
Introduction
SARS-CoV-2 is a β coronavirus responsible for over 578 million infections and over 6.9 million deaths world-wide. Although no longer a pandemic classification, its current circulating variants continue to pose threats through potential resistance to existing therpaeutics and vaccines. β coronaviruses represent one (B) of four genera (A, B,C, and D) of RNA positive-sense viruses in the Nidovirales order1,2. SARS-CoV-2 is the latest in human viral outbreaks of this genera being preceded by the Middle Eastern Respiratory Coronavirus (MERS-CoV) and the SARS-CoV outbreak of 2002.
Quantitatively, each SARS-CoV-2 virion carries approximately 100 Spike proteins per virion3. The prefusion Spike protein (S) is a large trimeric protein where each protomer may be in a so-called Up state or Down state, depending on the configuration of its receptor binding domain (RBD). In the “Up-state” the (prefusion) protein can efficiently bind to human epithelial cells expressing Angiotensin Converting Enzyme 2 (ACE2) on their surface and infect via a transformation to its fusion state. These cells include Type I and II pneumocytes; alveolar macrophage; nasal mucosal cells; and a large range of vascular epithelial cells throughout the body (kidney, heart, etc.)4. In addition, interaction of the Spike protein with TMPRSS2 and possibly other receptors may be another important route to viral entry5. However, ACE2 has been shown to play the critical role for viral entry5,6.
In addition, it is well-known that this family of β coronaviruses has the capability of recombination within animal hosts, leading to potential zoonotic transmission of recombinant viruses7,8. For example, it has been recently discovered in bats that a variant of MERS-CoV uses the ACE2 receptor, like SARS-CoV-1 and SARS-CoV-2, to infect human cells9,10. Other human coronaviruses also use the ACE2 receptor for cellular entry11. The disturbing situation is the broad range of potential viral threats from this family of viruses because of the “soup” of variations and recombination’s possible in the environment, and the potential subsequent transmission to humans.
The concept of pan-coronavirus vaccines and therapeutics looks to exploit the common features of viruses that can be used as conserved molecular targets across viral variations. It is clear that families of viruses use specific human cell receptors for their entry, and the development of cell receptor molecular decoys represents one possible path to pan-coronavirus therapeutics. Table 1 S (Supplementary) summarizes some of the viruses and their known human cell receptor host molecules. Human cell receptors have been used to develop decoy therapeutics, such as sialic acid mimetics for Influenza A12, ACE2 for SARS, and others such as ICAM-113. Human cell receptors typically have key binding sites that may be conserved across viral mutants, variations, and recombination events.
In this study, the key binding segment of human ACE2 is exploited to develop an optimized peptide that efficiently mimics the key binding segment of ACE2 with the receptor binding domain (RBD) of the spike protein of SARS-CoV-2. Although ACE2 derived peptides have been proposed and studied as SARS-CoV-2 therapeutics14,15, these studies are often based on the use of large compound libraries and random structure variations (high throughput screening). Note that the native α1 helix was studied some time ago as a potential therapeutic to SARS-CoV-116 and recently to SARS-CoV-217. A review of the development of ACE2 derivatives against SARS-CoV-2 was given in 202318. Small segments of the native peptide (~ 6 residues in length) have also been studied as SARS-CoV-2 viral inhibitors, such as E37-Q42 hACE2 segment15. Note that no attempt is made here to review the vast number of purely computational approaches to ACE2 mimetics. Here a rational approach for optimizing the native peptide was taken in response to the pandemic, with experimental verification, based on understanding of binding physics, helical structure/stability, and peptide solubility. Along these same lines, we note that improvement of the of the isolated α-helix as a therapeutic to SARS-CoV-2 was also demonstrated via rationally based Alanine or Leucine substitutions to improve helicity and simultaneously including the effects of antigenicity19. These more rational methods, including the ones outlined here, may also be transferable to other targets and biological therapeutics as an alternative to, or in hybrid with, large compound libraries and high throughput analyses, which may be costly and time-consuming.
