
Introduction
Many of the same properties that make plastics attractive in use–e.g., durability, non-reactivity, chemical, light, and thermal resistance–allow them to persist and accumulate in the environment1. Coupled with low recycling rates2 (i.e., high discard rates into landfills and the environment) and limited end-of-life pathways3,4,5, plastic pollution has become a cause for global concern, with mounting recognition of its environmental and health effects6,7,8.
Chemical recycling offers an alternative end-of-life pathway for plastics, in contrast to conventional, mechanical recycling9,10. In mechanical recycling, old plastics can be reformed to new materials, but characteristically, with diminished properties, and with a limit on the number of times they can be mechanically recycled11. Mechanical recycling typically involves the steps of: sorting, washing, shredding/grinding, and melting, followed by re-extrusion of new materials12. In chemical recycling, plastics are broken down to constituent chemical species, either monomers or oligomers, which can then be used to create like-new plastics, or as feedstocks for plastics upcycling13,14. Chemical recycling methods include glycolysis, methanolysis, pyrolysis, gasification, and hydrolysis12,13.
Enzymatic recycling (or biodegradation), via enzymatic hydrolysis, is a promising route for plastic recycling. Of particular promise is using enzymatic recycling for contaminated or colored plastic waste streams, as enzymatic recycling is more agnostic to feedstock quality vs. other methods, such as mechanical recycling15,16. Thus, enzymatic recycling could complement other types of recycling by creating and expanding value in waste feedstocks. Recent years have witnessed the discovery of naturally-occurring enzymes capable of breaking down manmade plastics to their building block, chemical constituents17,18. Hence, enzymatic recycling has gained increased attention, leading some to envision a biorefinery approach to dealing with post-consumer plastic waste streams13,18. Arguably the most successful and prolific efforts in enzymatic plastic recycling have been with poly(ethylene terephthalate) (PET)–common in single-use packaging and textiles, and one of the most-produced plastics–as the target17,19. The first observations of enzymes (in this case, the thermophilic soil bacterium Thermobifida fusca20, which can efficiently degrade plant cell walls21) degrading PET (Fig. 1a) were reported almost 20 years ago; since then, PET depolymerization with enzymes has increased orders of magnitude in efficiency and speed, from reactions taking several weeks to several hours19.

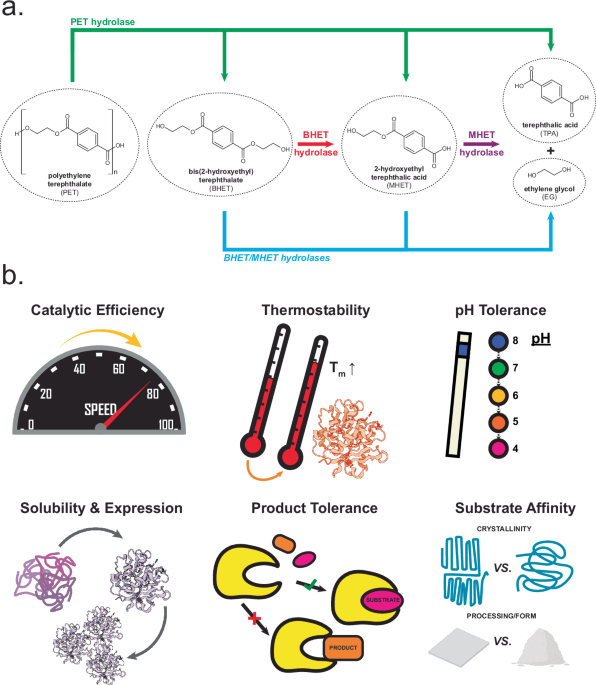
a. Reaction scheme for PET breakdown. Multiple classes of enzymes can participate in enzymatic breakdown of PET. PET hydrolases depolymerize PET to BHET, MHET, terephthalic acid (TPA), and ethylene glycol (EG), though may not effectively use intermediate products as substrates. BHET hydrolases, MHET hydrolases, and dual-function BHET/MHET hydrolases are active on intermediate substrates and may facilitate full conversion to TPA and EG products. b. Examples of protein engineering goals for enzymes involved in PET breakdown. Fulfillment of these engineering goals could improve the cost and environmental impact competitiveness of enzymatic PET degradation.
However, issues remain with enzymatic recycling. Natural enzymes are, generally, not yet optimized for industrial scale biodegradation17,18,19. Several properties lacked by these enzymes that will be essential to their industrial adoption include: high catalytic activity, high substrate and product tolerances, high thermostability, high expression and solubility, and acidic pH tolerance (Fig. 1b).
Recent efforts have been made to relieve the shortcomings of these enzymes by protein engineering. In this Review, we first overview the state of industrial enzymatic PET recycling, to frame and motivate efforts in enzyme engineering. Next, we highlight the current state-of-the-art approaches used to engineer enzymes toward improved PET depolymerization efficiency, focusing on PET hydrolases (also called PETases), bis(2-hydroxyethyl) terephthalate (BHET) hydrolases (also called BHETases), and 2-hydroxyethyl terephthalic acid (MHET) hydrolases (also called MHETases) (Fig. 1a). Further, we discuss future opportunities for researchers, toward advancing protein engineering of these classes of enzymes past existing bottlenecks. We anticipate this summarization and guidance will facilitate continuing work in the field, toward improved, economically- and environmentally-competitive industrial enzymatic PET deconstruction and recycling.
Prospects for industrial enzymatic PET recycling
Current approaches for chemical PET recycling
Viable chemical recycling approaches for PET include glycolysis, methanolysis, and enzymatic hydrolysis12,13. As previous Reviews, such as the thorough analysis by McNeeley and Liu22,23 have thoroughly discussed current chemical recycling approaches, and are not the focus of this Review, we will provide only an abridged overview here. Briefly, glycolysis uses ethylene glycol (EG), in the presence of a catalyst, to break down PET to BHET, which can then be repolymerized to PET12,22. Industrial glycolysis reactions are performed at 170–300 °C and 1–5 atm, with a typical solvent to PET ratio of 1:1 to 10:1 (w/w), for ~1–3 h, to recover ~60–90% BHET (of purities of 63–99%)12,22. Methanolysis uses vaporized methanol, in the presence of a catalyst, to break down PET to dimethyl terephthalate (DMT) and EG, which can then be repolymerized to PET using a multi-step process12. Industrial methanolysis reactions are performed at 240–300 °C and 10–200 atm, with a typical solvent to PET ratio of 2:1 to 8:1 (w/w), for ~1–10 h, to recover ~70–99% DMT12,22. The Technology Readiness Levels (TRLs) of glycolytic and methanolytic PET depolymerization are 5 and 7, respectively12. These are in comparison to mechanical recycling (typically used in municipal recycling programs) with a TRL of 912.
Enzymatic hydrolysis uses PET hydrolase enzymes (which have been discovered from a diversity of hosts and metagenomes24,25,26,27,28) to hydrolyze the ester bonds in PET to yield MHET and EG (Fig. 1a). MHET can be further hydrolyzed by MHET hydrolases to yield the monomers, terephthalic acid (TPA), and EG (Fig. 1a). PET oligomers or small molecules, such as BHET, may accumulate as byproducts, requiring the use of other enzymes, such as BHET hydrolases, for complete hydrolysis (Fig. 1a). PET depolymerizations with merely a PET hydrolase may directly yield TPA as a product and MHET may self-hydrolyze to TPA and EG. Enzymatic hydrolysis reactions are performed at 40–72 °C and 1 atm and a solvent (aqueous buffer) to PET ratio of 4:1 (w/w), for ~10–48 h, to recover 90% or more pure TPA12,22,29. Enzymatic PET hydrolysis has a TRL of 412.
As stated, a thorough comparison of current chemical recycling approaches are not the focus of this Review. We refer readers to the work of McNeeley and Liu22,23, who particularly highlight the specific unit operations and advantages and disadvantages of each process, and Uekert, Singh, and colleagues12,15,16, who perform technoeconomic analyses (TEAs) and life cycle analyses (LCAs) on the processes and provide recommendations for improvement, particularly for enzymatic PET hydrolysis. However, briefly, enzymatic hydrolysis is attractive because it i) uses relatively mild process conditions, ii) the enzymes are selective for the PET ester, and iii) the product stream (predominantly TPA and some MHET) is of high purity and can be directly incorporated into polymerization processes. Further, this TPA is relatively easy to recover by crystallization23. However, the enzymatic reactions require significantly longer residence times than other chemical recycling approaches, it is difficult to recover the EG product, and the process requires large amounts of water, base, and acid to ensure stability of the enzymes and precipitate products12,15,16,23. These issues present motivation for improvements via protein and process engineering.
Process development for industrial enzymatic PET hydrolysis
In a proposed enzymatic PET recycling process by Singh and collegues15, post-consumer PET (typically, as shredded flakes) is first pre-treated, where it is amorphized and its size is reduced, decreasing the polymer crystallinity and increasing its surface area–altogether, increasing its ability to be broken down by the enzyme29,30,31. The PET is then depolymerized in a stirred-tank, batch reactor, with the enzyme, in aqueous buffer15. Base addition (i.e., NaOH) is necessary to maintain the pH of the reaction, as generation of TPA causes acidification, while the optimal pH of many promising PET hydrolases is around 8 to 924,25,26. Following depolymerization, the hydrolysate is purified of remaining solids, enzymes, and other contaminants by passing through filters and columns. Acid (i.e., H2SO4) is then added to drop the pH to ~2.5 to precipitate the TPA, which can be passed through a continuous crystallizer, yielding high purity TPA. To recover the other reaction products, a base is added to neutralize the liquor, which then passes through a membrane unit. The retentate, which contains EG and the salt of the conjugate acid and base (i.e., Na2SO4) is then sent to a continuous crystallizer to recover the salt as a co-product for sale. The permeate is sent to wastewater. Finally, the EG is recovered by distillation of the EG/water liquor (with some distilled water sent to recycle in the depolymerization reactor)15.
