
Introduction
Brucellosis, a globally prevalent zoonotic disease caused by Brucella spp.1, can be transmitted to humans via contact with infected livestock or their products2, and has been listed by the World Health Organization (WHO) as a priority zoonotic disease for prevention and control3.
According to the latest epidemiological data, the incidence of this disease in regions such as the Middle East4 and Central Asia accounts for over 45% of the global total5,6, with a large number of reported cases annually in Inner Mongolia7 Xinjiang and other regions of China8.The incidence shows an upward trend year by year, posing a dual threat to public health security and the development of animal husbandry.
For livestock, Brucella can cause up to 60% abortion in pregnant females, leading to over $20 billion in annual global livestock economic losses9,10. In humans, symptoms include long-lasting fever (2–3 months on average) and joint pain. About 15% of patients develop neurological or cardiovascular issues, and the recurrence rate after antibiotic treatment remains at 10%-15%, showing the need for urgent prevention11,12.
At present, the prevention and control methods of brucellosis have obvious limitations: in terms of treatment, although the combination of doxycycline and rifampicin is the first-line option, the course of treatment is as long as 6–8 weeks13, and the detection rate of drug-resistant strains has increased from 3% in 2010 to 9% in 202414. In terms of vaccines, existing veterinary vaccines (such as S19 strain, RB51 strain) have biosafety risks for humans, while the research and development of human vaccines is still in the blank stage. Against this background, the design strategy of reverse vaccinology combined with bioinformatics has become an important direction for the research and development of new Brucella vaccines, relying on advantages such as efficient screening of antigenic epitopes and reducing virulence risks15,16.
In recent years, the research on Brucella subunit vaccines has made significant progress, with many related articles published, providing new ideas for the prevention and control of infectious diseases17,18. These subunit-based approaches, including MEVs, have been explored for their ability to accurately integrate multiple immunologically active epitopes while avoiding the side effects of whole-bacteria vaccines. Previous studies have confirmed that MEVs designed with bacterial membrane proteins as targets can achieve specific clearance of pathogens by activating humoral and cellular immune responses19,20. In the early stage of this study, through bioinformatics tools, dominant epitopes were screened from three membrane proteins of Brucella (CcmA, ccmC, BepC), and an MEV containing adjuvant HMGN1 (alarmin, which can promote dendritic cell maturation) and universal epitope PADRE (enhancing helper T cell activation) was constructed. The antigenicity (score 1.2542) and safety (non-toxicity, non-allergenicity) of the MEV were verified by tools such as ProtParam and VaxiJen.
However, the transformation of in silico prediction results to experimental verification still faces multiple challenges: first, the recombinant MEV expressed in vitro may have reduced epitope exposure efficiency due to changes in folding mode, affecting the actual immunogenicity; second, the immune response mode of animal models (such as mice and guinea pigs) is different from that of humans, and cross-validation is required to evaluate the vaccine protection efficacy; third, the binding stability between MEV and host receptors such as TLR4 and MHC needs to be further verified through protein interaction experiments, rather than relying solely on molecular docking scores21,22,23.
Collectively, the findings of this study suggest that the selected proteins and the constructed multi-epitope vaccines hold promise as effective targets for the development of a universal vaccine. Nevertheless, the functional efficacy of this candidate vaccine still warrants further experimental validation to confirm its practical application value.
Materials and methods
Target protein selection
First, we used the UniProt database to search and obtain protein sequences24. The sequences of these proteins were saved in FASTA format to facilitate subsequent analysis and research. Then, when selecting proteins to ensure that the proteins have strong antigenicity,we used VaxiJen v2.0 (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) -an antigen prediction software to evaluate the antigenicity of proteins by setting the threshold to 0.425. Also, as a candidate protein for vaccines, it is important to ensure that the protein is non-allergenic and non-toxic AllergenFP v.1.1(https://ddg-pharmfac.net/AllergenFP/) does a good job of analyzing whether a protein is an allergen26. ToxinPred2 (https://webs.iiitd.edu.in/raghava/toxinpred2/index.html) can be used to predict the toxicity of related proteins27.The physical and chemical properties of the target proteins, including amino acid number, isoelectric point, instability index, aliphatic index, and GRAVY, were evaluated using the ProtParam tool (https://web.expasy.org/protparam/), which provides a comprehensive analysis of proteins28.The sequences of Heme Exporter Protein C (ccmC) (UniProt ID: Q8YEM7), CcmA (UniProt ID: Q8YEM5), and BepC (UniProt ID: Q8G0Y6) were obtained from UniProt.
Prediction of protein signal peptides
Signal peptides (SPs) are short amino acid sequences that control protein secretion and translocation and contribute to the cotranslational translocation of nascent polypeptides into the endoplasmic reticulum29. When analyzing protein structure and predicting epitopes, signal peptide sequences should be removed first, because removing signal peptides prevents proteins from being mis-targeted to the endoplasmic reticulum or other organelles, and can reduce the likelihood of folding and localization errors that can affect the accuracy and reliability of subsequent epitope prediction. Therefore, we decided to use SignalP 6.0 (https://services.healthtech.dtu.dk/services/SignalP-6.0/) to predict the signal peptide30,31.
Prediction of protein epitopes
Prediction of T-cell epitopes
Recognition of antigenic peptides presented by MHC class I and MHC class II molecules classifies T cells into cytotoxic T cells (CD8 + T cells) and helper T cells (CD4 + T cells). This also categorizes T cell epitopes into cytotoxic T lymphocyte (CTL) epitopes and helper T lymphocyte (HTL) epitopes32. To efficiently predict T-cell epitopes of the target proteins, we used the high-frequency alleles HLA-A0201, HLA-A0301, and HLA-A1101 for CTL epitope prediction in the Xinjiang region of China, and HLA-DRB1*0301, HLA-DRB1*0701, and HLA-DRB1*1501 for HTL epitope prediction33.
For CTL epitope prediction, we used EpiJen34 (https://ddg-pharmfac.net/epijen/EpiJen/EpiJen.htm),NetMHCpan-4.135 (https://services.healthtech.dtu.dk/services/NetMHCpan-4.1/) with a peptide length of 9. We compared the prediction results of both tools and selected the high scoring epitopes (the likelihood of a peptide being an MHC ligand) for further analysis. For MHC-II binding prediction, we used NetMHCIIpan-4.3 (https://services.healthtech.dtu.dk/services/NetMHCIIpan-4.3/) with a peptide length of 15 and default parameters36,37.We then selected high scoring epitopes for further analysis.We filtered epitopes using the %Rank score provided by the software. For class I, the thresholds were %Rank < 0.5% for strong binders (SB) and %Rank < 2% for weak binders (WB). For class II, the thresholds were %Rank < 2% for SB and %Rank < 10% for WB36.