Results
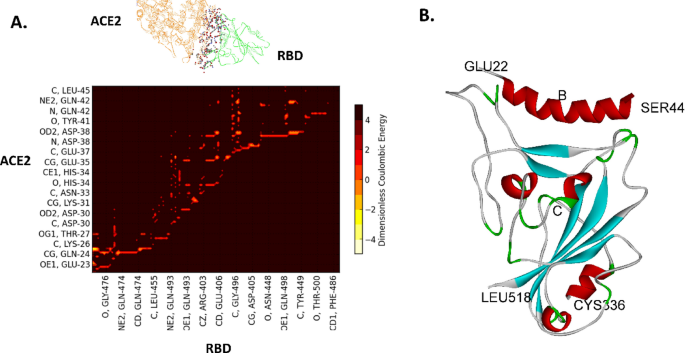
In order to generate optimized peptide mimetics to the RBD of the Spike protein, all-atom, ab initio molecular dynamics were initially carried out over a 0.5 microsecond total time based on the published structure of the RBD of SARS-CoV-2 Spike protein (S) with ACE220 (PDB ID 6M17; B and E chains), as described previously21. The average conformation of the binding complex near the end of the simulation is used to map the dominant energetic atom-atom interactions between ACE2 and the RBD as shown in Fig. 1. The dominant interactions in this case are the partial charge interactions between ACE2 and RBD residues among both backbone and side chain atoms of the amino acid residues, as shown in Fig. 2. There are two distinct binding domains to ACE2 : residues 22–41 (alpha helix 1) and residues 322–357 (loop domain).

Dominant energetic mappings for the development of peptide biomimetics from ACE221. A. Dominant Energetic Mappings or “Glue Points” of the Interaction of the RBD (green) of SARS-CoV-2 with full length ACE2 (orange); dominant atom-atom interactions are shown in “ball and stick” format. Not all residues are shown; see S2. B. Binding of ACE2 mimetic peptide (B) with the RBD of SARS-CoV-2 (C). Complete quantitative information, including van der Waals interaction energy values, is given in Supplementary Data S2.
Specific energetic and proximal contact points (atom-atom interactions) for helical binding typically form a distinct pattern of 1 residue “on” (meaning that is in closest contact with target), 2 residues “off” (most distal from target), 2 on, and 2 off. The pattern repeats approximately every 7 residues owing to the helical geometric requirement of 3.4 residues per turn, as shown in Fig. 2. (Also, see the helical wheel of Fig. 4).

Binding of SARS-CoV-2 to human ACE2 showing the two distinct binding regions: α1 helix (residues 23–42) and loop domains (324–357). Binding energies are normalized by kT, where k is Boltzmann’s constant and T is absolute temperature. The corresponding target residue numbers are given in the Supplementary information (S2).
Unfortunately, the α1 helix of ACE2 unfolds in isolation from its parent protein and no longer maintains its helical structure, and it is a weak binder to spike RBD in isolation22. There can also be solubility issues owing to the relatively large number of negatively charged groups (see Materials and Methods). In the native state, the so-called non-binding residues (with respect to the RBD of Spike) in Fig. 2 strongly interact with the α2 helix of ACE2 helping to stabilize its helical structure. In addition to improving binding to the Spike RBD through rational examination of single residue substitutions for binding residues, non-binding residue substitutions can be used to promote helicity and solubility. Table 2 summarizes the results of the rational strategy to optimize the 23-residue helical peptide (residues 22–44), Fig. 3 shows a side-by-side computational comparison of the optimized to native helical binding behavior, and Fig. 4 shows a helical wheel comparison. Note that molecular dynamics shows that the 23-residue helix does not significantly change its bound state conformation to the RBD from the full ACE2 bound state conformation (Supplementary S3).

Right: Partial charge interaction energies of the native residues α1 helix segment of hACE2 (B residues) with the RBD residues of SARS-CoV-2. Only the hACE2 residues are shown. Left: Significantly improved interactions with Spike RBD after optimization for the isolated, 23-residue peptide (native residues 22 through 44). Some substituted residues are associated with improved helical stability and solubility. Helical residues (< ~ 30 residues in length) form a unique 7 residue repeat sequence due to the requirement of 3.4 residues per turn which is exploited in the optimization process (Fig. 4). Note that the corresponding RBD residues are given in the Supplementary data (S2, S3).

Helical wheel representation of native and optimized sequence. Binding domain versus non-binding domain is shown in the optimized representation. (Helical wheels generated by NetWheels23.