The modeled process by Singh, et al., envisioned using a set of seven 950 m3 reactors at 60 °C and pH 8 with 5 mg enzyme per g of PET with 15% PET solids loading for 96 h to achieve a depolymerization of 90%15. Here, 1 kg of PET (consuming 0.17 kg H2O) yields 0.78 kg TPA and 0.29 kg EG (with 0.1 kg leftover PET), with the conceptual plant able to process ~50 million kg of PET per year. This is the approximate capacity of the PET biorecycling plant currently under construction by Carbios32, with Carbios currently operating a demonstration plant with a 20 m3 reactor able to process 2000 kg (~100,000 bottles) per cycle33. Process parameters may change from those modeled by Singh, et al., depending on optimal enzyme conditions and emerging recommendations to increase economics. Currently, small-scale (lab- and pilot-scale) bioreactors in literature are run at 65–72 °C and pH 7–8 with 0.5-3 (typically 1) mg enzyme per g of PET with recommended PET loadings of 16.5-20% (w/v), altogether ideally achieving ≥90% depolymerization in ≤12 h31,34. These lab-scale bioreactors are typically 250–500 mL29,31,34,35, although could be miniaturized for inexpensive validation testing in ~30 mL bioreactors36. Small-scale laboratory tests, meanwhile, are typically performed on the 100–1000 µL scale34,35.
Process considerations for enzymatic PET recycling
Current and proposed pilot- and industrial-scale enzymatic PET recycling processes include PET pre-treatment steps15,29,30,31,37,38. Post-consumer PET should be pre-treated to achieve a high efficiency of depolymerization. Pre-treatment involves steps for amorphization, whereby the crystallinity of the polymer is reduced, followed by steps to increase the specific surface area (SSA) of the PET22,30,37,38, typically by size reduction (e.g., pelletization29,30 and/or micronization by cryo-grinding/milling15,30) or foaming22,29,37. Typical particle sizes are 200–500 µm17,29,34,35, while pellets are <1 mm diameter34,38. Increasing the surface area of the polymer increases enzymatic reaction rates39, while amorphization is critical to overcoming PET’s recalcitrance to enzymatic hydrolysis29,40. Post-consumer PET is characteristically semi-crystalline–containing regions of highly-ordered, tightly-packed polymer chains40,41,42–which impedes entry of enzymes and the hydrolysis reactant, water. While crystallinity is important for some PET material properties (such as gas permeability), it severely inhibits enzyme accessibility40,41. The crystallinity of PET packaging can be ~1–6%41, although regions of PET bottles can be upwards of 34%41, while PET fibers from textiles can be ~16–40%43. PET hydrolases are significantly more efficient in depolymerizing amorphous PET, in which the polymer chains are disordered and randomly-oriented17,29,42. Amorphization is performed by melting the PET (above 260 °C) in an extruder, allowing the polymer chains to randomly disorder, then rapidly cooling it (below the glass transition temperature, Tg, ~70 °C) to preserve the amorphous structure30,40. The amorphized polymer can then be pelletized, ground, and/or milled. Patents assigned to Carbios describe a process whereby PET, melted in an extruder, is mixed with a foaming agent–a pressurized gas, volatile liquid, or gas-releasing chemical–then quickly quenched in water to create a PET foam with high surface area and low crystallinity37. A recent report describes a scalable melt fiber spinning system that rapidly extrudes and spools PET fibers with increased surface area and decreased crystallinity that does not require active cooling44.
Industrially-relevant enzymatic PET depolymerization reactions are typically run at temperatures of 65–72 °C17,29,30,45,46. At these elevated temperatures, using thermostable enzymes, these reactions have been most optimal17,27,45,46. This is, in part, due to the reaction temperature approaching the Tg, the temperature at which the polymer transitions from a hard and brittle state to one that is rubbery and viscous47, of PET: in the range of 65–81 °C, although water-soaking PET has been reported to decrease the surface (interfacial) Tg to as low at 40 °C48. Near the Tg, the polymer chain mobility is increased for enzymatic accessibility. However, increasing the temperature over Tg will induce crystallization, as polymer chains are able to reorder, which will impede significant PET hydrolysis17,29,45,46,49. Tournier, et al., reported that incubating post-consumer PET at 72 °C for ∼15 h increased its crystallinity from ∼15% to ∼40%, while this happened in less than 6 h at 75 °C and over 24 h at 70 °C29. Thus in PET enzymatic hydrolysis, there is a competition between depolymerization and recrystallization, the bounds of which depend on varied factors including: temperature, enzyme loading, enzyme stability/deactivation, PET particle size or film thickness, polymer molecular weight, amorphization/extrusion parameters, and the availability of water/buffer45,48,49. All things considered, the need for polymer chain mobility, enzyme activation and adhesion, and propensity for recrystallization, must be balanced and the optimal temperature, Topt, may vary based on the enzyme and process conditions. Notwithstanding, the Topt for many recent studies reporting high-performance enzymes is 68–70 °C29,31,35,50,51,52,53.
Process innovations for improved enzymatic PET recycling
Process innovations have the potential to increase the efficiency and cost-competitiveness of enzymatic PET hydrolysis. Kaabel, et al., for instance, developed a moist-solid reactor system, whereby highly-crystalline, non-pre-treated PET flakes were added at a high (40%) solids loading to a reactor with enzyme and minimal buffer, which were then mixed in a ball mill, then incubated for 7 days at 55 °C54. They found high selectivity for TPA over MHET. In this process, TPA precipitates and no pH control is necessary. Overall, moist-solids reactions could offer a route to more environmentally benign enzymatic PET recycling16.
Enzyme immobilization has not seen widespread use with PET degradation, but could offer improved enzyme longevity by mitigating the effects of extreme temperature and pH and allowing enzymes to be reused16,55. Major concerns are limited substrate accessibility and mass transport to/from an immobilized enzyme and detrimental effects to the enzyme caused by conjugation. López-Teijeiro, et al., recently demonstrated the immobilization of a PET hydrolase in protein nanospheres, which allowed activity at a broader range of temperature and pH vs. the non-immobilized enzyme and reusability for up to 10 cycles, with little loss to enzyme activity56. Fritzsche, et al., attempted a suite of immobilization strategies–fixation to solid carriers, formation of cross-linked enzyme aggregates, and attachment to stimulus-responsive polymers–and found that linkage to a pH-responsive polymer resulted in recyclability over 5 cycles with meager decreases in activity57. Schwaminger, et al., additionally reported that the attachment of a PET hydrolase to an iron oxide nanoparticle through a His-tag allowed for magnetic separation and reuse over 10 cycles with up to 50% decrease in activity58.
Likewise, the use of continuous reactor schemes has not seen significant use in enzymatic PET recycling, although continuous reactors generally offer superior yields, process control, scalability, and throughput vs. batch reactors. For industrial enzymatic PET recycling, a continuous reaction scheme could remove reaction products that drop reaction pH and/or inhibit enzyme hydrolysis, which could improve efficiency of reactions and reduce base additions required for pH maintenance. Toward continuous PET depolymerization, Barth, et al., demonstrated that using ultrafiltration to remove inhibitory PET depolymerization reaction intermediates improved the hydrolysis of PET by 70% over 24 h compared to a standard batch reactor59. Recently, Ayafor, et al., employed a membrane reactor for in situ product removal and showed over 2-fold improvements in PET depolymerization and TPA yields. However, this depended upon using a high enough enzyme loading that would significantly drop reaction pH and, further, they demonstrated the need for optimization of how much the reaction is diluted60. As it is posited that at least some PET hydrolases first break down PET to small oligomers, then break down these soluble oligomers to monomers61,62, removing oligomers from the reaction before full conversion can decrease yields of TPA60.
Life-cycle and technoeconomic analyses of enzymatic PET recycling processes
Performing TEA, LCA, and socioeconomic impact analyses on their modeled enzymatic PET recycling process, Singh, et al., reported a minimum selling price (MSP) of $1.93/kg for recycled TPA (rTPA), compared to virgin TPA (vTPA), which fluctuated in 2010–2020 from ~$1-1.5/kg15. The key cost drivers for the process were feedstock cost and electricity, which was dominated by the pre-treatment steps for extrusion and cryo-grinding. Other major cost drivers included the costs of base and acid for hydrolysis reactor pH maintenance and TPA crystallization, as well as water/steam usage for EG recovery via distillation. Sensitivity analyses indicated that variability of the MSP of rTPA was most affected by solids loading and extent of conversion. Enzyme loading, enzyme cost, and reaction residence time were less impactful. In the most demonstrative case, a process without the need for pre-treatment steps would reduce the MSP by 12%. With that said, Singh, et al., reported the rTPA process would have up to an 83% lower energy use and produce up to 43% less greenhouse gases (GHGs) and an economy-wide assessment showed the process would generate 45% more socioeconomic benefits and reduce environmental impacts by 95%, all compared to vTPA.
A follow-up in-depth LCA by Uekert, et al., using an altered methodology and more comprehensive system boundaries and parameters, however, found that the modeled rTPA process and rPET process (involving the closed-loop manufacturing of PET using rTPA) were generally worse than their virgin counterparts (excepting fossil fuel use and ecotoxicity), with up to 17-fold higher impacts to analyzed natural environment, natural resource, and human health metrics16. Similar to the study by Singh, et al., major impact drivers were base consumption (for pH maintenance) and electricity, water, and acid use15,16, as well as collection and shredding of PET and discarding of waste (including unconverted PET)16. Sensitivity analyses indicated that variability of the impacts were most affected by solids loading, PET depolymerization extent, sorting yield, and TPA recovery, with enzyme loading, pH, residence time, and temperature having lesser effects15.
A recent study by Murphy, et al.63, motivated by the shortcomings of the rPET process identified by Singh, et al.15, and Uekert, et al.16, showed that several process changes to the original process modeled by Singh, et al.15, could decrease the MSP of rPET to $1.51, compared to a 5-year U.S. average of $1.87 for vPET. An LCA of the altered process also showed reductions in GHG emissions, ecotoxicity, fossil resources use, and water use compared to the original rPET process and vPET63. Namely, Murphy, et al., modified (and experimentally-validated) the process by: implementing pre-treatment to be extrusion followed by rapid quenching and palletization, rather than extrusion and cryo-milling; using a fed-batch reactor scheme, where enzyme and PET were periodically added to existing reactor liquor, to concentrate EG prior to distillation, enabling economical EG recovery; using mechanical vapor recompression in the distillation; and using NH4OH, rather than NaOH, for base additions, forming (NH4)2TPA, which can be thermally cracked to NH3 and TPA, eliminating the need for stoichiometric acid/base additions. The fed-batch scheme also caused the precipitation of TPA salts in the depolymerization reactor, allowing for easier separation63.