Prediction of linear epitopes and conformational epitopes in B-cells
B cell epitopes, as part of the antigen recognition region, can bind to immunoglobulin molecules to stimulate B cell responses and thereby elicit humoral immunity38. Structurally, B cell epitopes can be divided into linear B cell epitopes (LBEs, continuous epitopes) and conformational B cell epitopes (CBEs, non-linear or discontinuous epitopes).Two prediction servers were used to predict linear B cell epitopes: SVMtrip (http://sysbio.unl.edu/SVMTriP/prediction.php)39 and ABCPred (https://webs.iiitd.edu.in/raghava/abcpred/ABC_submission.html)40.The peptide length was set to 16,while all other settings were left at their default values. By performing a comprehensive analysis and comparing the prediction results of both servers, we selected peptides with higher scores (> 0.8). This approach can reduce false positives and increase the reliability of the predictions. It is well known that approximately 90% of B cell epitopes (BCEs) are discontinuous41. In the analysis of B cellconformational epitopes, we used the ElliPro tool (https://tools.iedb.org/ellipro/)42and maintained the default threshold throughout the analysis to predict B cell conformational epitopes (CBEs), which are formed by folding proteins to bring distant residues closer together. Identification of B-cell epitopes is critical for vaccine design; by recognizing and exploiting B-cell epitopes, more effective vaccines can be designed with reduced immunogenicity and side effects43.
Epitope selection
In the epitope analysis described above, we did not consider the signal peptide structure of the proteins. The various T-cell and B-cell epitopes obtained were then evaluated for antigenicity, allergenicity, and toxicity using VaxiJen V2.0 (> 0.4), AllergenFP v.1.1 and ToxinPred2 (default threshold), respectively. Through this comprehensive analysis, we selected peptide sequences that exhibited strong immunogenicity, high antigenicity, non-allergenicity, and non-toxicity. These were ultimately selected as the cell-dominant epitope.
Docking of T-cell Epitopes and HLA alleles
Molecular docking was performed using the HDOCK server (http://hdock.phys.hust.edu.cn/) selected HLA class I (HLA-A02:01) and HLA class II (HLA-DRB1*0701) alleles for molecular docking with CTL and HTL epitopes, respectively, to analyze the interactions between the alleles and T-cell epitopes44.
Construction of MEV
In the construction of MEV, the selected CTL, HTL, LBE, and CBE epitopes were linked together using appropriate linkers. Appropriate linkers can connect amino acid residues to each other, thus avoiding many adverse outcomes, such as misfolding of fusion proteins, low protein production, and impaired biological activity45. In the design of multi-epitope vaccines, the selection and application of adjuvants is one of the key factors in enhancing immune responses. HMGN1 is an alarmin molecule that can mediate dendritic cell maturation through TLR4 and induce antigen-specific immune responses in the presence of antigens46. The adjuvant HMGN1 was linked to the pan-HLA-DR epitope (PADRE) sequence via a rigid linker EAAAK47, which activates helper T cells and thereby enhances the immunogenicity of the multi-epitope vaccine. The PADRE sequence was then linked to the CTL epitope using the stable and commonly used flexible linker GGGS. Meanwhile, HTL epitopes were linked via GPGPG and all B cell epitopes were linked via the double lysine (KK) linker to increase their flexibility and antigen presentation efficiency48. A polyhistidine tag, such as a His tag, was added to the C-terminus to facilitate recognition and capture of the vaccine during subsequent expression and purification processes49.
Evaluation of MEVs
The physical and chemical properties of multi-epitope vaccines (MEVs), including molecular formula, amino acid number, isoelectric point (pI), instability index, and generalized average of hydrophobicity (GRAVY), can be rapidly predicted using the ProtParam tool. These predictions are important for understanding the stability, solubility, and behavior of MEV in different environments and help in further experimental design and optimization. The antigenicity of MEV was then predicted using VaxiJen 2.0 with the threshold set to 0.4. To ensure that the designed vaccine would not cause cross-reactivity leading to sensitization, the sensitization of MEV was evaluated using AllergenFP v.1.1. Good solubility not only helps MEV express normal function, but also improves the efficiency and yield of MEV purification, optimizes protein engineering, and increases the bioavailability and stability of MEV.
Therefore, we used SOLpro(https://scratch.proteomics.ics.uci.edu/) to predict the solubility of the engineered MEV when overexpressed in E. coli50. ToxinPred was also used to determine the potential toxicity of MEV.
To ensure that the multi-epitope vaccine (MEV) does not induce an autoimmune response, we used the BLASTP tool (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastp&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome) provided by NCBI to align theMEV sequence with the entire human proteome.This comparison.
allows us to determine whether the MEV sequence is homologousto human proteins. If the comparison results show that the e-value of MEV is less than 0.005 and the homology to human proteins does not exceed 35%, MEV can be considered non-human homologous, thus reducing the risk of triggering an autoimmune response51.
Secondary and tertiary structure prediction of the vaccine
The secondary structure of MEV was analyzed using the online software SOMPA(https://npsa.lyon.inserm.fr/cgi-bin/npsa_automat.pl?page=npsa_sopma.html)52, which includes α-helix, β-sheet, β-turn, and random coil. The tertiary structure was predicted using the Robetta website (https://robetta.bakerlab.org/). Subsequently, the quality and validation of the tertiary structure model of the vaccine were evaluated using the UCLA-DOELAB-SAVES v6.1 (https://saves.mbi.ucla.edu/)53 and,PDBsum(https://www.ebi.ac.uk/thorntonsrv/databases/pdbsum/Generate.html) servers. The geometric conformation of amino acid residues was analyzed using Z-scores and Ramachandran plots (< 2% outliers in disallowed regions)to evaluate the protein structure.
The GalaxyWEB (http://galaxy.seoklab.org/)54,55 server was used to refine and optimize the tertiary structure. The optimized tertiary structure was then re-evaluated using UCLA-DOE LAB—SAVES v6.1 and PDBsum.
Molecular docking
Molecular docking can be used to predict the structure and stability of protein complexes and to elucidate the mechanisms of protein–protein interactions. To understand the affinity between the vaccine protein and the immune cell receptors, we selected the HDOCK server to perform docking of the optimized MEV with the immune cell receptors TLR4 and MHC-I56. The tertiary structures of both immune cells can be found in the PDB. The best model was then selected from the top ten models based on the docking scores. Additionally57, PyMOL was used to visualize the docking results.
Molecular dynamics simulation
Molecular dynamics simulations were performed using GROMACS 2024.258.The protein was modeled using the amber14sb_parmbsc1.ff force field and the TIP3P59 water model. A box boundary was set at a distance of 1.0 nm from the system atoms.The simulation was conducted at 298.15 K,1 atmosphere pressure, and physiological salt concentration.The process began with energy minimization, setting emtol = 100.0 kJ mol⁻1 nm⁻1. Subsequently, restrained dynamics were applied to the protein atoms for 300 ps with a time step of 1 fs. The formal simulation time was set to 100 ns with a time step of 2 fs. During the simulation, Coulomb interactions were handled using the PME method with a cutoff radius of 1.0 nm. Van der Waals (vdW) interactions were treated with a cutoff method, setting the vdW cutoff radius to 1.0 nm. The pressure coupling algorithm was set to Parrinello-Rahman. GROMACS built-in tools were used to calculate the ligand’s RMSD, root mean square fluctuation (RMSF), and radius of gyration (RG).