Helicity strategy follows the study of Facchiano et al.24 and Creamer and Rose25 Here, E-K at positions (i, i + 3) or (i, i + 4) (charge “staple”), L-L and L-F at (i, i + 3) or (i, i + 4) (non-polar “staple”), and branch removal were used for this particular native sequence. Rational, in-silico single residue substitutions to improve helicity were computationally verified using Agadir26. Note that N-terminal acetylation and C-terminal amidation were included. It was found here that E-R can be a slightly more effective staple then E-K for this sequence perhaps due to the small rotational change allowed for the protonated amine side chain of arginine. Fortunately, the three binding improvements are shown in Table 2. also resulted in improved stapling effects in this case. Binding improvement strategy involves improving the specific charge or partial charge interactions of the helix with the RBD, such as H34E shown in Table 2, which was strategized through atom-atom interaction mappings and verified computationally (see Methods).
The water solubility, in this case, was addressed through reduction in the total number of charged residues from a −3 native charge to a −2 charge in the optimized helix and, at the same time, keeping the number of polar and charge groups greater than the number of hydrophobic residues (see Fig. 4). Note that excess positive or negative charges can sometimes lead to lack of dissolution and aggregation due to alterations in the local solvent structure27,28.
Computationally optimized peptides were then physically synthesized for in-vitro studies. In this case, two peptides with and without K26R were studied (Peptide A: EQQLKYFLERFAEQLEQLFYQSS.
and AK26R: EQQLRYFLERFAEQLEQLFYQSS). In the end, AK26R, experimentally showed slightly better helicity and authentic viral inhibition, but both peptides were effective at Spike RBD binding, dissolution in water, and significantly improved helical structure. Note that the experimental results reported here were obtained in real-time over the course of the pandemic. Because of the frequency of change, studies on all of the different variants of concern (VOC) that appeared during the pandemic are beyond the scope of this work.
Experimental CD measurements of optimized peptides demonstrated excellent helicity (68.6% AK26R; 58.5% A), as shown in Fig. 5. The small overall charge reduction from − 3 to −2 had a significant effect on improving solubility. Although not quite fully soluble in nanopure water, the addition of as little as 4% of a 1.0 M solution of NaOH completely dissolved the optimized peptides in water.

Experimental Circular Dichroism (CD) measurement of (A) AK26R peptide. (B) A peptide (Graph and analysis from BestSel29.
Experimental binding studies of optimized peptides were carried out over a range of SARS-Cov-2 variants during the course of the pandemic. This is illustrated in Fig. 6 for a set of variants prior to the appearance of the Omicron variant (see Supplemental S4 for additional SPR studies using full length hACE2 as a positive control). Equilibrium dissociation constants, KD =(kd/ka, dissociation/association kinetic rate constants) demonstrated the effectiveness of the optimization strategy using ACE2 α1-helix, which all variants must use for host cell entry.

(A) Binding of Peptide A to RBD-mutant (K417N, E484K, and N501Y) associated with a number of SARS-CoV-2 variants of concern during the course of the pandemic, as measured by Surface Plasmon Resonance (SPR; see Methods). (B) Peptide A binding to the Delta variant as measured during the pandemic. SPR data of hACE2 binding to the Omicron variant as positive control is given in Supplementary S4.
Authentic viral inhibition studies were subsequently carried out during the course of the pandemic in a BSL3 facility with the two lead, optimized peptides against Wuhan WT and what became the dominant circulating variant: the Omicron variant. As shown in Fig. 7, peptide A was effective against the WT Wuhan SARS-CoV-2 strain (TI > ~ 4). Peptide AK26R was also effective against Omicron BA.2 with a Therapeutic Index > ~ 36 (Supplemental S5). Again, these were the experimental activities carried out in real-time during the pandemic. Optimized peptides have also been exposed to other cell lines, such as A549 human lung cells (Supplemental S6), with no cellular toxicity out to millimolar levels of peptide concentration leading to much larger TI’s.

Authentic Viral Cell Challenge Assays as measured during the pandemic. Peptide A was tested against Wuhan WT (USA_WA1/2020) (Therapeutic Index > ~ 4) (Red). Peptide AK26R was tested against Omicron BA.2. (Therapeutic Index > ~ 36) (Supplemental S5). No toxicity to cells by either peptide in the absence of virus, was observed. (Yellow). The coefficient of variation (COV) across peptide A inhibition studies averaged ~ 60%, whereas AK26R inhibition studies averaged ~ 40%; all toxicity values reported were ~ 10% COV (Supplemental S5).