Engineering PET hydrolases
Design goals for PET hydrolase engineering
Most of the work in protein engineering toward enzymatic PET depolymerization has focused on PET hydrolases (Fig. 1a). Informed by process goals and challenges and lessons learned from TEA/LCA15,16 (vide supra), key engineering goals for PET hydrolases include: increased activity, thermostability, expression/solubility, and improved pH and substrate/product tolerance (Fig. 1b)17,19,31,35. Meeting these goals will allow for efficient depolymerization in reactions with high PET loadings to high extents, while minimizing enzyme and waste costs, which have been identified as cost and impact drivers of modeled processes. An ideal PET hydrolase would exhibit high thermostability, allowing for longevity in reactions and potential reuse, and could hydrolyze varied substrate types and forms, including semi-crystalline PET and un-treated/minimally-treated post-consumer PET, e.g., as shredded flakes. Resistance to product inhibition would increase depolymerization efficiency while also facilitating new process innovations, such as fed-batch reactions, while stability and activity at a range of pH, down to acidic pHs, could dramatically improve process costs by reducing the needs for pH maintenance.
Rational design strategies
A selection of the rationally-designed PET hydrolases discussed in this Review are shown in Table 1, while a structural depiction of the rational design strategies that have been used to engineer PET hydrolases is shown in Fig. 2a. Additionally, an overview of key PET hydrolase functional domains and residues is shown in Fig. 3. Regions are classified according to Richter, et al.50. Subsite I refers to residues that directly interact with the substrate and residues that comprise a second interaction sphere that influences the interacting residues. Subsite II is suggested to contribute to PET binding and guidance toward the active site50.

Strategies are selected from those discussed in this Review for engineering. a PET hydrolases and b MHET hydrolases. Modeled structures are shown only to be representative, with lines indicating approximate locations on the enzyme scaffold (or analogous location mapped from homologous scaffold) where mutation(s) would be made. The modeled PET hydrolase is LCC-ICCG (PDB: 6THT29) and the modeled MHET hydrolase is IsMHETase (PDB: 6QZ473). Color(s) of boxes and text indicate the design goal the mutation(s) aim to achieve; two colors indicate the mutation(s) may affect multiple properties.

a Annotated multiple sequence alignment (MSA) of IsPETase24, LCC25, and PHL726. In each panel, an MSA is shown for the 3 protein sequences performed by Clustal Omega157 on the EMBL-EBI Webserver. The 5 panels span the entire protein sequences, with residue numbers at the right. Above the MSAs are images showing the secondary structure, performed using ESPript 3.0158, from the crystal structures for IsPETase (PDB: 5XJH84), LCC (PDB: 4EB025), and PHL7 (PDB: 7NEI26). Nomenclature for the MSA is standard: * denotes identical residues, : denotes conserved residues, and . denotes semi-conserved residues. Images for secondary structure represent: α-helices as numbered α/squiggles, 310 helices as numbered η/squiggles, β-strands as numbered β/arrows, and β-turns as TT. Residues are colored as follows: orange for native cysteine residues (which are part of native disulfide bonds), green for residues corresponding to subsite I (classified by Richter, et al.50), cyan for residues corresponding to subsite II (classified by Richter, et al.50), red for the catalytic triad, blue for the extended loop region, and magenta for residues that could form disulfide bonds or salt bridges with other magenta residues, as discussed in the text. b Modeled structures of IsPETase (magenta), LCC (cyan), and PHL7 (green) overlaid and docked to the PET3mer. c A surface representation of PHL7 docked to the PET3mer, focusing on the substrate binding groove, with protein regions highlighted according to the colors scheme in (a) and residues labeled.
Economically viable PET hydrolases must be thermostable at high temperatures for long reaction times (Fig. 1b), but not at the expense of enzyme activity. Efforts have been made to increase enzyme thermostability of varied natural PET hydrolase scaffolds, with the goal to confer wild-type PET hydrolases with increases in melting temperature (Tm)17,19,27. A recommended guideline is the enzyme having a Tm that is at least 10 °C higher than the reaction temperature19. A rational design strategy has been used in several scaffolds successfully–based off early work in Thermobifida fusca (T. fusca) cutinase II (TfCut2), where Ca2+ binding sites were modified to install salt bridges64 or disulfide bonds65 (Fig. 2a)–to increase Tm by up to 25 °C. Analogous salt bridges and disulfide bonds have been created in homologous PET hydrolases based on existing metal binding sites, in Ideonella sakaiensis (IsPETase)41,52, leaf-branch compost cutinase (LCC)29,41, Polyester Hydrolase Leipzig #7 (PHL7)50,51, and others66,67. Son, et al., pursued structure-guided rational engineering to target a flexible, high B-factor region away from the active site, of IsPETase (Fig. 2a). The S121E/D186H mutations designed to stabilize the loops in the region were found to result in a Tm increase of over 7.2 °C and up to 6-fold higher activity than wild-type. The mutations R280A, S121E, D186H compose the mutant ThermoPETase68.
Fusion of polymer binding domains to PET hydrolases have been used as a means to increase enzyme activity, by increasing the rates of enzyme adsorbing to the hydrophobic polymer. This has been extensively reviewed by Wei, et al.19, and Tournier, et al.17. Natural and engineered cellulose-binding domains have been fused to the C-termini of PET hydrolases (Fig. 2a) for increased degradation activities69,70,71. However, at higher, more industrially-relevant PET solids loadings, in levels past the adsorption-limited regime, the beneficial effects of the cellulose-binding domain were observed to be diminished by Graham, et al.72. Thus, further exploration is necessary to determine if domain fusions are necessary to increase the activity of PET hydrolases in industrial settings. However, tethering of IsPETase to Ideonella sakaiensis MHETase (IsMHETase) to maintain the proximity of the enzyme active sites (Fig. 2a) showed significantly improved catalytic turnover vs. the simple addition of both enzymes separately to the same reaction73. Fusions with hydrophobins74,75 and zwitterionic peptides76 (Fig. 2a) are two additional methods shown to be successful in increasing substrate affinity.
Tuning enzyme surface properties has been proposed as an additional solution to enhancing PET hydrolysis, via improved association with the hydrophobic, noncharged polymer17,19. Creation of hydrophobic patches near the active site by, for example, truncating one of the termini (Fig. 2a), has been demonstrated successfully, even conferring observable activity when the wild-type enzyme showed none77. Concerning surface electrostatics, a study of two highly related PET hydrolases, Thc_Cut1 and Thc_Cut2, revealed that mutating a residue away from the active site from a charged to a noncharged residue (Fig. 2a) resulted in increased hydrolysis78. Considering that, during PET enzymatic hydrolysis, the polymer becomes locally negatively charged due to anionic carboxyl groups, the use of surfactants have been proposed as a way to mitigate charge repulsion between the polymer and enzyme79. However, from a purely protein engineering perspective, introduction of positively charged residues (Fig. 2a) can increase the binding constant of the enzyme to PET, as demonstrated in PET2, a thermostable enzyme found in a metagenomic library67.
Alteration of the binding cleft (Figs. 2a and 3) has been shown to affect the catalytic properties of PET hydrolases. In Fusarium solani cutinase (FsC), enlarging the active site (Fig. 2a) was shown to have 4- to 5-fold increases in activity on PET fibers compared to wild-type80. In IsPETase, conversely, structure-guided mutagenesis of residues to narrow the opening of the substrate binding site were shown to decrease activity81. However, narrowing of the binding cleft (Fig. 2a) also in IsPETase by introducing a set of mutations, S238F/W159H to mimic T. fusca cutinase, was shown by Austin, et al., to increase both catalytic activity and reduction in crystallinity of the substrate82. The reduction in crystallinity is presumed to be due to surface erosion and pitting of the plastic by the enzyme, which disrupts ordered crystalline structures, leading to a relative increase of amorphous regions82. Notwithstanding, this finding by Austin, et al., is perhaps contrary to the thought that a larger binding cleft may allow funneling of larger polymer chains, but was explained through more favored aromatic interactions of the substrate with F238, whereby the polymer is better positioned and distanced from the catalytic triad (Fig. 3c)82. Hence, activity could decrease if substrate affinity decreases at the expense of larger binding cleft. In LCC and TfCut2, there were likewise increases in activity from mutation of the residue analogous to S23829,83. Joo, et al. observed, in IsPETase, that polar R280, in the binding cleft, may inhibit large-chain substrate binding and saw that making the R280A mutation increased activity over 30% compared to the wild-type84. Similarly, in IsPETase, W185 can wobble, adopting multiple conformations to facilitate ligand-induced opening and widening of the substrate-binding groove, permitted by the small side chain of S21482,84,85. Mutation of this Ser to a His (i.e., S214H) dramatically decreased activity85–corroborated by the observation that the equivalent His residue in homologs packs against the Trp residue, preventing its rotation85. However, the equivalent residue to IsPETase S214, in PHL7, H185, when mutated to a serine (i.e., H185S) resulted in a dramatic decrease of protein stability and activity50. Mutation to an asparagine, meanwhile (i.e., H185N) resulted in increased activity53. Efforts to transplant the mechanism of the wobbling W185 in mesophilic IsPETase to thermophilic LCC and TfCut2 by mutation of the surrounding, smaller residues mimicking IsPETase S214/I218 (i.e., creating LCC-H218S/F222I) (Fig. 2a) showed that activity was increased at low temperatures, but not at higher temperatures86. With that said, transferal of the unique IsPETase S214/I218 to several other PET hydrolases which natively have His/Phe at the analogous sites were reported to enhance activity for those that function at less than 60 °C86. It is postulated that the unique Ser/Ile in heat-labile68 IsPETase contributes to the flexibility of the substrate-binding pocket, allowing improved binding to PET, although this flexibility may be detrimental at higher temperatures86. Taken together, this indicates that key residues, located proximal to the PET active site and suspected to play important roles in the mechanism of PET binding and catalysis, may not yet be optimized for PET degradation, and it may be difficult to map homologous residues across different scaffolds (notably, between mesophilic vs. thermophilic enzymes). Each enzyme scaffold may require its own specific tuning.