Using this server, we simulated the TLR4 protein and its complex with MEV (TLR4-MEV) to gain a deeper understanding of how MEV binding affects the structure and stability of the TLR4 protein.
Immune stimulation
Immunostimulation simulation allows the simulation of the immune system’s response to foreign antigens (e.g. vaccine components) on an online tool to predict the immunogenicity and protective efficacy of a vaccine prior to experimentation. This approach can significantly reduce the time and cost of vaccine development. It is an essential step in vaccine development.
We use C-ImmSim (https://kraken.iac.rm.cnr.it/C-IMMSIM/), a simulation tool that simultaneously simulates three separate anatomical regions of the mammalian bone marrow, thymus and lymph nodes60. This server is used to describe the process by which the immune system recognizes and responds to pathogens. Since the minimum clinically recommended interval between two vaccine doses is four weeks, the simulation time for each region was 1, 84, and 168 time units, respectively. We focused on and selected high-frequency alleles such as HLA-A1101(13.46%),HLA-A0201(12.50%), HLA-B5101(8.17%), HLA-B3501(6.73%),HLA-DRB1*0701(16.35%),HLA-DRB1*1501 (8.65%) for analysis in the Xinjiang population. During the simulation process, all parameters were used with the default settings of the software: random seed was 12345, simulation volume was 50, and simulation step size was 105061.
Codon optimization and molecular cloning
To improve the translation efficiency and expression levels of the protein and to facilitate successful cloning, we used the online codon optimization tool ExpOptimizer to optimize the codons of the vaccine construct (MEV) for improved expression efficiency in Escherichia coli. During the optimization process, two restriction enzyme sites (XhoI and BamHI) were excluded to avoid potential interference in subsequent cloning steps. The optimized MEV DNA sequence was evaluated using the codon adaptation index (CAI)62 and GC content. A CAI score of 1 indicates that the codons of the target gene are perfectly matched to the high-frequency codons of the host63. In addition, since GC pairs are linked by three hydrogen bonds, the GC content directly affects the binding stability of the DNA sequence and the annealing temperature. Both excessively high and low GC content can lead to overly stable or unstable mRNA secondary structures, thereby affecting translation efficiency. A GC content between 30 and 70% is considered an ideal optimization result64.
The MEV gene was successfully cloned using the pET-28a( +) vector as the expression carrier. The MEV DNA sequence was subsequently imported into SnapGene 8.0.1 for primer design, which adhered to the following principles: primer length of 15 to 30 base pairs (bp), Tm value of 60 °C, and GC content between 40 and 60%. These parameters were meticulously designed to ensure the specificity and amplification efficiency of the primers, while circumventing the formation of primer dimers or hairpin structures.Subsequently, an XhoI restriction enzyme site was incorporated at the 5′ end of the forward primer, and a BamHI restriction enzyme site was added to the 5′ end of the reverse primer.The MEV DNA sequence was then amplified using SnapGene 8.0.1. The MCS region of the pET-28a( +) vector was then analyzed to identify suitable XhoI and BamHI restriction enzyme sites, after which the amplified MEV target gene sequence was inserted into these sites, thus completing the cloning process.
Agarose gel electrophoresis of MEV
To predict experimental outcomes in advance, avoid operational risks, and optimize experimental design, this study employed SnapGene 8.0.1 software to simulate the 1% agarose gel electrophoresis process, focusing on the electrophoretic behavior of PCR products (MEV target gene), pET-28a( +) empty vector, and recombinant plasmid (pET-28a( +)-MEV).
Results
Selection of target proteins
The antigenicity of the candidate proteins was initially assessed using VaxiJen v2.0, with all results surpassing the threshold of 0.4, demonstrating robust antigenic potential. Subsequently, ProtParam was employed to compute key physicochemical properties of the target proteins, including molecular weight, theoretical isoelectric point (pI), grand average of hydropathy (GRAVY), and instability index. Furthermore, analyses conducted with AlgPred 2.0 and ToxinPred2 confirmed that the selected proteins are devoid of allergenic and toxic properties. Detailed results are summarized in (Table 1).These results indicate that the selected proteins can serve as immunogenic candidates for further research.The antigenicity of MEV, at 1.2542, is significantly higher than that of previously reported Brucella multiepitope vaccines.
Prediction of signal peptides
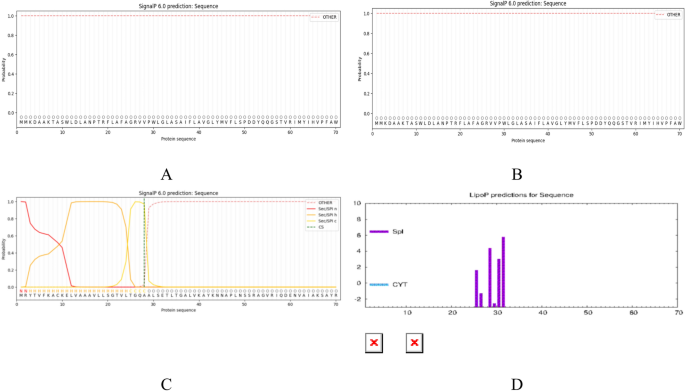
The signal peptides of the three proteins were predicted using SignalP6.0 and LiPOP1.0. The results of these two prediction tools revealed the absence of a signal peptide for both proteins ccmC (Fig. 1A) and CcmA (Fig. 1B). Conversely, the prediction results of BepC indicated the presence of a signal peptide, which might be located within the range of amino acid residues at positions 1–32 (Fig. 1C).LiPOP1.0 suggested that 25–31 peptides might function as signal peptides (Fig. 1D). Ultimately, peptide sequences that were entirely devoid of signal peptides were selected by combining the results of the two software tools, thereby enhancing the accuracy and reliability of the subsequent epitope prediction.

(A–D) SignalP 6.0 and LiPOP1.0 provides predictions regarding the signal peptide of the target protein. (A) Presents the prediction results for the CcmC signal peptide. (B) Presents the prediction results for the ccmA signal peptide, (C) Displaying BepC signalling peptide predictions in SignalP 6.0. (D) Provides the prediction results for the BepC signal peptide in LiPOP1.0.
Prediction of protein epitopes
T-cell epitope prediction results
The prediction of cytotoxic T lymphocyte (CTL) epitopes was accomplished through the utilization of a combination of EpiJen and NetMHCpan-4.1, while the identification of helper T lymphocyte (HTL) epitopes was achieved by employing EpiJen and NetMHCIIpan-4.3. Candidate epitopes with higher prediction scores were selected and subsequently evaluated for antigenicity, allergenicity, and toxicity using VaxiJen 2.0, AllergenFP v1.0, and ToxinPred, respectively. This comprehensive analysis ensured that the selected epitopes exhibited optimal antigenic properties, lacked allergenic potential, and were non-toxic. Following this rigorous screening process, 11 CTL epitopes and 9 HTL epitopes were chosen for the design of a multi-epitope vaccine. The final dominant CTL and HTL epitopes are summarized in Tables 2, 3, respectively.