Repeating of authentic viral inhibition tests are currently planned against circulating Omicron sub-lineages and for testing effectiveness in pre-clinical animal models. Note that authentic viral inhibition tests were also done in parallel using the known IV therapeutic, Remdesivir, as a comparison at the time of these studies (Supplemental S5). Because all variants use the ACE2 receptor, it is expected that the optimized peptides would continue to remain effective across future strains and possibly other emerging viruses that use the human ACE2 receptor, as discussed more fully below. Note that the pharmacokinetic properties and in-vivo stability are additional factors not considered here. (Also, see Discussion below.)
Discussion
Here we have demonstrated a rational approach to the optimization of a helical biomimetic of the ACE2 receptor domain for possible use as a therapeutic agent in the treatment of COVID-19 and other viruses that use human ACE2 for cellular entry. The importance of seeking small peptide therapeutics or prophylactics is centered on ease and rapidity of world-wide manufacturing, ease of administration, such as intranasal for airborne viruses, and the peptides demonstration of extremely low toxicities. The optimization method begins with large scale, ab initio molecular dynamic computations and the mapping of specific atom-atom interactions between the ACE2 receptor and spike RBD that are responsible for the binding behavior. Helical peptides have a binding domain and a non-binding domain with a specific pattern owing to the regular geometry of 3.4 residues per turn. The non-binding residues may be changed to promote helicity according to known residue interactions that promote helical formation. Solubility issues can also be addressed via residue substitutions in the non-binding domain, as demonstrated here. Since all variants use the ACE2 receptor for entry, the approach is demonstrated to withstand spike RBD variants. Although not guaranteed, the α1 segment of ACE2 is the exposed native binding segment of this receptor and may be likely to remain so for future variants or viral recombination.
Viruses recombine through genetic exchange mechanisms that allows mixing and shuffling of genetic material, leading to the emergence of new viral strains7,8,30. Recombination can occur between different viral strains of the same type or differing viral types, in either case with the same host cell. The primary modes of recombination in RNA viruses include template shifting in non-segmented genomes and reassortment in segmented genomes8. It is noted that SARS-Cov-2 is a non-segmented genome, whereas Influenza A is an 8-segmented genome. In non-segmented RNA viruses, recombination primarily occurs through a mechanism called “copy choice recombination,” where the RNA polymerase, during replication, switches from one RNA template to another8. Reassortment involves mixing of the genome segments between two different strains or two different types of viruses during host cell viral reproduction7,30. Recombination plays a crucial role in viral evolution, immune evasion, and cross-species transmission, contributing to the emergence of novel and potentially more virulent strains.
Although most viral recombination effects are deleterious in nature, it is reasonable that non-deleterious recombination would likely conserve human cell surface host receptors in order to be effective at human transmission. Additionally, viral surface protein mutations alone could lead to zoonotic transmission to human cell receptors, for example, “Bird Flu” or H5N1 influenza is currently one mutation away from switching from its current avian sialic acid α2–3 receptor to a human α2–6 sialic acid receptor31 (Table S1); again, giving credence to using forms of human cell receptors as therapeutic or prophylactic agents.
Coronaviruses and Influenza A viruses appear to present the highest likelihood for recombination events that could lead to pandemics due to their genetic recombination mechanisms, frequent zoonotic events, and capacity for human-to-human transmission32. Other viruses, like Paramyxoviruses (Nipah), Orthomyxoviruses (Thogotovirus), and Flaviviruses (Dengue and Zika), also have the ability to infect humans and co-circulate in high-risk areas where recombination may produce new pathogenic variants33. Development of cell receptor decoys may represent a forward-looking and robust approach to viral therapeutics beyond coronaviruses, for example, as recently demonstrated for the Respiratory Syncytial Virus (RSV) human cell receptor, ICAM-113. Note that ICAM-1 is also used by the Rhinovirus (Table S1).