Additionally, there exist tradeoffs in altering the substrate binding cleft size and affinity. While a larger substrate binding cleft (Fig. 3c) may, for example, increase catalytic rates via improved PET substrate binding, it may also affect specificity, affinity, and/or stability. For example, the I224A mutation in Cut190*, attempting to improve substrate binding by affecting hydrophobicity, was shown to increase activity (higher Vmax and kcat) but decrease affinity (higher KM) toward the PET model substrate poly(butylene succinate-co-adipate) (PBSA)87. The I224A/Q138A double mutant showed a further increase in KM and a lower Vmax than the I224A single mutant, indicating potentially excessive space near the active site, at the detriment of activity and affinity. Molecular modeling showed that the I224A and Q138A mutants indeed had lower binding energies and increased binding surface areas compared to wild-type Cut190*87. Conversely, binding affinity that is too high can impede enzyme activity by the Sabatier principle: if the enzyme complexes too tightly to the PET interface and cannot dissociate, there is a cost to turnover88. Arnling Bååth, et al., found that LCC and TfC may bind PET too tightly for maximum activity and turnover could be increased by adding small amounts of surfactant. Presumably, mutations that enhance binding affinity could decrease activity; an intermediate binding affinity could be optimal88. On the other hand, enhanced activity and thermostability could be at the expense of one another. The Y87E mutation in IsPETase, which was made to a loop adjacent to the binding site, showed a 12 °C increase in Tm, but the mutant showed practically no enzyme activity, suggested to be due to unfavorable structural rigidity89. As discussed, increasing the flexibility of the binding cleft may decrease stability at high temperatures86. Increased affinity may more directly correlate with thermostability, as PET hydrolases are thought to be more stabilized upon substrate binding, as aromatic residues around the catalytic site associate with aromatic PET moieties84,85. Substrate selectivity could also be affected through altering substrate binding cleft size and affinity (vide infra).
Examining the binding of a model substrate to LCC using molecular docking and enzyme contact-surface analysis, Tournier, et al. identified 15 residues in the first contact shell, near the active site (Fig. 3a, c). Upon mutating 11 of the residues using site-saturation mutagenesis, it was found that most mutations either decreased activity, or did not increase it more than 48%29. However, several variants were discovered with increased PET depolymerization activity or increased Tm. Several of these mutations, T96M, Y127G, F243I, F243W (Fig. 3a), were combined with a rationally-designed disulfide bond, D238C/S283C (Fig. 3a), to create a handful of high performance PET hydrolases, including LCC-ICCG29. Other PET hydrolase scaffolds have been subjected to mutations of their analogous residues, showing significant increases in catalytic activity. For example, the PHL7-L93F/Q95Y variant, which showed over a 2-fold increase on PET substrates over wild-type, was created by mutation at these hotspots51, as have other PHL7 variants50, to resemble LCC-ICCG29 or DuraPETase90 (which is discussed below). Such mutagenesis is typically a good starting point for improving activity, especially considering that PET hydrolases generally share high structural and sequence homology (Fig. 3b) and minimal knowledge of enzyme structure is required. In a recent study, Zeng, et al., used the structure of LCC-ICCG co-crystallized with a PET analog to guide the engineering of hydrophilic interactions on the protein surface and internal hydrophobic interactions to yield LCC-RIP, increasing Tm to nearly 99 °C and activity by up to 100% vs. LCC-ICCG in reactions with reinforced PET over a week at 85 °C91. However, it is questionable whether reaction at such high temperatures is advantageous; highly-active enzymes would be required to outcompete presumably rapid re-crystallization at 85 °C.
Product (i.e., MHET, BHET, TPA, EG) accumulation has been shown to inhibit depolymerization in PET hydrolase scaffolds including IsPETase92, TfCut293, FsC94, and PHL753 and will likely impede industrial use of these enzymes31 (Fig. 1b). Rational design of the substrate binding site to lower affinity of breakdown products has been proposed as one method to relieve inhibition. Wei, et al., saw up to a 2.7-fold increase in activity over wild-type by exchanging binding site residues in TfCut2 with those of LCC, whereby the G62A mutation was made. From molecular modeling, it was found that the G62A decreased the binding strength interaction (Fig. 2a) between the residue and the PET dimer by 5.5-fold95. Creation of tethered chimeras of PET hydrolase with an enzyme able to efficiently break down intermediates has been shown to increase depolymerization greater than the two enzymes alone (Fig. 2a), as discussed73,96, potentially, in part, due to relieved substrate inhibition via rapid elimination of inhibitory intermediates. In another study, Avilan, et al.97, discovered that the IsPETase W159H/S238F variant created by Austin, et al.82, showed relieved product inhibition compared to wild-type, with the suggestion that these mutations may stabilize a key loop, and thereby reduce the flexibility, where the active site (catalytic) histidine is located (Fig. 3a). The IsPETase HotPETase variant52 shares this relieved product inhibition, potentially due to fewer cavities and decreased flexibility near the active site97. Hence, Avilan, et al., argued that increasing enzyme thermostability may also help to relieve product inhibition97.
Emerging computational design approaches
Computationally-aided design has recently made significant headways into improving PET hydrolases toward a variety of design targets. Here, we aim to highlight several recent methods that function by a range of diverse approaches. A selection of the computationally designed PET hydrolases discussed in this Review are shown in Table 1 and an overview of the strategies is shown in Fig. 4.

Any sequences, mutations, or structures are only for demonstration and are not specifically taken from the study in question. a GRAPE90 method, which could feature a language model with a Transformer encoder prior to the GRAPE workflow102. b MutCompute41. The representative structure is LCC-ICCG (PDB: 6THT29). c MDL105. The representative structure is LCC-ICCG (PDB: 6THT29). d ADD106. The representative structure is the PET 3mer docked to PHL7 (PDB: 7NEI26). e Premuse107. The WebLogo was based on ref. 159. f Optimal temperature ML and evolutionary analysis algorithms110. g CAM111. The representative structures are from LCC-ICCG29 and PHL726 (overlaid) and PHL7 docked to the PET 3mer. h Landscape profiling method112.
Cui, et al., developed a methodology termed GRAPE90 (GReedy Accumulated strategy for Protein Engineering) toward the goal of discovering the most efficient paths of accumulated mutations–namely, that avoid uncooperative epistatic effects, while identifying coupled synergistic effects (Fig. 4a). First, mutations predicted to be stabilizing in IsPETase were generated in silico (from ABACUS98, FoldX99, Rosetta_ddg100, and Skylign32101) which were then down-selected to avoid any observed biophysical pitfalls (e.g., mutations creating hydrophobic surface residues). Potential candidates were then experimentally examined and mutations of variants that expressed well were clustered according to their positions and presumed effects, reducing the total number of combinations. A workflow then tested and accumulated mutations in each cluster before adding the mutations in other clusters, systematically combining mutations until the final variant, DuraPETase was discovered90. DuraPETase had a 31 °C higher Tm and over 300-fold higher activity compared to wild-type on high-crystallinity PET powder90. In a second study, Cui, et al., discovered TurboPETase102 (based on the BhrPETase scaffold103) by first employing an artificial intelligence (AI) based language model with a Transformer encoder trained on datasets of homologous PET hydrolase sequences to predict amino acid variation probability across the evolutionary landscape. Positions that the wild-type residues fit less well than substitutions were identified, mutated, and experimentally tested. After finding that some accumulated mutations beneficial for activity had decreased Tm, they employed GRAPE. TurboPETase achieved nearly complete depolymerization of 20% (w/w) PET in 8 h102.
Lu, et al., developed a self-supervised, structure-based machine learning (ML) 3D convolutional neural network, MutCompute (Fig. 4b)41. MutCompute was trained on 19,000 structures from the Protein Data Bank (PDB) to identify stabilizing mutations in the IsPETase/ThermoPETase scaffold based on those wild-type residues not optimized for their microenvironments. Predictions were ranked by predicted probabilities and mutations were combined stepwise, with variants that showed higher activity and thermostability explored further. After identifying four mutations which showed the highest improvements (S121E, T140D, R224Q, and N233K), they experimentally tested the combinations to discover the best variant, FAST-PETase (S121E, R224Q, and N233K)41. FAST-PETase had up to 38-fold higher activity compared to ThermoPETase41. Meng, et al., later employed MutCompute to identify residues in TfCut2 to increase hydrolytic activity and stability104. Stepwise recombination and verification gave new enzyme variants with 2.9-fold and 5.3-fold improvements in activity on amorphous PET film and crystalline PET powder, respectively, and a Tm increase of 5.7 °C compared to the wild-type104.
Qi and colleagues used ML methods to mine molecular dynamics (MD) trajectories in a method they termed MDL (Fig. 4c) to engineer TfCut2105. They used MD to simulate the structural changes in the enzyme at reaction conditions, coupled with ML, to analyze relationships between the enzymes’ characteristics and their thermostability. After verifying that the MD simulations provided sufficient structural features for ML algorithm input–from negative and positive data–and the regression results performed well, they predicted variants with improved thermostability. Their TfCut2 variant had a Tm that was 9.3 °C higher and an activity that was increased by over 45-fold compared to wild-type105. They then extended this idea to develop a PET ligand affinity analysis based on dynamic docking (termed ADD) workflow (Fig. 4d) to improve the activity of LCC-ICCG over 3 rounds of engineering. Their top variants were named LCC-A2 and LCC-A3106. ADD involves analyzing the ligand affinity energy of enzymes associating with PET chains by molecular docking and examining dynamic protein conformations to determine hotspot residues for mutation. A hotspot residue would have longer duration of binding to the substrate under dynamic conformational sampling106.
The Premuse tool was designed to integrate sequence alignments and quantitative selection of consensus-preferred mutations based on natural sequence evolution (Fig. 4e)107. Using pairwise sequence alignments and calculated position-specific amino acid probabilities, the IsPETase W159H/F229Y variant was identified, which was conferred with over a 10 °C increase in Tm compared to the wild-type enzyme107. In another attempt to engineer LCC-ICCG, Ding, et al., pursued two strategies using an ML optimal temperature prediction tool, Preoptem108, combined with co-evolutionary sequence analysis, EVcouplings109, (Fig. 4f) to create LCCICCG_I6M, which showed 3.6-fold higher activity than LCC-ICCG on high-crystallinity, untreated PET110. Zheng, et al., improved the activity of LCC-ICCG by 2.1-fold to create LCC-YGA by using a cross-correlation-based accumulated mutagenesis (CAM) strategy (Fig. 4g)111. With the goal to mitigate epistatic effects in combinatorial mutation sets, they devised a strategy to first explore single mutations via remodeling of the binding pocket and incorporating mutations from homologs, then exploring combinations of beneficial mutations, before finally accumulating additional mutations with little dynamic correlation. In this way, accumulated mutations can increase activity until nonadditive effects are seen; then, additional mutations with low cross-correlation are added. Mutations with low-cross correlation can mitigate epistatic effects, as they can rescue conflicting dynamics between protein residues111.