Prediction of B-cell epitopes
The linear B-cell epitopes of the target proteins were identified and analyzed by combining SVMTriP and ABCpred software. This was followed by analysis and screening for epitopes that were antigenic, non-sensitizing, and non-toxic. The final selection comprised nine B-cell linear epitopes, as outlined in (Table 4).
Prediction of B-cell Conformational EpitopesThe Ellipro online server was utilized to predict discontinuous B-cell epitopes of the three target proteins. Following a comprehensive evaluation, two conformational epitopes were identified in CcmC in (Table 5).
Structural association analysis and docking evaluation of HLA alleles and T-cell epitopes
Figure 2A illustrates the docking results of HLA-A*02:01 with CTL epitope with a docking score of -272.45, confidence level of 0.9205 and RMSD value of 58.60. Figure 2 B depicts the docking results of HLA-DRB1*0701 with HTL epitope with a docking score of -240.69, confidence level of 0.8598, and RMSD value of 91.99. These results indicate that the molecular docking complexes have strong binding affinity.

The results of the study are as follows: T-cell epitopes and HLA alleles were analyzed. (A) Demonstrates the outcomes of the docking process for HLA alleles and CTL, (B) Presents the results of the docking process for HLA alleles and HTL.
Construction of MEV
After selecting high-quality CTL/HTL/B-cell epitopes, we assembled them into a multi-epitope vaccine (MEV) using linkers .As illustrated in Fig. 3, epitopes were linked together using suitable linkers.

Epitopes are connected using appropriate linkers to construct the MEV.
Comprehensive characterization of MEV
The physicochemical properties of MEV were evaluated using ProtParam, revealing that MEV comprises 638 amino acids with a molecular formula of C2912H4682N858O883S₁₃, a molecular weight of 66,257.21 Da, and a theoretical isoelectric point (pI) of 9.97. The instability index (II) of MEV was determined to be 26.80, below the stability threshold of 40, indicating high structural stability. The GRAVY value of -0.524 classifies MEV as a hydrophilic protein. Furthermore, VaxiJen 2.0 predicted the antigenicity of MEV to be 1.2542, well above the threshold of 0.4, demonstrating robust antigenic potential. AllergenFP v1.0 classified MEV as “likely non-allergenic,” suggesting negligible cross-reactivity in vivo. ToxinPred analysis confirmed the non-toxic nature of MEV. SOLpro assessed the solubility of MEV at 0.896683, exceeding the threshold of 0.5, indicative of excellent solubility. NCBI BLASTP analysis further confirmed minimal homology between MEV and the human proteome, with an e-value significantly below 0.005, establishing its non-human homology. Collectively, these findings validate the rational and feasible design of MEV. A detailed summary is provided in (Table 6).
Prediction results of protein secondary structure
The secondary structure of MEV was predicted by SOPMA, and the results indicated that α-helix accounted for 11.29%, β-turns accounted for 6.90%, random coils accounted for 59.09%, and stretched chains accounted for 22.73% (see Fig. 4A,B).Alpha helices enhance protein stability via intra-chain hydrogen bonds, whereas β-sheets orient key residues outward, maximizing epitope solvent accessibility. The content of α-helices is not high, but other data are still needed to determine the properties of the protein.


(A,B) Secondary structure prediction of MEV; (C) Ramachandran plot of MEV tertiary structure; (D) Ramachandran plot of MEV tertiary structure after refinement; (E) Tertiary structure prediction of MEV; (F) Optimized tertiary structure; (G) Z-score plots obtained from ProSA-web; (H) Protein structure residue scores.
Evaluation and optimisation of tertiary structure
The tertiary structure of MEV was evaluated for quality using Procheck.The results of the Ramachandran plot (Fig. 4C) showed that 85.0% of the residues were located in the most acceptable region, while 13.0% were located in the additional permissive region. Subsequently, GalaxyRefine was used to optimize the tertiary structure of the subdivided MEV, and the optimized tertiary structure was selected. Subsequent quality assessment was performed using Procheck, revealing that the optimized tertiary structure (Fig. 4D) contains 89.5% of the residues within the most acceptable region and 8.7% within the additional permissive region, a proportion of 1.2% in the disallowed region is acceptable. The pre- and post-optimized tertiary structures were then visualised (Fig. 4E,F), and the z-values and local model quality (shows that most of the sequence positions are negativemaps)were obtained by analyzing the post-optimized tertiary structures using ProSA-web (Fig. 4G,H). Consistent with the parameters of a typical proteinThese structures collectively demonstrate that the quality of the tertiary structure of the MEV after refinement using GalaxyRefine is satisfactory and can be utilized.
Molecular docking analysis
Following physicochemical characterization of the MEV, we evaluated its binding to TLR4 molecules via molecular docking to assess immunogenic potential. The top 10 models generated by the HDOCK server were evaluated, and the model exhibiting the most negative docking score was selected for detailed analysis of the interactions between the receptors (MHCI and TLR4) and the ligand (MEV). The docking results revealed a score of -256.97 for the MHCI-MEV complex, with a ligand RMSD of 194.14and a confidence score of 0.8947.For the TLR4-MEV complex, the docking score was -312.79, with a ligand RMSD of 97.79and a confidence score of 0.9629 Subsequently, PyMOL was utilized to visualize the three-dimensional docking structures and intermolecular interactions. As shown in (Fig. 5).

PyMOL display of molecular docking results. (A) Structural diagram of the MEV-MHCI complex drawn using PyMOL; (B) Analysis of the interaction of the MEV-MHCI complex and its 3D image taken using PyMol. (C) Structural diagram of the MEV-TLR4 complex drawn using PyMOL; (D) Analysis of the interaction of the MEV-TLR4 complex and its 3D image taken using PyMol.
Molecular dynamics simulations
Molecular dynamics simulations of the complex were performed to further validate the stability of the binding. .In this simulation, we calculated the RMSD of TLR4 protein and TLR4-MEV complex and plotted the RMSD plot (Fig. 6A).

(A–F) (A) RMSD diagram of TLR4 and TLR4-MEV; (B–D) The RMSF locus of acceptor-ligand (E) HBond diagram of the TLR4 and TLR4-MEV. (F) Rg diagram of the TLR4 and TLR4-MEV.
The RMSD value reflects the fluctuation of the protein conformation and can be used to assess the stability of the ligand binding to the target protein: the smaller the value, the more stable the binding. In the simulation of protein–ligand binding, the RMSD value of the complex tends to stabilize. By superimposing the protein backbone and calculating the ligand’s RMSD, a smaller ligand RMSD indicates less positional change of the ligand relative to the protein. In this simulation, the ligand’s RMSD value has stabilized, indicating stable binding of the ligand to the protein.RMSF reflects the fluctuation of amino acid residues in proteins. The greater the flexibility of the protein, the higher the RMSF value. The RMSF values of each chain of the protein during the simulation are as follows (Fig. 6B,D). Their The RMSF value changes in a similar trend, and the overall structure of the complex is relatively stable. It fluctuates roughly between 0.2 and 0.35 nm.
