ReBi-S/ReRcBi-L/2001/CPCSEA.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Singh, V.K., Gupta, V., Das, C. et al. Polymerase spiral reaction assay for rapid visual detection of Mycobacterium avium subsp. paratuberculosis in fecal samples. Sci Rep 15, 27149 (2025). https://doi.org/10.1038/s41598-025-12435-3
-
Received:
-
Accepted:
-
Published:
-
DOI: https://doi.org/10.1038/s41598-025-12435-3