In a recent study, Seo, et al., developed a landscape profiling method (Fig. 4h) to determine those enzymes with the highest fitness for PET degradation activity and thermostability112. First, putative, natural sequences across the polyesterase-lipase-cutinase families were collected and clustered into related neighborhoods. Then, representative nodes (sequences) from clusters were sampled for fitness on the basis of activity (in terms of monomer release from bottle-derived PET powder at 30 °C), Tm, and production yield. This indicated which clusters had the highest fitness. Finally, high-fitness clusters were thoroughly sampled to identify high-fitness nodes112. Analytical testing identified two high-fitness nodes (sequences/enzymes), Mipa-P and Kubu-P. Following discovery of these two enzymes, cross-template engineering was performed, whereby rationally-designed mutations were chosen for the two scaffolds by alignment to homologous high-fitness nodes. This yielded the final variants, Mipa-PM19 and Kubu-PM12. Notably, Kubu-PM12 exhibited significantly enhanced PET degradation activity compared to LCC-ICCG at high PET loadings (20% and 30% (w/w)) and increased tolerance to reactions in ethylene glycol112.
Collectively, computational approaches show promise in significantly enhancing PET hydrolase activity for industrial use. Recent methods have demonstrated successfully engineering PET hydrolases with improved thermostability and/or activity, as has been common with laboratory-based rational design, although some recent studies have begun considering additional goals, including protein production112, as additional selection criteria. Other goals, such as function at a range of pH, may be amenable future targets for computational design. Gado, et al., recently reported EpHod, a tool to predict the optimal pH of an enzyme113. EpHod showcases language model embeddings and optimized hyperparameters for training, allowing it to learn complex structural and biophysical features–such as proximity of residues from the catalytic center and accessibility of solvent–from sequence data113. Tools such as EpHod may be impactful when used to complement other discussed methods–i.e., those targeting traditional activity and thermostability goals. Further, these tools span a range of principal approaches, including: systematic workflows to accumulate functional mutations90,102,111; AI/ML methods to determine amino acid variation across evolutionary landscapes102, analyze MD simulations105, design for optimal temperature110, and optimize residues’ structural microenvironments41,104; analyze dynamic binding of ligand to enzyme via docking simulations106; sequence-based consensus design and co-evolutionary analysis107,110; and thorough sequence fitness landscape profiling for enzyme identification and engineering112.
Screening methods for PET hydrolase engineering: from rational to semi-rational design
Past screening methods for PET hydrolase engineering have varied significantly in scale and scope—e.g., from assays meant to characterize single rational design mutants (Fig. 5a), to microtiter plate assays sufficient to screen semi-rational design site-saturation libraries (Fig. 5a) over several days. Weight loss of polymer before vs. after reaction (gravimetric analysis)26,51,64 and release of fluorogenic probes from polymer structures114,115 were some of the first screening assays developed—and have been employed to characterize ~10 enzyme variants at a time (Fig. 5b).

a Rational design, semi-rational design, and directed evolution are the three key laboratory approaches to engineering PET hydrolases. However, a screening approach to evaluate the library associated with the method is required. b Screening methods used to evaluate PET hydrolase libraries.
Microtiter plate-based screens (Figs. 5b and 6a) have been used to indirectly measure reaction products (or characteristic byproducts), enabling the efficient screening of up to 103 enzyme variants at a time. Belisário-Ferrari, et al., developed a plate-based assay to measure turbidity changes from breakdown of the 2PET model substrate in suspensions116. Lusty Beech, et al., developed an assay to measure activity with colorimetric readout by detecting pH changes from the H+ released by PET hydrolysis117. Arnling Bååth, et al.118, and Zhong-Johnson, et al.119, developed plate-based screens to measure aggregate aromatic breakdown products from enzymatic PET hydrolysis via UV absorbance readout. Work by Wei and colleagues demonstrated use of reaction products with hydroxyl radicals to generate the fluorophore 2-hydroxyterephthalic acid to quantify TPA in microtiter plates120,121. Similarly, Shi, et al., demonstrated using the hydrolysis of the model substrate bis (2-hydroxyethyl) 2-hydroxyterephthalate (BHET-OH), which PET hydrolases can convert to the fluorescent 2-hydroxyterephthalate (TPA-OH), to measure activity (Fig. 6a). They then screened error-prone PCR libraries of IsPETase over 3 rounds of directed evolution to yield DepoPETase, which had an over 1,400-fold increase in PET hydrolysis products compared to wild-type at 50 °C and a 23.3 °C higher Tm122. Others have used direct product quantification using ultra high performance liquid chromatography (UHPLC) (Fig. 5b)27,52. Using a high-throughput reaction setup and HPLC screening workflow in microtiter plates, Bell, et al., significantly improved the activity and thermostability of IsPETase through targeted, semi-rational directed evolution (Fig. 6b)52. Screening approximately 2,000 variants per round over 6 rounds of increasing selection pressures, mutational hotspots from literature were site-saturated, and mutations from the best variants from each round (based on improved monomer product release) were combined to finally discover HotPETase. HotPETase showed a Tm increase of 37.5 °C and approximately a 3.5-fold increase in activity over ThermoPETase52,68.

a Microtiter plate-based assays. The overview is based on the screen by Shi, et al., to engineer DepoPETase122, but similar workflows could be applied to using absorbance (change in turbidity)116, color (change in pH)117, or UV absorbance (aromatic product release)118,119 as readouts. b UHPLC screening52. c FADS129. Note: the authors did not use the screen to engineer enzymes, but the screen could be adapted to screen enzymes using directed evolution similarly to the other methods. A similar workflow could be used with a TPA biosensor132, where the cells would need to be encapsulated for the reaction and the TPA would need to be imported. d Yeast display coupled with FACS133. e Coupled BHET and split-GFP screens on agar plates35,53,135.
High-throughput screens to enable directed evolution of PET hydrolases
Until recently, PET hydrolase engineering focused exclusively on rational and semi-rational design17,19, (Fig. 5a) in which, single mutations (~101 variants) and small mutant libraries (<103 variants) were constructed based upon structural inspection, homology, domain swapping, and modeling. While directed evolution is a powerful technique for engineering enzymes (and other proteins) with improved and/or new functions123,124, it has lagged behind rational design for engineering PET hydrolases. The key limitation has been in the lack of high-throughput screening assays that can effectively evaluate the large enzyme libraries required for directed evolution by random mutagenesis (Fig. 5b)19. Indeed, directed evolution is able to better probe a protein’s sequence space–in scale and scope–for rare, beneficial, and surprising mutations than more constrained libraries123, (Fig. 5a) and thus future advances in PET hydrolase engineering may be enabled or accelerated via directed evolution. Rational design may be insufficient to, for example, enhance multiple catalytic steps simultaneously19, account for epistatic effects and incompatible mutations, and requires extensive structural information and a rational basis for engineering. Now, screening assays have been developed that can evaluate 105 or more variants in several days (Fig. 5b). A selection of the PET hydrolases engineered using high-throughput screening, as well as the semi-rationally designed PET hydrolases, discussed in this Review are shown in Table 1 and an overview of screening assays used to evaluate libraries for semi-rational design and random mutagenesis are shown in Fig. 6.
Agar plate-based screening methods are one possibility to increasing the screening throughput to 105 variants at a time (Fig. 5b). Substrates can be embedded in agar plates, with visible signals indicating breakdown of the substrate. PET nanoparticle suspensions have been used in several studies, where halos, or clearing zones, indicate that the variant can hydrolyze the PET125,126,127,128. However, the need for high hydrolytic activity can render identification of improved enzyme variants at the colony-level difficult on PET substrates.
Fluorescence-activated droplet sorting (FADS) approaches129 offer one high-throughput alternative (Fig. 5b). Qiao, et al., developed an ultra-high-throughput method where bacteria expressing PET hydrolases were enclosed in droplets, then the droplets were injected with the model substrate fluorescein dibenzoate (FDBz) (Fig. 6c). Breakdown of FDBz by a PET hydrolase creates fluorescent products, which allows the droplets to be sorted and bacteria to be isolated. Recent advances demonstrate that biosensors could provide for ultra-high-throughput screening approaches, as well130,131,132. Pardo, et al., developed a TPA-responsive biosensor, which can sense and respond to levels of TPA to express green fluorescent protein (GFP). Fluorescent activated cell sorting (FACS) of libraries of cells encapsulated with extracellularly-expressed PET hydrolases that can then import the TPA (as demonstrated in the work) could rapidly screen 108 variants or greater at a time132.
A recent report by Cribari, et al., developed a novel, ultra-high-throughput screening platform for PET hydrolase engineering using yeast display with FACS (Fig. 5b)133. On cell surfaces tagged with probes comprised of an aromatic ester and biotin, a PET hydrolase was expressed (Fig. 6d). In this screening assay, if a PET hydrolase was able to efficiently cleave the aromatic ester, then biotin, which is later detected with a fluorescent streptavidin conjugate, was present in lower amounts. Hence, cells expressing a high-activity PET hydrolase will exhibit low fluorescence, which can be isolated with FACS. This screening assay could process 107 variants or greater. From 3 rounds of sorting of error-prone PCR libraries, the screen discovered LCC variants with enriched mutations at H218, with H218Y being the exemplary variant. LCC-ICCG-H218Y exhibited 2.6-fold higher activity compared to LCC-ICCG. The authors postulated that mutation at H218 could improve substrate binding, as H218 is adjacent to W190 (Fig. 3a), which helps form part of the substrate binding pocket. W190 and the W185 analog in IsPETase are, as mentioned, critical for activity82,84,85,133,134. While the authors screened 27 million variants in their work, however, a downside is that the screening must be performed at ambient temperature, where yeast grows, so it is not yet optimized to screen for thermostable variants. The same issue would present with the FADS approach by Qiao, et al., using bacteria in microdroplets129. An ideal screening assay for PET hydrolases should screen for improved variants with a selection pressure mandating activity near the Topt of PET, about 70 °C.