Hydrogen bonds are one of the strongest non-covalent interactions. The number of hydrogen bonds formed during the simulation reflects the binding strength (Fig. 6E). The number of hydrogen bonds remains above 10.This indicates that MEV-TLR4 interaction has good characteristics and stability.
The radius of gyration is a global property of protein structure, representing the average distance between the protein’s center of mass and its individual atoms. In this study, we analyzed the radius of gyration (Rg) of the TLR4 protein and the TLR4-MEV complex, as shown in (Fig. 6F).
Immunostimulation
The C-ImmSim server was utilized to simulate the immune response subsequent to three injections of MEV. As illustrated in Fig. 7A, the specific antibodies produced by the body increased after three MEV injections, with IgM + IgG antibody levels peaking after the third immunisation and gradually decreasing thereafter. While it is well established that B cells are critical for both humoral immunity and immune response, Fig. 7B demonstrates that total B cells and memory B cells declined with increasing vaccine doses, reaching their nadir after the third immunisation. Furthermore, Fig. 7C demonstrates that helper T cells increased after three MEV stimulations, forming progressively higher peaks, with the overall trend approximating a similar trend to that of cytotoxic T cells (see Fig. 7D,E). Furthermore, Fig. 7F demonstrates that the number of activated macrophages remained stable for the three vaccinations, but rapidly decreased after 100 days of initial vaccination. Furthermore, Fig. 7G,H illustrate that dendritic cells and CD4 + T cells exhibited stability in their levels. Figure 7I demonstrates that the immune response and stimulation by the vaccine resulted in a significant increase in the cytokines IFN-γ, TGF-β, IL-10, IL-12, and IL-2.The immune simulation shows increased antibody levels but a slight decrease in total B cells. This is likely because activated B cells differentiate into plasma cells (which produce antibodies) rather than remaining as naive B cells—plasma cells were not separately quantified in the simulation, leading to the apparent contradiction.

(A) The immunoglobulin production after antigen injection. (B) The B cell population after three injections. (C)The helper T cell population after three injections. (D) The cytotoxic T cell population after three injections. (E) Number of cytotoxic T cells in each state after three injections. (F) Number of macrophages in each state after three injections. (G) Number of dendritic cells in each state after three injections. (H) The EP Population per state after three injections. (I) Cytokine and interleukin levels and Simpson’s index of immune response.
Codon optimization and in silico cloning
The MEV FASTA sequence was back-translated into a 1914-base nucleotide sequence in the codon optimization tool ExpOptimizer. The optimized MEV DNA sequence exhibited a codon adaptation index (CAI) of 0.80and a GC content of 56.90%, indicating that MEV in E. bacillus was designed according to the principles of primer design with a forward primer (5′-CTCGAGATGCCGAAACGCAAAGTTTCCTC-3′) with a length of 29, a Tm value of 65, and a GC content of 52%. The reverse primer (5′-GGATCCGTGGTGATGGTGGTGGTGAC-3′), with a length of 26, a Tm value of 65, and a GC content of 62%, was used for the amplification of the target gene in a polymerase chain reaction (PCR). As shown in Fig. 8A, the primer design was carried out using the software SnapGene8.0.1. Subsequently, the MEV-amplified target gene sequence was inserted into the multiple cloning site (MCS) domain of the vector pET-28a( +), completing the in silico cloning, as shown in (Fig. 8B).

(A) simulated mev vaccine amplification plot; (B) simulated cloning product plot; (C) simulated agarose gel experiment results (1.0% agarose).
The 1% agarose gel electrophoresis simulation revealed that the MEV DNA sequence was 1914 bp in length, while the post-PCR MEV DNA sequence measured 1926 bp. Additionally, the pET-28a( +) DNA sequence was 5369 bp, and the recombinant plasmid DNA sequence was 7249 bp. See (Fig. 8).
Discussion
Brucellosis poses significant threats: In terms of human health65,66, the infection progresses through latent, acute, and chronic phases. If left untreated, it can easily lead to chronic complications such as endocarditis and arthritis. The chronic phase may last for over half a year, causing severe organ damage and even death. On the socioeconomic front, as a zoonotic disease, the actual number of cases is underestimated due to inadequate diagnosis, monitoring, and reporting. This places a heavy burden on livestock production and the public health system67.
Diagnosis and treatment of human brucellosis face multiple challenges68. In terms of diagnosis, the nonspecific clinical manifestations often lead to misdiagnosis. Laboratory tests each have their limitations69,70: although bacterial culture is the gold standard, it is slow-growing, high-risk, and difficult to isolate in the later stages71. Traditional serology cannot distinguish the state of infection, while new detection methods, although superior, are not widely available due to high costs72,73. In terms of treatment, the combination of multiple antibiotics cannot eradicate the disease. Long-term use of drugs can lead to resistance, further increasing the physical, mental, and economic burden on patients74.
Given the deficiencies in diagnosis and treatment, prevention is crucial, and vaccine development is urgent. Currently, only attenuated live vaccines for animals (such as B. abortus S19 and B. melitensis Rev1) are available, while human vaccines have not been approved75. Although these animal vaccines are inexpensive and effective, they have drawbacks such as antibiotic resistance, interference with serological diagnosis, and residual virulence. In contrast, subunit vaccines, with their high safety, non-infectious nature, strong immunogenicity, and potential for optimized design, are considered more promising candidates76. In this study, a multi-epitope vaccine was designed for Brucella abortus using reverse vaccinology and bioinformatics methods to select cytotoxic T-cell epitopes, helper T-cell epitopes, and B-cell linear/conformational epitopes.
In order to enhance the development of novel multi-epitope vaccines (MEVs), the selection of proteins is paramount. In this study, reverse vaccinology was used to screen Brucella membrane proteins for potential vaccine candidates with the goal of developing a subunit multiepitope vaccine. Bioinformatics tools were used to perform computational identification and screening of multiple antigenic epitopes, ultimately leading to the construction of a novel vaccine candidate capable of eliciting the desired immune response77. BepC, an outer membrane protein of Brucella spp., functions as a Type IV Secretion System (T4SS)-secreted effector protein and is critically involved in the pathogen’s virulence mechanisms. With high antigenicity, BepC may disrupt host cellular autophagy to regulate bacterial intracellular survival, making it a promising candidate target for developing vaccines against brucellosis78. In the context of bacterial pathogens, the ability to acquire sufficient iron within the iron-limited environment of a host typically acts as a critical determinant of their survival and competitive fitness in a specific ecological niche79. Studies have shown that Brucella spp. utilize heme as an iron source in vitro80. Heme exporter protein C can transport heme into the cytoplasm as a source of iron for bacteria. As a membrane protein, it has been screened by software to have high immunogenicity and can be used as a potential vaccine protein. Cytochrome c plays a key role in cellular respiration and apoptosis, and abnormal synthesis of cytochrome c is associated with multiple diseases. As a part of the ABC transporter complex CcmAB, CcmA is involved in the biosynthesis of c-type cytochromes81. Similarly, through bioinformatics software analysis, we found that it has characteristics such as high immunogenicity and stability, and can be used as a candidate protein for vaccines. Previous studies have demonstrated that proteins with strong antigenicity are instrumental in the development of effective MEVs. The three candidate proteins were analyzed by bioinformatics methods, and the results indicated that their predicted antigenicity values were higher than the threshold of 0.4, suggesting that these proteins possess considerable antigenicity. Furthermore, a comprehensive evaluation of the physicochemical properties, sensitization, and toxicity of these proteins revealed that they possess adequate stability and are not associated with allergic reactions or toxicity concerns. Given the absence of studies related to the construction of MEVs based on these three proteins, they have been selected as potential candidate proteins for vaccine development.