From a practical bent, it is noted that peptides less than 30 residues in length can be manufactured relatively quickly for clinical use in large scale peptide synthesizers (non-biologic). Those synthesizers are available world-wide, which affords world-wide therapeutic production without supply chain issues, which is of great importance during world-wide viral outbreaks. Single residue substitutions of decoy peptides can also be readily invoked to improve binding against any emerging variants. Specific in-vivo therapeutic delivery methods are not addressed here, but intranasal, inhalation, oral, and subcutaneous pathways are well-known, for example, as in the current popular GLP-1 agonist drugs (Victoza, Wegovy, etc.)34. GLP-1 (Glucagon Like Peptide 1) is a small helical peptide that is subcutaneously administered with improved pharmacokinetics and protection from proteases by the addition of a fatty acid group. Other known, successful in-vivo delivery methods involve complexing peptides with nanolipids and liposomes, not reviewed here. Thus, pharmacokinetic issues with peptides as therapeutics have also been addressed over the years. Furthermore, developing many potential lead candidates for future clinical study, rather than just one or two in-vitro “winners”, would appear prudent given the variability in patient response to any one therapeutic.
Materials and methods
Peptides and proteins
Peptides were synthesized to 95+% purity by GenScript (NJ). SARS-CoV-2 spike proteins were purchased through ACRO Biosystems (DE). Lyophilized peptides (~ 10 mg) were dissolved in 1.5 ml of nanopure water as recommended by the manufacturer. Note that fully soluble peptides will leave the nanopure water or solvent crystal clear.
SPR kinetic binding studies
Kinetic binding assays were performed using a Reichart Surface Plasmon Resonance (SPR) instrument (Reichart, NY) according to the manufacturer’s guidelines using a Streptavidin (SA) immobilization chip. RBD WT and VOCs are synthesized and biotinylated at the C-term end (ACROS Biosystems) to avoid any blocking of the binding domains. Briefly, the RBD WT or RBD VOCs were immobilized to the SA chip surface at a concentration of 25 µg/ml and flow rate of 10 µl/min in running buffer. For peptide kinetic binding measurements to RBD, peptide at various concentrations in running buffer were injected over immobilized RBD at a flow rate of 25 µl/min for 3 min with dissociation time of 20 min. Because of the slow off-rate, strong peptide binding, and despite repeated attempts using a suite of suggested regeneration buffers, the “CLAMP” method had to be employed for all runs due to the difficulty of removing adsorbed peptide without damaging the RBD protein35. All data was reference subtracted and fitted to 1:1 Langmuir binding model in “CLAMP” to obtain the association rate constant (ka), dissociation rate constant (kd) and the equilibrium dissociation rate constant (KD).
Circular Dichorism (CD) and Peptide Helical Content. Peptide helical content was experimentally measured using a Jasco J-1500 CD spectrometer according to the manufacturer’s guidelines. Briefly, samples were measured at concentration of 0.1 mg/ml in a 1 mm path length quartz glass cuvette. Background (dilution buffer) readings are reference subtracted and helical content is determined in the wavelength range of 175 to 300 nm using the BESTSEL software29 (https://bestsel.elte.hu/index.php). Computationally, helical peptide content was estimated from the AGADIR software (https://agadir.crg.es), which has been shown to compare well to experimental helical content (2%±6%)36.
Authentic viral inhibition studies
Authentic viral inhibition studies were carried out in BSL3 facilities (ImQuest Biosciences, Inc.). Optimized peptides were tested for antiviral activity against SARS-CoV-2, including pre- exposure of virus to samples for 1 h in serum-free media prior to adding to cell plates. SARS-CoV-2 variants are tested in Vero E6 cells (ATCC) in MEM’s (Cytiva) supplemented with 50 µg/mL gentamicin (Sigma) and 2% FBS (Cytiva). Virus was diluted in serum-free test media to achieve a MOI of 0.001 for antiviral tests. Test peptides were serially diluted using eight 2-fold dilutions in serum-free test media. Serum-free was necessary to avoid peptide non-specific binding to components of FBS. Each dilution was added to 5 wells of a 96-well round- bottom plate containing no cells. Three wells of each dilution were infected with virus, and two wells remain uninfected as toxicity controls. Six wells were infected and untreated as virus controls, and six wells are uninfected and untreated as cell controls. Sample dilutions and virus are incubated together for 1 h at 37 C prior to adding to cell plates. Controls are tested in parallel. Following the pre-exposure of sample and virus, plate contents (100 µl per well) are transferred to plates containing 80–100% confluent Vero E6 cells with 100 µL MEM with 4% FBS and 50 µl/mL gentamicin (final culture media was MEM with 2% FBS and gentamicin on plate). Plates are incubated at 37 C, 5% CO2. On day 4 post-infection, once untreated virus control wells reached maximum cytopathic effect (CPE), plates were stained with neutral red dye for approximately 2 h. Supernatant dye was removed and wells rinsed with PBS, and the incorporated dye extracted in 50:50 Sorensen citrate buffer/ethanol for > 30 min and the optical density was read on a spectrophotometer at 540 nm. Optical densities were converted to percent of cell controls and normalized to the virus control. The concentration of test compound required to inhibit CPE by 50% (EC50) was calculated by regression analysis. The concentration of compound that would cause 50% cell death in the absence of virus was similarly calculated (CC50). The therapeutic index (TI) was the CC50 divided by EC50.
