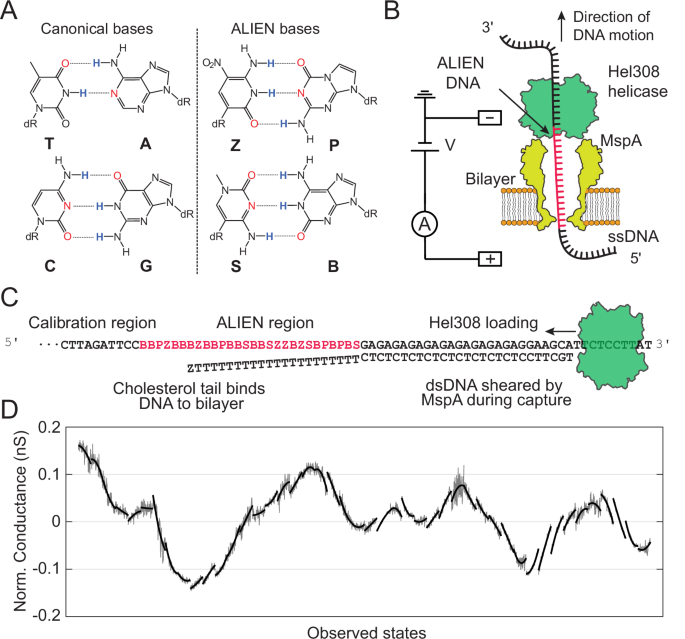
Introduction
DNA forms the informational basis of Terran life through its ability to store, transmit, and evolve genetic information. Two features let it do so: 1) rules of base pairing (i) size complementarity and (ii) hydrogen bond complementarity provide a direct mechanism for information-conserving replication, and 2) a repeating backbone charge. The repeating backbone charge is required for any informational biopolymer to support evolution under the “Polyelectrolyte Theory of the Gene”1 because it controls strand-strand interactions, but also allows informational units to be exchanged without dramatically changing the overall biophysical properties.
Many research groups have added new nucleotides and new nucleobase pairs to the four canonical nucleobases in standard Terran (A, C, G, T) DNA2,3,4,5,6. Some of these retain both pairing rules4,5. Others retain size complementarity, while dispensing with inter-base hydrogen bonds3. Others retain intra hydrogen bonds, but dispense with size7. In all cases, however, anthropogenic pairs are sparsely introduced into double helices that are largely stabilized by standard A:T and G:C pairs. For anthropogenic pairs that are not joined by inter-base hydrogen bonds, this sparseness is a requirement for a functioning informational biopolymer.
ALternative Isoinformational ENgineered (ALIEN) DNA moves beyond this constraint. Here, the goal is an entirely anthropogenic informational system, without any Terran nucleotides at all. Recently, this was done using four synthetic nucleotides:
B: isoguanine 6-amino-9[(1′-β-D-2′-deoxyribofuranosyl)-4-hydroxy-5-(hydroxymethyl)-oxolan-2-yl]-1H-purin-2-one, which pairs with
S: 3-methyl-6-amino-5-(1′-β-D-2′-deoxyribofuranosyl)-pyrimidin-2-one, and
P: 2-amino-8-(1′-β-D-2′-deoxyribofuranosyl)-imidazo-[1,2a]-1,3,5-triazin-[8H]-4-one, which pairs with
Z: 6-amino-3-(1′-β-D-2′-deoxyribofuranosyl)-5-nitro-1H-pyridin-2-one.
ALIEN DNA retains the standard deoxyribose-phosphate polyelectrolyte backbone of standard DNA, and uses both size and hydrogen bonding complementarity to form specific pairs8,9,10 (Fig. 1A). It is “isoinformational”, since each site holds two bits of information, the same as with standard DNA. Further, ALIEN DNA can form the same A and B double helical forms as standard DNA. Both the polyelectrolyte backbone and rule-base pairing allow ALIEN DNA to meet the requirements for Darwinian evolution1. Anthropogenic genetic systems including P and Z bases have been shown to evolve medically useful receptors, ligands, and catalysts with high affinity11,12,13,14,15,16, support highly multiplexed PCR17, and are components of commercial diagnostics products18.

A ALternative Isoinformational ENgineered (ALIEN) DNA (right) consists of four anthropogenic bases (Z, P, S, B) that form orthogonal base pairs (Z:P and S:B) through size and hydrogen bond complementarity. ALIEN DNA is structurally similar to canonical ACGT-DNA (left) and is “isoinformational”, also carrying two bits of information at each site. B A single Mycobacterium smegmatis porin A (MspA) nanopore (yellow) is embedded in a lipid bilayer. A single DNA molecule bound by a Hel308 helicase (green) is drawn into the pore by an electric field. The ion current through the pore is modulated by the bases in the pore constriction and is measured to sequence the strand. C Schematic for the DNA design used throughout this manuscript. Each strand had a 5′ region composed of ACGT DNA designed for ion current calibration and 3′ region designed for motor enzyme loading. The GA tandem repeat on the 3′ end was selected to aid in sequence-to-conductance alignment. We found previously that every 4th step, on the ATP-independent step, Hel308 helicase has increased propensity for backstepping. We used these backsteps in the raw data to help ensure correct registration of the DNA bases to the measured conductance curves. D A representative variable-voltage nanopore trace of a DNA strand section containing 28 consecutive ALIEN bases. Conductance (current/voltage) is displayed instead of ion current for variable-voltage traces due to the variation of the applied voltage. Each curve segment represents one enzyme step of the Hel308 helicase, which feeds DNA through the pore in two steps per nucleotide, roughly equal in length. For ease of visualization, enzyme backsteps have been filtered and recombined in software (Supplementary Information).