In the analysis of the Brucella multi-epitope vaccine (MEV) designed in this study, the basic physicochemical properties were first clarified: the vaccine protein has a molecular weight of 66.26 kDa, which is within the ideal range of < 110 kDa82; the theoretical isoelectric point (pI) is 9.97, and the total number of amino acids is 638. These parameters clearly reflect the basic characteristics of the vaccine construct. Additionally, the stability of the vaccine protein was further verified by the instability index (26.80) and the grand average of hydropathy (GRAVY) value. The antigenicity and toxicity assessment results showed that the vaccine has high antigenicity (1.2542), and is non-toxic and non-allergenic. Overall, the vaccine construct designed in this study has the core advantages of being stable, soluble, hydrophilic, highly antigenic, non-toxic, and non-sensitizing, meeting the key basic requirements for vaccine development.
In the structural prediction and analysis of the vaccine protein, the secondary structure prediction results showed that beta turns account for 6.90% and random coils account for 59.09%. Previous studies have indicated that a high proportion of beta turns and random coils in MEVs is conducive to the formation of antigenic epitopes83. Although the low proportion of alpha helices in this vaccine may theoretically have some impact on solubility, the final protein properties still need to be further verified in combination with other parameters and subsequent experiments. In terms of tertiary structure quality assessment, the results showed that the MEV has high precision and good approximation factors, with a reliable and high-quality overall structure. Although 1.2% of the structural regions are in the disallowed range, this value is below the acceptable threshold of 2% and does not affect the overall structural validity.
In the analysis of immune mechanisms, the strong interaction between the antigen molecule (MEV) and the immune receptor molecule (TLR4) is a key prerequisite for initiating the immune response in the host84. Toll-like receptor 4 (TLR4), as a pattern recognition receptor, often plays an important role in the construction of MEVs85. Therefore, the study conducted protein–ligand docking analysis of the MEV-TLR4 complex to evaluate the stability of their binding. The atom interaction diagram shows strong interactions between MEV and TLR4 molecules. This stable binding ability ensures that the two can be effectively transported in the host body, laying the foundation for the initiation of subsequent immune responses.
During the immune simulation of the vaccine, the levels of B cells and T cells in the body increased with each dose of the vaccine, reaching a peak after the third injection. Moreover, the MEV increased the levels of cytokines and antibodies. Surprisingly, the increase in the cytokine IFN-γ was significant. The substantial elevation of IFN-γ indicates that the vaccine can play an important role in killing pathogens86. We also found that the total number of B cells decreased after a certain period but remained at a certain level, which led to the maintenance of antibody levels. These results show that the vaccine has elicited a good humoral and cellular immune response, and we believe that the constructed vaccine can be considered a candidate for a Brucella vaccine.The optimized codon GC content (56.90%) and codon adaptation index (CAI = 0.80) also indicate good expression levels of the vaccine protein in the E. coli host.
In summary, this study has employed a variety of bioinformatics methods to combat Brucella infection, providing a theoretical basis for future laboratory testing. The constructed MEV exhibits favorable physicochemical properties and immune responses. Further molecular dynamics simulations have demonstrated the high stability of the MEV. However, key analyses such as epitope prediction, molecular docking, and immune simulation all rely on computational models, which inherently have uncertainties and false-positive rates. Therefore, we need to proceed to the next step of in vitro and in vivo experiments to verify whether the designed multi-epitope vaccine (MEV) can be successfully expressed in vitro and maintain solubility and stability, and whether it can induce protective immunity in vivo. These all require experimental validation.
Data availability
The authors confirm that the data was derived from public domain information: NCBI database (https://www.ncbi.nlm.nih.gov/) and PDB library (https://www.rcsb.org/). The data that support the findings of this study are available in the methods.
References
-
Liu, Z., Gao, L., Wang, M., Yuan, M. & Li, Z. Long ignored but making a comeback: a worldwide epidemiological evolution of human brucellosis. Emerg. Microb. Infect. 13, 2290839 (2024).
-
Ko, J. & Splitter, G. A. Molecular host-pathogen interaction in brucellosis: Current understanding and future approaches to vaccine development for mice and humans. Clin. Microbiol. Rev. 16, 65–78 (2003).
-
Laine, C. G., Johnson, V. E., Scott, H. M. & Arenas-Gamboa, A. M. Global estimate of human brucellosis incidence. Emerg. Infect. Dis. 29, 1789–1797 (2023).
-
Bagheri Nejad, R., Krecek, R. C., Khalaf, O. H., Hailat, N. & Arenas-Gamboa, A. M. Brucellosis in the middle East: Current situation and a pathway forward. PLoS Negl. Trop. Dis. 14, e0008071 (2020).
-
Pal, M., Gizaw, F., Fekadu, G., Alemayehu, G. & Kandi, V. Public health and economic importance of bovine brucellosis: An overview. AJEID 5, 27–34 (2017).
-
Rubach, M. P., Halliday, J. E. B., Cleaveland, S. & Crump, J. A. Brucellosis in low-income and middle-income countries. Curr. Opin. Infect. Dis. 26, 404–412 (2013).
-
Liu, Z. et al. In vitro antimicrobial susceptibility testing of human Brucella melitensis isolates from Ulanqab of Inner Mongolia, China. BMC Infect. Dis. 18, 43 (2018).
-
Zhu, X. et al. Brucella melitensis, a latent “travel bacterium”, continual spread and expansion from Northern to Southern China and its relationship to worldwide lineages. Emerg. Microb. Infect. 9, 1618–1627 (2020).
-
Pizarro-Cerdá, J., Moreno, E. & Gorvel, J. P. Invasion and intracellular trafficking of Brucella abortus in nonphagocytic cells. Microb. Infect. 2, 829–835 (2000).
-
Khairullah, A. R. et al. Brucellosis: Unveiling the complexities of a pervasive zoonotic disease and its global impacts. Open Vet. J. 14, 1081–1097 (2024).
-
Atluri, V. L., Xavier, M. N., de Jong, M. F., den Hartigh, A. B. & Tsolis, R. M. Interactions of the human pathogenic Brucella species with their hosts. Ann. Rev. Microbiol. 65, 523–541 (2011).
-
Franco, M. P., Mulder, M., Gilman, R. H. & Smits, H. L. Human brucellosis. Lancet Infect. Dis. 7, 775–786 (2007).