Molecular dynamics
Explicit solvent molecular dynamics (MD) simulations of the novel coronavirus Spike protein were performed using the NAMD2 program36. We used the CHARMM-GUI37 with the CHARMM36m force field along with TIP3P water molecules to explicitly solvate proteins and add any missing residues from the experimental structure files. Disulfide bonds and glycosylated sites were all included. Simulations are carried out maintaining the number of simulated particles, pressure, and temperature (the NPT ensemble) constant with the Langevin piston method specifically used to maintain a constant pressure of 1 atm. Periodic boundary conditions were employed and initial equilibration for a water box simulation. For water, the particle mesh Ewald (PME) method was used with a 20 A cutoff distance between the simulated protein and water box edge. The integration time step was 2 femtoseconds with our protein simulations conducted under physiological conditions (37 C, pH of 7.4, physiological ionic strength with NaCl ions, LYS and ARG were protonated and HIS was not). All mutations associated with Spike VOC’s and peptide residue substitutions associated with optimization were added via the CHARMM-GUI during the course of the pandemic37. However, experimental Spike VOC structure files were used when available, such as Omicron PDB ID: 7T9L (Supplemental S6)38. The closeness of computationally generated structure versus experimental structure has also been recently demonstrated39. Our studies show that 100 nsec. was required to reach an equilibrium from mutated states from the WT structure files40. The total simulation time was therefore taken as 0.5 microseconds (250 million-time steps). All MD results given here were also repeated several times in order to help confirm trends in data.
All-Atom energetic mappings- opencontact
Previously,41 we analyzed the complete inter- and intra-protomer interactions across two independently published structure files (PDB ID: 6VSB and 6VYB; one up and two down protomers) for SARS-CoV-2 trimeric Spike protein using the open source energy mapping algorithm developed by Krall et al. 42. This spatial and energetic mapping algorithm efficiently parses the strongest or most dominant non-covalent atom-atom interactions (charge and partial atomic charge, Born, and van der Waals forces), according to empirically established parsing criteria, based on the ab initio AMBER03 force field model. Following our previous studies, the parsing criteria was taken as the upper limit of −0.1 kT units for Lennard-Jones (van der Waals) criteria and − 0.3 kT units for Coulombic interactions, although lower values can also be specified in the analysis part of the mappings in order to further refine the results41. Note that in the all-atom analysis dominant van der Waals interaction forces are commonly associated with nonpolar atom-atom interactions and hydrophobic protein interaction regions, whereas the Coulombic partial charge and charge interactions are commonly associated with hydrophilic protein interaction regions and can include hydrogen bonding and backbone atom partial charge interactions.
Data availability
Peptide sequence information is given explicitly in the manuscript.
References
-
Shereen, M. A., Khan, S., Kazmi, A., Bashir, N. & Siddique, R. COVID-19 infection: Origin, transmission, and characteristics of human coronaviruses. J. Adv. Res. 24, 91–98 (2020 July).
-
Letko, M., Marzi, A. & Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 5 (4), 562–569 (2020).
-
Bar-On, Y. M., Flamholz, A., Phillips, R. & Milo, R. SARS-CoV-2 (COVID-19) by the numbers. eLife 9, e57309 (2020).
-
Li, M. Y., Li, L., Zhang, Y. & Wang, X. S. Expression of the SARS-CoV-2 cell receptor gene ACE2 in a wide variety of human tissues. Infect. Dis. Poverty. 9 (1), 45 (2020).
-
Xia, X. Domains and functions of Spike protein in SARS-Cov-2 in the context of vaccine design. Viruses 13, 109–125 (2021).