In two other recent works, Groseclose and colleagues developed a high-throughput coupled plate-based screening assay (Fig. 5b) to identify improved PET hydrolases from directed evolution (Fig. 6e). The assay can evaluate variants simultaneously for activity, thermostability, and protein solubility/expression35,53,135. In their screening, libraries of Escherichia coli (E. coli) containing variants of PET hydrolases (105 or greater at a time) with C-terminal GFP11 tags136,137,138 were grown on semi-permeable membranes. The libraries were then induced to express the enzyme by moving the membrane to a plate with isopropyl β-d-thiogalactopyranoside (IPTG). The membrane was then again moved to an agar plate containing the BHET model substrate, where the cells were partially lysed, and the membrane was removed. The BHET plate was then incubated at the reaction temperature (~70 °C). Appearances of halos, or clearing zones, indicated enzyme variants active on BHET. Following this, the GFP11 tag on the enzymes was complemented by incubation with GFP1-10 solution on the plates, showing green fluorescent signal based on the amount of enzymes from the lysed colony (indicating solubility/expression). Thermostability was reported to act as a selection pressure via heat treatment prior to reaction, or reaction at high temperature.
Following the co-screening assay, potential hits were validated by small-scale expression and reaction with PET substrates in solution, measuring aromatic products in throughput of ~102 with UV absorbance screens118,119. After rounds of directed evolution, with libraries diversified from random mutation and shuffled from previous, validated hits, final enzymes were expressed, purified, and tested in vitro. An advantage of the assay is that it allows normalization of activity to solubility/expression through the high-throughput screening steps, without laborious, low-throughput expression, purification, and quantification. Further, it uses only widely-accessible reagents and equipment and is adaptable to a variety of selection pressures (e.g., pH, temperature, PET substrate type)135. Groseclose and colleagues used the high-throughput screening platform to engineer LCC35 and PHL753 using directed evolution, with evolved LCC-LANL exhibiting 11% higher conversion after 48 h in a bioreactor with 16.5% amorphous PET loading vs. LCC-ICCG35 and evolved PHL7-Jemez exhibiting 270% higher conversion after 48 h in a bioreactor with 20% amorphous PET loading vs. PHL7-WT53. Final enzyme variants had a diversity of mutations, at least 3 amino acid mutations each, after at least 3 rounds of directed evolution, with increasing stringency of thermostability and activity selection pressures35,53.
Considerations for PET hydrolase screening
A key consideration in high-throughput screening is the evaluation metric. That is, the criteria for which an enzyme variant is chosen from the library to be characterized further or proceed to additional rounds of directed evolution. This is typically a lesser consideration with rational design, as the pools of variants are significantly smaller and most/all of the variants are pursued through thorough screening and characterization. The evaluation metric could also relate to how many rounds of directed evolution are undertaken: How improved must potential enzyme variants be before they are declared final? Commonly, performances of PET hydrolases engineered in the lab are reported in terms of fold change for activity (e.g., 3-fold higher products released by a certain time) and change in Tm temperature for thermostability (e.g., +3 °C). Evaluation on PET is commonly performed in small-scale (<1500 µL), either in test tubes or 96-well plates, using commercially-available (i.e., from Goodfellow) PET substrates, in sheet or powder forms, of different percent crystallinities (from ~9% sheets35, product ES301445, to ~42% powders139, product ES306031), at <10% solids loading34–but typically lower, from 0.4%52 to 2.9%27,35,53. Toward improved transparency and reproducibility in this rapidly-growing, global field of research, it is critical for researchers to report standardized details of their assessment methods, ultimately permitting fair comparison between enzymes and groups34. This includes source, property, and treatment parameters of PET34,139, as well as enzyme characteristics and reaction conditions: notably, enzyme and substrate loading34. Mass and/or concentration of enzyme and substrate should be disclosed, not simply volume of enzyme or piece or size of substrate, as has been common in previous studies (Table 1).
Often the key evaluation metric for PET hydrolase screening is increased depolymerization activity–i.e., of a set PET substrate type and loading–under the relevant reaction conditions–i.e., at a set temperature and pH–at a certain endpoint–e.g., 48 h. However, it may be desired to also select enzyme variants that display some interesting, emergent property depending on engineering goals, not merely those variants that have higher fold changes in depolymerization activity (in terms of higher UV absorbance35,118,119 or monomer release via HPLC52) vs. the starting point enzyme. For illustration, a researcher may choose to investigate a variant with a high initial rate (within 4 h reaction) that plateaus (after 12 h) or a variant that exhibits lower, but sustained activity (over 24 h and beyond). These variants could provide breakthrough insights into engineering faster or more temperature- and pH-stable enzymes. Even if these variants may not be optimized themselves, combining these interesting mutations with others (e.g., using DNA shuffling140) can provide paths to enzymes that simultaneously achieve many identified engineering goals (Fig. 1b). This also motivates potential advantages of taking different measurements to allow for data-rich decision-making. Rather than merely measuring end-point activity at 48 h, reactions can be measured over time, or parallel reactions can be set up at high temperature or low pH to use more extreme conditions as selection pressures. Ultimately, evaluation metrics may vary somewhat depending on specific engineering goals and the number of enzyme variants a researcher or team can reasonably handle.
To provide a sense of throughput and evaluation metrics, we will focus on two recent screening methods developing high-performance PET hydrolases: Bell, et al., using high-throughput UHPLC screening52 (Fig. 6b) to develop HotPETase52 and Groseclose, et al., using a high-throughput plate-based screening assay35,135 (Fig. 6e) to develop PHL7-Jemez53. Bell, et al. performed 6 rounds total of evolution. Each round, 1,930 to 2,000 variants were screened and 3 to 13 best variants (bearing single mutations) were selected. Within rounds, up to 3 identified mutations were shuffled together. Selected variants had 1.13- to 2.96-fold increases in activity compared to their parents at reaction conditions of increasing pressure (in terms of increasing heat treatment temperature, reaction temperature and time, and decreasing enzyme volume). In each round, ~3% of the top hits were assessed as purified proteins, and in Rounds 5 and 6, the top 3% of variants were selected to be tested on crystalline PET (only amorphous PET used in Rounds 1–4). As mentioned above, the final HotPETase variant showed an approximate 3.5-fold increase in activity over ThermoPETase52. Groseclose, et al. performed 4 rounds total of evolution. Each round, approximately 20,000 variants were screened on BHET agar plates, of which roughly 25 to 50 were selected for screening on PET. In Rounds 1–3, 8 to 14 variants were selected in reactions of increasing pressure (in terms of increasing reaction time and substrate loading, with increasing BHET concentration and higher/longer heat treatment temperatures used in the high-throughput screening stage), yielding variants with 1.03- to 12.74-fold increases in activity and up to 3.51-fold higher expression vs. PHL7-WT. The final four enzyme variants from Round 4 had 2.0- to 3.2-fold higher activities and 1.7- to 3.1-fold higher expression levels compared to PHL7-WT. Groseclose, et al., stopped evolution after Round 4 as screened enzyme variants were achieving approximately 2-fold higher activities and expression levels under relevant reaction conditions compared to the starting parent. The exemplary variant, PHL7-Jemez, showed approximately a 4-fold higher product release compared to PHL7-WT in small-scale reactions. As a heuristic, Bell, et al., and Groseclose, et al., were selecting the top 0.05% to 0.5% of their variants in their successful directed evolution campaigns.
In comparison, the large-scale, computationally-guided rational design study by Cui, et al., began with 253 mutations predicted to be stabilizing, which were down-selected to 85 to avoid biophysical pitfalls90. These 85 mutations were further filtered to 21 variants (mutations) that expressed and increased stability ( ≥ 1.5 °C increase in Tm). Then, in each of 10 rounds of shuffling, 4 to 12 new variants were made by creating combinations of single mutations with the best parent from the previous round. One best variant was selected per round (based off of degradation performance), but mutations that were not selected in a round were allowed to appear in later rounds. In each round, degradation performance was increased by up to 2-fold, with 65 total combination mutants tested. The final DuraPETase mutant, as mentioned, had a 31 °C higher Tm and over 300-fold higher activity compared to IsPETase90.
Engineering BHET and MHET hydrolases
Design goals for MHET and BHET hydrolases
While PET hydrolases appear to exhibit promiscuous activity to hydrolyze the intermediate products BHET and MHET, MHET hydrolases and, recently, BHET hydrolases, have been shown to increase the efficiency of PET depolymerization reactions when coupled with PET hydrolases (Fig. 1a)141,142.
An ideal process scheme would involve the simultaneous hydrolysis of all PET-derived substrates to TPA and EG in one pot. One advantage of MHET and BHET hydrolases is that they exhibit significantly higher turnover on their small-molecule, partially-soluble MHET and BHET substrates compared to PET hydrolases on PET. Additionally, due to their small-molecule substrates, MHET and BHET do not possess many of the properties that could inhibit PET hydrolase activity, such as crystallinity, limited interfacial substrate surface area, and a heterogeneous interface. However, to date, no MHET or BHET hydrolase has been discovered that can survive at the optimal temperatures of PET reactions, ~70 °C. Hence, a critical design goal for these enzymes is increasing their thermostability so they can be used in tandem reactions near the Topt of PET hydrolases (Fig. 1b).
Moreover, MHET hydrolases express in significantly lower amounts than PET hydrolases–and its low expression may currently hinder industrial application143. Although concentrations of MHET hydrolases in reactions could likely be permitted to be lower compared to PET hydrolases due to their increased turnover, the expression of these enzymes must be significantly increased to improve enzyme cost and process economics (Fig. 1b). This could be mitigated with improved enzyme activity, a perennial design goal for industrial enzymes.