-
Bosilkovski, M., Keramat, F. & Arapović, J. The current therapeutical strategies in human brucellosis. Infection 49, 823–832 (2021).
-
Ivanov, A. V., Salmakov, K. M., Olsen, S. C. & Plumb, G. E. A live vaccine from Brucella abortus strain 82 for control of cattle brucellosis in the Russian Federation. Anim. Health Res. Rev. 12, 113–121 (2011).
-
Rappuoli, R. Vaccines: science, health, longevity, and wealth. Proc. Natl. Acad. Sci. USA 111, 12282 (2014).
-
Rappuoli, R., Bottomley, M. J., D’Oro, U., Finco, O. & De Gregorio, E. Reverse vaccinology 2.0: Human immunology instructs vaccine antigen design. J. Exp. Med. 213, 469–481 (2016).
-
Shi, H. et al. Development of innovative multi-epitope mRNA vaccine against central nervous system tuberculosis using in silico approaches. PLoS ONE 19, e0307877 (2024).
-
Luo, J.-R. et al. Design of a multi-Epitope mRNA vaccine against Brucella type IV secretion system using reverse vaccinology and immunogenicity approaches. Sci. Rep. 15, 30698 (2025).
-
Delany, I., Rappuoli, R. & Seib, K. L. Vaccines, reverse vaccinology, and bacterial pathogenesis. Cold Spring Harb. Perspect. Med. 3, a012476 (2013).
-
Zhang, L. et al. The role of outer membrane protein 16 in brucella pathogenesis, vaccine development, and diagnostic applications. Vet. Sci. 12, 605 (2025).
-
Wei, Y. et al. Advances of computational methods enhance the development of multi-epitope vaccines. Br. Bioinform. 26, bbaf055 (2024).
-
Vittorio, S. et al. Addressing docking pose selection with structure-based deep learning: Recent advances, challenges and opportunities. Comput. Struct. Biotechnol. J. 23, 2141–2151 (2024).
-
Anderson, L. N. et al. Computational tools and data integration to accelerate vaccine development: challenges, opportunities, and future directions. Front. Immunol. 16, 1502484 (2025).
-
UniProt Consortium. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 47, D506–D515 (2019).
-
Doytchinova, I. A. & Flower, D. R. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformat. 8, 4 (2007).
-
Dimitrov, I., Naneva, L., Doytchinova, I. & Bangov, I. AllergenFP: allergenicity prediction by descriptor fingerprints. Bioinformatics 30, 846–851 (2014).
-
Gupta, S. et al. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE 8, e73957 (2013).
-
Wilkins, M. R. et al. Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 112, 531–552 (1999).
-
Liaci, A. M. & Förster, F. Take me home, protein roads: Structural insights into signal peptide interactions during ER translocation. Int. J. Mol. Sci. 22, 11871 (2021).
-
Tian, T. et al. FcRn-Driven nanoengineered mucosal vaccine with multi-epitope fusion induces robust dual immunity and long-term protection against Brucella. Vaccines (Basel) 13, 567 (2025).
-
Teufel, F. et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 40, 1023–1025 (2022).
-
Tsurui, H. & Takahashi, T. Prediction of T-cell epitope. J Pharmacol. Sci. 105, 299–316 (2007).
-
Shen, C. et al. Allele polymorphism and haplotype diversity of HLA-A, -B and -DRB1 loci in sequence-based typing for Chinese Uyghur ethnic group. PLoS ONE 5, e13458 (2010).
-
Doytchinova, I. A., Guan, P. & Flower, D. R. EpiJen: a server for multistep T cell epitope prediction. BMC Bioinformat. 7, 131 (2006).
-
Stranzl, T., Larsen, M. V., Lundegaard, C. & Nielsen, M. NetCTLpan: pan-specific MHC class I pathway epitope predictions. Immunogenetics 62, 357–368 (2010).
-
Reynisson, B., Alvarez, B., Paul, S., Peters, B. & Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 48, 4W49-W454 (2020).
-
Nilsson, J. B. et al. Accurate prediction of HLA class II antigen presentation across all loci using tailored data acquisition and refined machine learning. Sci. Adv. 9, eadj6367 (2023).
-
Getzoff, E. D., Tainer, J. A., Lerner, R. A. & Geysen, H. M. The chemistry and mechanism of antibody binding to protein antigens. Adv. Immunol. 43, 1–98 (1988).
-
Yao, B., Zheng, D., Liang, S. & Zhang, C. SVMTriP: A method to predict B-cell linear antigenic epitopes. Methods Mol. Biol. 2131, 299–307 (2020).
-
Shen, W. et al. Predicting linear B-cell epitopes using amino acid anchoring pair composition. BioData Min. 8, 14 (2015).
-
Ferdous, S., Kelm, S., Baker, T. S., Shi, J. & Martin, A. C. R. B-cell epitopes: Discontinuity and conformational analysis. Mol. Immunol. 114, 643–650 (2019).
-
Ponomarenko, J. et al. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinformat. 9, 514 (2008).
-
Lo, Y.-T. et al. Conformational epitope matching and prediction based on protein surface spiral features. BMC Genom. 22, 116 (2021).
-
Li, M. et al. Design of a multi-epitope vaccine candidate against Brucella melitensis. Sci. Rep. 12, 10146 (2022).
-
Chen, X., Zaro, J. L. & Shen, W.-C. Fusion protein linkers: property, design and functionality. Adv. Drug Deliv. Rev. 65, 1357–1369 (2013).
-
Yang, D. et al. High-mobility group nucleosome-binding protein 1 acts as an alarmin and is critical for lipopolysaccharide-induced immune responses. J. Exp. Med. 209, 157–171 (2012).
-
Tarrahimofrad, H., Rahimnahal, S., Zamani, J., Jahangirian, E. & Aminzadeh, S. Designing a multi-epitope vaccine to provoke the robust immune response against influenza A H7N9. Sci. Rep. 11, 24485 (2021).
-
Fan, Y. et al. Design of a novel EmTSP-3 and EmTIP based multi-epitope vaccine against Echinococcus multilocularis infection. Front. Immunol. 15, 1425603 (2024).
-
Liu, C., Chin, J. X. & Lee, D.-Y. SynLinker: an integrated system for designing linkers and synthetic fusion proteins. Bioinformatics 31, 3700–3702 (2015).
-
Magnan, C. N., Randall, A. & Baldi, P. SOLpro: accurate sequence-based prediction of protein solubility. Bioinformatics 25, 2200–2207 (2009).
-
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
-
Geourjon, C. & Deléage, G. SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput. Appl. Biosci. 11, 681–684 (1995).
-
Morris, A. L., MacArthur, M. W., Hutchinson, E. G. & Thornton, J. M. Stereochemical quality of protein structure coordinates. Proteins 12, 345–364 (1992).
-
Ko, J., Park, H., Heo, L. & Seok, C. GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Res 40, W294-297 (2012).
-
Heo, L., Park, H. & Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 41, W384-388 (2013).
-
Yan, Y., Tao, H., He, J. & Huang, S.-Y. The HDOCK server for integrated protein-protein docking. Nat. Protoc. 15, 1829–1852 (2020).