-
Chen, B., Farzan, M. & Choe, H. SARS-CoV-2 spike protein: structure, viral entry and variants. Nat Rev Microbiol [Internet]. May 6 [cited 2025 May 14]; (2025). Available from: https://www.nature.com/articles/s41579-025-01185-8
-
Sanjuán, R. & Domingo-Calap, P. Mechanisms of viral mutation. Cell. Mol. Life Sci. 73 (23), 4433–4448 (2016).
-
Simon-Loriere, E. & Holmes, E. C. Why do RNA viruses recombine? Nat. Rev. Microbiol. 9 (8), 617–626 (2011).
-
Xiong, Q. et al. Close relatives of MERS-CoV in bats use ACE2 as their functional receptors. Nature 612 (7941), 748–757 (2022).
-
Chen, J. et al. Bat-infecting merbecovirus HKU5-CoV lineage 2 can use human ACE2 as a cell entry receptor. Cell 188 (6), 1729–1742e16 (2025).
-
Noettger, S. et al. Role of N-linked glycosylation sites in human ACE2 in SARS-CoV-2 and hCoV-NL63 infection. Liu SL, editor. J. Virol. ;e02202–e02224. (2025).
-
Heida, R. et al. Advances in the development of entry inhibitors for sialic-acid-targeting viruses. Drug Discov Today. 26 (1), 122–137 (2021).
-
Battles, M. B. & McLellan, J. S. Respiratory syncytial virus entry and how to block it. Nat. Rev. Microbiol. 17 (4), 233–245 (2019).
-
Panda, S. K., Sen Gupta, P. S., Biswal, S., Ray, A. K. & Rana, M. K. ACE-2-Derived biomimetic peptides for the Inhibition of Spike protein of SARS-CoV-2. J. Proteome Res. 20 (2), 1296–1303 (2021).
-
Larue, R. C. et al. Rationally designed ACE2-Derived peptides inhibit SARS-CoV-2. Bioconjug. Chem. 32 (1), 215–223 (2021).
-
Han, D. P., Penn-Nicholson, A. & Cho, M. W. Identification of critical determinants on ACE2 for SARS-CoV entry and development of a potent entry inhibitor. Virology 350 (1), 15–25 (2006 June).
-
Oliveira, E. H. et al. A mimetic peptide of ACE2 protects against SARS-CoV-2 infection and decreases pulmonary inflammation related to COVID-19. Antiviral Res. 229, 105968 (2024 Sept). PMID: 39004311.
-
Zhang, H. et al. Advances in developing ACE2 derivatives against SARS-CoV-2. Lancet Microbe. 4 (5), e369–e378 (2023). PMCID: PMC10019897.
-
Karoyan, P. et al. Human ACE2 peptide-mimics block SARS-CoV-2 pulmonary cells infection. Commun. Biol. 4 (1), 197 (2021). PMCID: PMC7881012.
-
Yan, R. et al. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 367 (6485), 1444 (2020).
-
Peters, M. H. Flap structure within receptor binding domain of SARS-CoV-2 Spike periodically obstructs hACE2 binding subdomain bearing similarities to HIV-1 protease flap. Sci. Rep. 2022 Sept 28;12(1):16236 .
-
Das, A. et al. Biophysical properties of the isolated Spike protein binding helix of human ACE2. Biophys. J. 120 (14), 2785–2792 (2021 July).
-
Mól, A. R., Castro, M. S. & Fontes, W. NetWheels: A web application to create high quality peptide helical wheel and net projections [Internet]. Bioinformatics; [cited 2025 Apr 24]. Available from: http://biorxiv.org/lookup/doi/ (2018). https://doi.org/10.1101/416347
-
Facchiano, A. M., Colonna, G. & Ragone, R. Helix stabilizing factors and stabilization of thermophilic proteins: an X-ray based study. Protein Eng Des Sel. Oxford University Press (OUP); Sept 1;11(9):753–760. (1998).
-
Creamer, T. P. & Rose, G. D. Interactions between hydrophobic side chains within alpha-helices. Protein Sci. Publ Protein Soc. 4 (7), 1305–1314 (1995 July). PMCID: PMC2143171.
-
Muñoz, V. & Serrano, L. Elucidating the folding problem of helical peptides using empirical parameters. Nat. Struct. Mol. Biol. 1 (6), 399–409 (1994 June).