Engineering MHET hydrolases
Palm, et al., were the first to determine the structure of IsMHETase, and established the foundation for MHET hydrolase (Fig. 1a) rational engineering. In general, they determined that IsMHETase and IsPETase are structurally conserved (Fig. 2), except for a large lid domain in IsMHETase that confers high activity on MHET73,144. They determined that IsMHETase specificity is highly dictated by contact between the substrate and lid domain, including critical residue R411, which coordinates with the carboxylic groups, and, further, that substrate binding is through an induced-fit mechanism facilitated by F415144. Palm, et al., then conducted mutagenesis of the active site residues, discovering abolishment of activity for several residues by mutation to alanine and a W397A variant that caused increased activity at high substrate concentrations, but lowered substrate affinity (decreasing the KM). Toward improved BHET hydrolysis by IsMHETase, Palm, et al., further discovered that the active site mutations S416A and S419G retained MHET activity while also allowing for BHET activity (Fig. 2b), possibly due to the increased flexibility of R411 to bind BHET. The F424Q and F424N mutations, which potentially confer more space within the active site and provide for hydrogen bonding partners, also increase BHET activity. Eliminating the positive charge of R411, either by mutating to alanine or glutamine, increased BHET activity, as well. As discussed with PET hydrolases, it should be acknowledged that altering the selectivity of an MHET hydrolase, for example by widening the active site binding cleft to better accommodate the BHET substrate, has potential tradeoffs: activity, affinity, and/or thermostability may be affected. A structural depiction of the rational design strategies that have been used to engineer MHET hydrolases is shown in Fig. 2b.
Knott, et al., further investigated active site and structural features of IsMHETase to confer high MHET activity. Motivated by sequence alignments with proteins across the tannase family, mutations were made to active site residues. While IsPETase has a serine at position 131, 91% of tannase family sequences have a glycine at the analogous position. However, they observed activity of the S131G mutant was decreased by 97% compared to wild-type73. Similarly, the F495I mutation, to mimic that of other tannase family members, showed significantly decreased activity, as did E226T. Collectively, as witnessed by mutagenesis of PET hydrolases, mutation of residues around the active site selected to enhance activity may not be outright predictable based on computational design or homology mapping. Knott, et al., additionally confirmed the necessity of the lid domain for MHETase activity. Addition of the IsMHETase lid domain to IsPETase conferred a small level of activity on MHET and the deletion of the lid domain from IsMHETase resulted in 1000-fold less activity than wild-type73.
Inspired by the work by Palm, et al., Sagong, et al., sought to increase the BHET hydrolysis activity of IsMHETase145. To affect BHET binding, they created an R411K mutant, aiming to allow more beneficial hydrogen bonding vs. R411, when BHET is the substrate (Fig. 2b). They also created F424N/H/D/E/T/V/L/I (mutation to smaller residues) mutants to enlarge the substrate-binding pocket to better accommodate the BHET substrate (Fig. 2b), as suggested by Palm, et al. The R411K mutant showed 1.7-fold higher activity on BHET than wild-type and the F424 mutants showed up to 3.9-fold higher activity (for F424N), and, further, combined mutants R411K/F424N, R411K/F424V, and R411K/F424I showed up to 11.1-fold higher activity. The authors also determined that the mutants had decreased activity for MHET, while several, including R411K/F424I and R411K/F424V, had higher activity against the PET5 oligomer. A summary of the engineered MHET hydrolases (specifically, IsMHETase) discussed in this Review is shown in Table 2.
Discovery and engineering of BHET hydrolases
Li, et al., discovered two BHET hydrolases (Fig. 1a), BsEst and ChryBHETase, via enzyme mining from environmental landfill samples and screening with stepwise straining with diethyl terephthalate (DET), PET powder, and MHET substrates141. Modeling of these two enzymes’ structures with substrates showed solvation of the substrate binding cleft, but no presence of BHET. Using a mechanism-guided approach, potential barriers to BHET access were removed, truncating identified regions with flexible glycine linkers. Following in silico scoring, two top variants, ΔBsEst and ΔChryBHETase, were identified. ΔBsEst hydrolyzed 100% of BHET in 3 h, a 2-fold improvement over wild-type, and ΔChryBHETase hydrolyzed 100% in 3 h less than its wild-type. MD simulations verified that the increased flexibility of the removed barriers in the enzymes allowed improved BHET substrate access. The BHET hydrolases additionally were conferred with increased thermostablity, presumably via creation of more favorable physiochemical properties including internal H-bonds and SASA. A summary of the engineered BHET hydrolases discussed is shown in Table 3.
Miao, et al., recently discovered an enzyme from the UniParc database they termed BMHETase for its ability to hydrolyze both MHET and BHET142. It was reported to have higher activity on BHET than both LCC-ICCG and FAST-PETase at temperatures of 50 to 70 °C. Rational engineering to mimic the residues in IsMHETase, to increase MHET activity, resulted in a 6 point-mutation variant that had 8-fold higher activity on MHET, 2-fold higher activity on BHET, and a Topt increased by 5 °C. It was posited that this was due to improved interactions with the MHET substrate and a larger substrate binding pocket. TfCa, a carboxylesterase from T. fusca, also has activity on both MHET and BHET, as well as a cyclic PET oligomer146,147. After determination of TfCa’s structure in complex with an MHET substrate analog, von Haugwitz, et al., conducted alanine scanning and site-saturation mutagenesis of identified residues to discover conferred improved activities on BHET by the R428A and V376A mutations, which may allow for more bulky substrate binding, and the I69W mutation, which may better interact with aromatic moieties on the substrate (Fig. 2a)148. A summary of the engineered MHET/BHET hydrolases discussed is shown in Table 3.
Screening methods for MHET and BHET hydrolase engineering
Given the prospect of MHET and BHET hydrolases to enhance the efficiency of enzymatic PET recycling processes and the growth of discoveries of novel MHET and BHET hydrolase scaffolds–such as the MHET hydrolase Mle046149, the BHET hydrolase ScLipA150, and those enzymes discussed in the previous sections–engineering of MHET and BHET hydrolases could lead to future improvements in enzymatic PET recycling. Just as with PET hydrolases, we anticipate that coupling rational design, computational design, and directed evolution approaches will prove to be successful in developing new MHET and BHET hydrolases.
With that said, the development of screening assays enabling directed evolution of MHET and BHET hydrolases has been scarce. Currently, the use of HPLC (Fig. 5b) in screening MHET and BHET hydrolase activity widely prevails over other methods73,141,142,144. However, many of the high-throughput screening assays developed for PET hydrolases could feasibly be co-opted for use with MHET and BHET hydrolases. Lusty Beech, et al., for example, designed their colorimetric microtiter plate assay to detect PET degradation through protons released by BHET and MHET hydrolysis (Fig. 5b)117.
One key consideration is that MHET hydrolases have high specificity toward MHET and little activity toward substrate analogs typically used for PET hydrolase engineering, such as p-nitrophenols144. As such, screening assays for MHET hydrolases should use MHET as the substrate to be most efficient. Recently, Saunders, et al., reported a medium-throughput screening assay for MHET hydrolases, whereby the enzymes are first screened on the model substrate 1-napthyl terephthalate (1NT). If the MHET hydrolase exhibits activity toward this substrate, 1-napthol and TPA are produced. Addition of the Fast Blue B Salt, which reacts with the 1-napthol, will produce a brown azo dye, which can be detected with absorbance at 610 nm143. Saunders, et al., then used consensus design of IsMHETase, where the most common (consensus) amino acids were introduced at various positions of the protein, which has been shown to improve protein stability, folding, and expression143,151. They saw an approximate order of magnitude increase in soluble expression, postulated to be due to improved protein folding. The variants did have reduced activity on MHET143, likely in part due to screening on 1NT.
Altering the model substrates used in PET hydrolase screening assays to MHET or BHET can give a direct route to screen for MHET and BHET hydrolases, provided that the assays can specifically detect the desired product. For this reason, using UV absorbance based screening, in which there would be high background from the soluble substrate, would cause it to be difficult to detect product species. However, the use of the screening assay developed by Shi, et al.122, (Fig. 6a) to screen for hydrolysis of BHET-OH to TPA-OH, could likely be employed to discover high activity BHET hydrolases. The TPA biosensor developed by Pardo, et al.132, could putatively screen for BHET or MHET activity, if there is minimal promiscuity of the biosensor to detect larger PET subunits. Likewise, the yeast-display screening assay pioneered by Cribari, et al.133, (Fig. 6d) could be used to detect BHET and MHET hydrolases in ultra-high throughput, as the model substrate to detect ester bond cleavage is universal across PET, BHET, and MHET. Additionally, agar plate based screening (Fig. 3b) could be adapted toward MHET and BHET hydrolase directed evolution, whereby the plates could be cast with MHET or BHET, rather than PET nanoparticles. By detecting halos on the plates, enzyme variants could be effectively identified. Adaptation of the platform by Groseclose, et al.35, (Fig. 6e) either by using the BHET agar plates disclosed, or instead using MHET as the substrate, could then additionally couple protein expression/solubility and thermostability screening to activity screening.
The evaluation criteria for MHET and BHET hydrolases will be generally similar to that for PET hydrolases (see above). That is, for rational design and small semi-rational design campaigns (<100 variants total), all variants will be thoroughly tested and characterized, with the best variants chosen to be reported. Multiple mutations can be combined (maintaining <100 variants screened), with engineering continuing until all combinations have been attempted or no further improvements are observed90. For larger libraries–large semi-rational design campaigns and random mutagenesis libraries for directed evolution (numbering >100 variants, and preferably 103 and greater)–the top 0.05%53 to 0.5%52 of variants should be chosen. However, this could depend on the ability to handle and characterize more (or less) variants and, as with PET hydrolases, we recommend selecting variants with any interesting properties for further study, even if they do not outright have higher activity under the measured conditions.
Concluding Remarks
This Review summarizes and discusses current state-of-the-art and emerging approaches for engineering enzymes involved in enzymatic PET degradation–namely, PET, MHET, and BHET hydrolases. The goal is to bridge i) lessons learned in the last ~10 years of rapidly-growing PET-degrading enzyme discovery and engineering–which has focused on PET hydrolases–with ii) next-generation directed evolution/high-throughput screening and computational design approaches, along with iii) the development of other classes of PET-degrading enzymes, MHET and BHET hydrolases. We motivate these engineering efforts with lessons learned from recent process modeling, technoeconomic, and life cycle analyses, to further bridge ongoing laboratory-based testing with considerations for scaleup and industrial deployment, as enzymatic PET recycling matures as a technology. Achieving these analysis-driven design goals will be critical to improving the competitiveness of enzymatic PET recycling vs. other PET recycling strategies.