-
Yan, Y., Zhang, D., Zhou, P., Li, B. & Huang, S.-Y. HDOCK: a web server for protein-protein and protein-DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 45, W365–W373 (2017).
-
Van Der Spoel, D. et al. GROMACS: fast, flexible, and free. J. Comput. Chem. 26, 1701–1718 (2005).
-
Abraham, M. et al. GROMACS 2024.2 Source code. Zenodo https://doi.org/10.5281/ZENODO.11148655 (2024).
-
Rapin, N., Lund, O., Bernaschi, M. & Castiglione, F. Computational immunology meets bioinformatics: the use of prediction tools for molecular binding in the simulation of the immune system. PLoS ONE 5, e9862 (2010).
-
Yin, Z. et al. Design of multi-epitope vaccine candidate against Brucella type IV secretion system (T4SS). PLoS ONE 18, e0286358 (2023).
-
Sharp, P. M. & Li, W. H. The codon Adaptation Index–a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 15, 1281–1295 (1987).
-
Fu, H. et al. Codon optimization with deep learning to enhance protein expression. Sci. Rep. 10, 17617 (2020).
-
Synonymous but not Silent: The Codon Usage Code for Gene Expression and Protein Folding – PMC. https://pmc.ncbi.nlm.nih.gov/articles/PMC8284178/.
-
Buzgan, T. et al. Clinical manifestations and complications in 1028 cases of brucellosis: a retrospective evaluation and review of the literature. Int. J. Infect. Dis. 14, e469-478 (2010).
-
Elfaki, M. G., Alaidan, A. A. & Al-Hokail, A. A. Host response to Brucella infection: review and future perspective. J. Infect. Dev. Ctries 9, 697–701 (2015).
-
Hartady, T., Saad, M. Z., Bejo, S. K. & Salisi, M. S. Clinical human brucellosis in Malaysia: a case report. Asian Pac. J. Trop. Dis. 4, 150–153 (2014).
-
Yagupsky, P., Morata, P. & Colmenero, J. D. Laboratory diagnosis of human Brucellosis. Clin. Microbiol. Rev. 33, e00073-e119 (2019).
-
Esel, D., Doganay, M., Alp, E. & Sumerkan, B. Prospective evaluation of blood cultures in a Turkish university hospital: epidemiology, microbiology and patient outcome. Clin. Microbiol. Infect. 9, 1038–1044 (2003).
-
Mesureur, J. et al. A MALDI-TOF MS database with broad genus coverage for species-level identification of Brucella. PLoS Negl. Trop. Dis. 12, e0006874 (2018).
-
Yagupsky, P., Peled, N., Press, J., Abramson, O. & Abu-Rashid, M. Comparison of BACTEC 9240 Peds Plus medium and isolator 1.5 microbial tube for detection of Brucella melitensis from blood cultures. J. Clin. Microbiol 35, 1382–1384 (1997).
-
Navarro, E., Serrano-Heras, G., Castaño, M. J. & Solera, J. Real-time PCR detection chemistry. Clin. Chim. Acta 439, 231–250 (2015).
-
Nimri, L. F. Diagnosis of recent and relapsed cases of human brucellosis by PCR assay. BMC Infect. Dis. 3, 5 (2003).
-
Maldonado-García, J. L. et al. Concomitant Treatment with Doxycycline and Rifampicin in Balb/c Mice Infected with Brucella abortus 2308 fails to reduce inflammation and motor disability. Pharmaceuticals (Basel) 17, 638 (2024).
-
Simpson, G. J. G. et al. Immunological response to Brucella abortus strain 19 vaccination of cattle in a communal area in South Africa. J. S. Afr. Vet. Assoc. 89, e1–e7 (2018).
-
Ficht, T. A., Kahl-McDonagh, M. M., Arenas-Gamboa, A. M. & Rice-Ficht, A. C. Brucellosis: the case for live, attenuated vaccines. Vaccine 27 (4), D40-43 (2009).
-
Jenner, D. C., Dassa, E., Whatmore, A. M. & Atkins, H. S. ATP-Binding Cassette Systems of Brucella. Comp. Funct. Genom. 2009, 354649 (2009).
-
Jalal, K., Khan, K. & Uddin, R. Immunoinformatic-guided designing of multi-epitope vaccine construct against Brucella Suis 1300. Immunol. Res. 71, 247–266 (2023).
-
Schaible, U. E. & Kaufmann, S. H. E. Iron and microbial infection. Nat. Rev. Microbiol. 2, 946–953 (2004).
-
Paulley, J. T., Anderson, E. S. & Roop, R. M. Brucella abortus requires the heme transporter BhuA for maintenance of chronic infection in BALB/c mice. Infect. Immun. 75, 5248–5254 (2007).
-
Christensen, O., Harvat, E. M., Thöny-Meyer, L., Ferguson, S. J. & Stevens, J. M. Loss of ATP hydrolysis activity by CcmAB results in loss of c-type cytochrome synthesis and incomplete processing of CcmE. FEBS J. 274, 2322–2332 (2007).
-
Barh, D. et al. Exoproteome and secretome derived broad spectrum novel drug and vaccine candidates in Vibrio cholerae targeted by Piper betel derived compounds. PLoS ONE 8, e52773 (2013).
-
Yu, M. et al. Design of a novel multi-epitope vaccine against Echinococcus granulosus in Immunoinformatics. Front. Immunol. 12, 668492 (2021).
-
Chaplin, D. D. Overview of the immune response. J. Allergy Clin. Immunol. 125, S3-23 (2010).
-
Bayani, F., Safaei Hashkavaei, N., Karamian, M. R., Uskoković, V. & Sefidbakht, Y. In silico design of a multi-epitope vaccine against the spike and the nucleocapsid proteins of the Omicron variant of SARS-CoV-2. J. Biomol. Struct. Dyn. 41, 11748–11762 (2023).
-
Dittmer, U. et al. Role of interleukin-4 (IL-4), IL-12, and gamma interferon in primary and vaccine-primed immune responses to friend retrovirus infection. J. Virol. 75, 654–660 (2001).
Acknowledgements
The authors are thankful to the State Key Laboratory of Pathogenesis, Prevention, Treatment of Central Asian High Incidence Diseases, The First Affiliated Hospital of Xinjiang Medical University, PR China.
Funding
This research was supported by the Natural Science Foundation of China (No. 82360394, 82460399), the Youth Science and Technology Top Talent Project (No.2022TSYCCX0112), the Outstanding Youth Project of the Autonomous Region (No.2023D01E12), the Central Government Guides Local Science and Technology Development Fund Project (ZYYD2025ZY19).
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chai, ZL., Qi, XX., Li, R. et al. Reverse vaccinology-driven construction and bioinformatics validation of a multi-epitope vaccine against Brucella spp.. Sci Rep 15, 36663 (2025). https://doi.org/10.1038/s41598-025-20507-7
-
Received:
-
Accepted:
-
Published:
-
DOI: https://doi.org/10.1038/s41598-025-20507-7