-
Raevsky, O. Physicochemical descriptors in Property-Based drug design. Mini-Rev Med. Chem. 4 (10), 1041–1052 (2004).
-
Tanford, C. Physical Chemistry of Macromolecules (Wiley, 1962).
-
Micsonai, A., Bulyáki, É., Kardos, J. & BeStSel, U. S. [cited 2021 Oct 25]. pp. 175–189. Available from: http://link.springer.com/ (2021). https://doi.org/10.1007/978-1-0716-0892-0_11
-
Lowen, A. C. It’s in the mix: reassortment of segmented viral genomes. Spindler KR. editor. PLOS Pathog 2018 Sept 13;14(9):e1007200 .
-
Lin, T. H. et al. A single mutation in bovine influenza H5N1 hemagglutinin switches specificity to human receptors. Science 386 (6726), 1128–1134 (2024).
-
Neumann, G. & Kawaoka, Y. Which Virus Will Cause the Next Pandemic? Viruses. ;15(1):199. PMCID: PMC9864092 (2023).
-
Madere, F. S. et al. Flavivirus infections and diagnostic challenges for dengue, West nile and Zika viruses. Npj Viruses. 3 (1), 36 (2025).
-
Knudsen, L. B. Inventing Liraglutide, a Glucagon-Like Peptide-1 Analogue, for the treatment of diabetes and obesity. ACS Pharmacol. Transl Sci. 2 (6), 468–484 (2019). PMCID: PMC7088919.
-
Karlsson, R., Katsamba, P. S., Nordin, H., Pol, E. & Myszka, D. G. Analyzing a kinetic Titration series using affinity biosensors. Anal. Biochem. 349 (1), 136–147 (2006).
-
Phillips, J. C. et al. Scalable molecular dynamics with NAMD. J. Comput. Chem. 26 (16), 1781–1802 (2005). PMCID: PMC2486339.
-
Jo, S., Kim, T., Iyer, V. G. & Im, W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 29 (11), 1859–1865 (2008). PMID: 18351591.
-
Mannar, D. et al. SARS-CoV-2 Omicron variant: antibody evasion and cryo-EM structure of Spike protein–ACE2 complex. Science 375 (6582), 760–764 (2022).
-
Peters, M. H. Mutations in the receptor binding domain of severe acute respiratory Coronavirus-2 Omicron variant Spike protein significantly stabilizes its conformation. Viruses 2024 June 4;16(6):912 .
-
Peters, M. H., Bastidas, O., Kokron, D. S., Henze, C. E. & Transformations Lineage Comparisons, and Analysis of Down-to-Up Protomer States of Variants of the SARS-CoV-2 Prefusion Spike Protein, Including the UK Variant B.1.1.7. Lee SC, editor. Microbiol Spectr [Internet]. 2021 Sept 3 [cited 2021 Oct 10];9(1). Available from: https://doi.org/10.1128/Spectrum.00030-21
-
Krall, A., Brunn, J., Kankanala, S. & Peters, M. H. A simple contact mapping algorithm for identifying potential peptide mimetics in protein-protein interaction partners: contact mapping for potential peptide mimetics. Proteins Struct. Funct. Bioinforma. 82 (9), 2253–2262 (2014 Sept).
Acknowledgements
BSL3 studies reported here were carried out through ImQuest Biosciences (NJ). Computations were carried out at the Center for High Performance Research Computing at VCU. Optimized peptides are associated with VCU Patent Applications: US 17/997,038 and PCT/US2021/024319. Special thanks to Mary Murphy (Reichart), and Mike Davis (VCU HPRC) for help and guidance.
Funding
This work was supported, in part, by Hoth Therapeutics Inc., New York. Some services in support of the research project were provided by the VCU Massey Cancer Center Flow Cytometry Shared Resource supported, in part, with funding from NIH-NCI Cancer Center Support Grant P30 CA016059.
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Peters, M.H., Pei, X.Y. Rationally designed, ACE2 mimetic binder to the SARS Cov-2 associated Spike protein for COVID-19 therapeutics and beyond. Sci Rep 15, 37120 (2025). https://doi.org/10.1038/s41598-025-21023-4
-
Received:
-
Accepted:
-
Published:
-
DOI: https://doi.org/10.1038/s41598-025-21023-4