In some cases, i.e., with clean, high-quality PET, mechanical recycling is the most energy-efficient, low-cost, and least environmentally-impacting recycling method (Table 4)12. However, there is some PET waste that should not be recycled mechanically and is currently landfilled12,152,153. These are materials such as thermoformed containers (for salads, berries, etc.), colored bottles, and textiles. Further, there is a limit on the number of times a material can be mechanically recycled11. Hence, in a future PET economy, there can be a niche for chemical recycling (Table 4). For example, enzymatic (hydrolytic) recycling is more agnostic to mixed and contaminated waste streams compared to mechanical recycling15,16. With that said, the chemical recycling approach that should be pursued will depend on technical constraints (e.g., feedstock quality) and process priorities (e.g., cost, environmental impact, quality of resin)12. Advanced chemical recycling technologies are active areas of scientific research and, as such, process innovations could significantly affect future decision-making13. A summary of the PET recycling strategies discussed in this Review are shown in Table 4.
Several challenges currently experienced by enzymatic PET recycling center around the PET pre-treatment process, base addition for pH maintenance in depolymerization reactions, and difficulty in EG/water distillation, leading to high costs (MSPs), energy and water use, emissions, and toxicity/waste (Table 4)15,16. While Murphy, et al., recently proposed key process improvements to mitigate these challenges and make the cost and impacts of rPET better relative to vPET63, opportunities remain in process and enzyme engineering to strengthen the industrial feasibility of enzymatic PET recycling on an industrial scale.
Toward industrially-relevant design goals for PET hydrolase enzyme engineering (Fig. 1b), researchers have traditionally relied upon rational design (Table 5) and screening of several variants at a time to confer improvements in properties such as: thermostability, via creation of disulfide bonds and salt bridges29,41,52,64,65,66,67; activity, via enhanced substrate binding29,67,80,83,84; and product inhibition, via tethering of a PET hydrolase and an MHET hydrolase73,96. More recently, the development of higher throughput screens has facilitated the screening of larger, semi-rational design libraries (Table 5), allowing researchers to improve activity by targeting residues surrounding active sites29 and mutational hotspots52 identified previously with a range of amino acid substitutions. Now, varied computational approaches (Table 5)–which may be guided by structure41,104, sequence107,110,112, or simulations105,106–are emerging to rapidly design potential enzymes prior to any laboratory testing. Likewise, as high-throughput screening assays (Table 5) are at last developed for PET hydrolase engineering35,53,129,133, directed evolution is now possible35,53,133, enabling the screening of large enzyme libraries to discover rare, highly-optimized enzyme variants. A summary of the enzyme engineering strategies discussed in this Review is shown in Table 5. In parallel, primarily rational73,144,145 and semi-rational148 design approaches have been used to increase improve the MHET and BHET hydrolysis activities of MHET and BHET hydrolases, although recent advances (i.e., in computational design and high-throughput screening) and lessons learned in PET hydrolase engineering could accelerate the engineering of these other classes of enzymes.
Outlook
Directed evolution efforts, either by large, semi-rational design libraries or random mutagenesis libraries, have lagged behind rational design efforts due to the lack of high-throughput screening methods that can operate near the optimal temperature of most efficient PET depolymerization reactions, ~70 °C. However, recent advances in the development of high-throughput screening assays, such as microtiter plate screens52,122, surface display technologies133, and coupled plate-based assays35,53,135, have successfully demonstrated the directed evolution on PET hydrolases (Fig. 5). Anticipated future developments, such as in ultra-high-throughput biosensor screening132, will accelerate PET hydrolase engineering further. Future advances in protein engineering of PET-degrading enzymes should also complement emerging process innovations. While the engineering goals discussed here (Fig. 1b) will be valuable for reducing costs of industrial enzymatic PET recycling processes, for instance, Murphy, et al., suggest that enzyme longevity, tolerance to EG, and ability to depolymerize alternative forms of pre-processed PET will be most impactful in their modeled process63. We posit these forward-looking engineering goals can be approached using strategies adapted from those discussed here.
As new PET hydrolases are discovered, especially those with interesting native features, such as preference for high-crystallinity substrates27–allowing them to serve as better footholds for engineering–we envision a workflow whereby rational design and semi-rational design would be the first focus. Here, site-saturation mutagenesis in proximity to the active site and mutation of other hotspot residues can be easily explored, via homology mapping of these highly-conserved enzymes based on literature, by structural inspection guided by previous successes, or computational approaches (Fig. 3). Directed evolution strategies can then be employed with these first-generation evolved PET hydrolases as templates, whereby larger sequence spaces can be explored to engineer the enzymes toward improved catalytic activity under specific selection pressures. Next-generation selection pressures should include screening for properties currently underexplored with PET hydrolases, but these could be aided by existing work with similar enzymes. Huang, et al., for example, engineered a lipase using error-prone PCR for high pH tolerance154. Identified hotspots through directed evolution may then be used in future rounds of rational or semi-rational design, with mutations fed into ML models, that would further accelerate discovery of high-performance PET hydrolases.
Toward the application of AI and ML in engineering new PET-degrading enzymes (Fig. 4), large quantities of high-quality sequence/function relationship data will be extremely critical in training and building models155,156. While both positive and negative enzyme performance data would be informative for training AI/ML models, current studies with PET-degrading enzymes focus primarily on mutations that improve the performance of the final enzyme variants. To increase the transparency of future studies and facilitate the success of forthcoming AI/ML assisted protein design, we encourage the community to openly present the results of their mutational studies including both positive and negative effects of single, double, triple, etc., mutations on the performance of enzyme variants. This type of data will become increasingly rich and valuable as future high-throughput screening and directed evolution studies generate large amounts of random and combinatorial mutation data for diverse enzyme scaffolds, although a wealth of data currently exists from dozens of studies on engineering these enzymes. A selection of mutational data is shown in Table 6, where several studies have identified single mutation and combinations of mutations that increase/decrease enzyme activity and/or thermostability. Other metrics, such as expression/solubility, may be beneficial to be considered as well. The high-throughput screening assay recently developed by Groseclose and colleagues is well-suited to acquire such multi-dimensional datasets, where one can examine multiple enzyme properties including hydrolytic activities, thermostability and expression/solubility levels without the need for enzyme purification35,53,135.
>With that said, this data should be gathered and reported using standardized protocols. To ensure reproducibility across research groups and the fair comparison of evolved enzymes, the complete details of reaction conditions and parameters must be disclosed. Arnal, et al.31, and Wei, et al.34 have recently proposed protocols and considerations to better standardize PET hydrolase testing. Proposals by Arnal, et al., suggest using a standardized substrate (cryo-milled Goodfellow amorphous powder), a fixed enzyme to substrate ratio, and fixed substrate loading for bioreactor experiments, with small-scale (laboratory) experiments measured by TPA formed after a certain time and activity reported as specific rates (i.e., gTPA,eq h-1 mgenzyme-1)31. Wei, et al. suggest reporting a range of polymer and enzyme properties and reaction parameters34.
Emerging methods in high-throughput screening and computational design may have the power to alleviate some bottlenecks in PET-degrading enzyme engineering. The additive (epistatic) effects of mutations in a protein can be difficult to discern and require trial and error, which may be intractable using low-throughput techniques for large mutational spaces. This is additionally convoluted as some mutations may decrease enzyme activity while increasing thermostability or vice versa (see Table 6). However, some combinations of mutations may be synergistic, while others are incompatible. Screening using high-throughput experimental and computational methods can enable the efficient testing of combinatorial mutation spaces and could help avoid rationally-designed dead-ends.
For instance, Cui, et al.90, in their development of DuraPETase, extensively experimentally tested the thermostability and activity of 85 single mutants, which were then combinatorially combined in several rounds. While P181A was identified as a potential beneficial mutation, it was discovered that this variant had increased thermostability, but decreased activity, and this mutation was not propagated in later rounds (Table 6). Son, et al.68 had previously rationally designed P181A to fix a disruption in a β-sheet and also found an increase in stability, but postulated that this came with a collapse of the active site. Interestingly, Bell, et al., discovered P181V in their final HotPETase variant52. Cui, et al., additionally discovered synergistic and incompatible mutations in their engineering90. While S188Q behaved synergistically with the IsPETase W159H/I168R/S214H mutation combination, they saw that while I168R/S214H and D186H/S214H improved activity and thermostability compared to IsPETase wild-type, the addition of D186H to I168R/S214H resulted in a lower-performing variant. Similarly, Tournier, et al.29, discovered that some single mutations in LCC, such as F243I and Y127G, displayed an improvement to either thermostability or activity (Table 6). However, upon combination of the mutations (along with a rationally-designed disulfide bond), synergistic effects gave highly improved enzymes such as D238C/F243I/N246M/S283C (LCC-ICCM) and Y127G/D238C/F243I/S283C (LCC-ICCG)29. Other combinations of the mutations, though, such as D238C/F243I/N246D/S283C, showed incompatibilities, resulting in reduced activity and stability (Table 6). In the PHL7 scaffold, it is notable that Pfaff, et al., discovered the PES-H1-L92F/Q94Y51 mutant by combination of the mutations L93F and Q95Y also identified by Richter, et al.50 as improving activity and thermostability (Table 6).
Although work on BHET and MHET hydrolases has been outnumbered by that for PET hydrolases, we anticipate that these enzymes will become increasingly important in industrial PET recycling. Fortunately, some of the structural and methodological insights gained from PET hydrolases seem to be able to be extended to BHET and MHET hydrolases (Fig. 2), including that many of the high-throughput screening assays for PET hydrolases can be adapted for these enzymes. This should enable the rapid engineering of BHET and MHET hydrolases toward industrial desired goals.
Similarly, adapting existing high-throughput screening assays for PET polymer and subunit hydrolysis to other plastic polymers targeted for enzymatic recycling (with hydrolyzable polymer backbones), such as poly(urethane) and nylon, will enable the rapid screening and engineering of enzymes degrading these polymers. Altering the model substrates in those high-throughput screening assays discussed here to include amide or urethane bonds will be the first step to development of new screening assays for these enzymes. Hence, as enzyme cocktails become developed that can break down mixed, post-consumer plastic waste streams in one pot reactions, a complete biorefinery may be developed where mixed plastic can be recycled, or valorized, enabling an alternative end-of-life strategy for plastics that may be more feasible than current, conventional approaches.