DNA built from B, P, S, and Z is intrinsically biocompatible because it retains the properties of canonical DNA but cannot hybridize to any biological DNA sequence8,9,10. Such DNA, completely orthogonal to biological DNA, has applications in synthetic biology such as expanded codon libraries19,20, chimeric PCR primers for cleaner multiplex PCR17, and rapid assembly of nanostructures14. Because P:Z and B:S pairs are both joined with three hydrogen bonds, PZBS DNA folds have higher melting points than comparable ACGT DNA.
ALIEN DNA oligonucleotides can be synthesized using standard solid phase phosphoramidite chemistry8,9,10,21,22,23. However, standard commercial sequencing platforms are incompatible with this material that is entirely anthropogenic. Transliteration techniques have enabled sequencing of six-letter ACGTPZ DNA24, by probabilistically converting P:Z pairs to A:T and C:G pairs. Comparison of multiple reads reveal the locations of sites that are consistently ACGT, while those which are read as sometimes T, sometimes C, are inferred to be Z, and those which are read as sometimes A, sometimes G, are inferred to be P. However this strategy relies on base-pair mismatching and only works if the anthropogenic components are relatively sparse. This strategy has not been demonstrated for 4-letter DNA consisting of B, P, S, and Z, and does not work well with dense, long tracts of non-canonical bases because of its use of base pair mismatches.
Realizing the full potential of ALIEN DNA requires sequencing technology that is routine, accurate, and agnostic to both the chemical identity of bases and alphabet size of anthropogenic genetic systems. Further, the development of anthropogenic genetic systems is ongoing, with the potential for new analogues of P, Z, S, and B optimized for chemically stability, biocompatibility, or with different chemical functional groups useful for aptamers and aptazymes12,15,25,26. Thus, it is important that any sequencing technology developed can be rapidly adapted to alternative alphabets with minimal cost of development.
Nanopore strand sequencing27,28,29 is a well-accepted strategy for sequencing DNA which requires no chemical label, has single molecule sensitivity, and needs no prior DNA amplification (Fig. 1B). Critically, unlike other techniques that are based on chemical recognition of DNA bases, nanopore sequencing identifies bases physically based on their interaction with an ion current passing through the nanopore. This makes nanopores sensitive to the detailed chemical structure of the base, and therefore easily adaptable to new bases and analytes. As examples, nanopore sequencing has been used to detect base modifications that include methylated cytosine30,31,32,33, base adducts34, and base isomers such as pseudouridine35. Nanopore techniques are even being developed for peptide sequencing36. We, in collaboration with others, have used nanopore sequencing for reference sequencing of the 8-letter system [A,C,G,T,P,Z,S,B]37, the 12-letter system [A,C,G,T,P,Z,S,B,J,V,K,X]38, DNA with negatively charged nucleobase (Z–)39, and a 6-letter system containing a base pair between two hydrophobic nucleotides40. Each of these works demonstrated that nanopores could identify isolated incorporations of many different non-canonical bases surrounded by ACGT DNA of known sequence. De novo sequencing of DNA with continuous tracts of non-canonical bases is a more challenging problem. Essentially, instead of finding and identifying deviations from what is expected of ACGT DNA, one has to produce an entire sequence blind.
In enzyme-actuated nanopore strand-sequencing27, a single nanoscale pore (nanopore) is incorporated into an artificial lipid bilayer membrane and electrically connects two electrolyte reservoirs (Fig. 1B, C). An ion current flows through the nanopore in response to a voltage applied across the membrane. This ion current is measured as the primary nanopore signal. Nucleic acid molecules coupled to a motor enzyme are captured by the electric field near the pore due to their negative charge. DNA is drawn into the pore until the enzyme comes to rest on the rim of the pore. DNA motion through the pore is controlled by the motor enzyme, which translocates along the DNA in discrete steps. For strand sequencing with the Hel308 helicase, the helicase is loaded on an 8-base overhang at a strand’s 3′ end while the strand’s 5′ end is threaded into the pore. As the DNA strand is pulled into the pore, the complementary strand is rapidly unzipped by the nanopore until the Hel308 helicase comes to rest on the pore rim. We find nearly all reads in this configuration begin with Hel308 helicase at the 3′ end of the DNA, suggesting that Hel308 helicase is not particularly good at initiating unwinding at ssDNA-dsDNA junctions41. With the complementary strand removed, Hel308 helicase begins translocating along the template strand from 3′ to 5′, feeding DNA through the pore in two ~half-nucleotide steps per ATP hydrolysed41,42.
Bases within the pore constriction block the ion current through the pore to differing degrees because each base has a unique shape and electrostatic characteristics that alter the flow of ions uniquely through the tight pore constriction. Even small molecular differences in the nucleotides, such as base methylation30 or isomeric nucleobases such as guanine and B37,38, produce detectable ion current differences that can be used to determine base identity.
Oxford Nanopore Technologies (ONT) produce commercial nanopore sequencers which have sufficient accuracy, high throughput, and are capable of long reads43. However, the inner workings of the ONT devices are proprietary and lack the experimental customizability required for sequencing more exotic analytes. Further, the 9-base k-mer of the current ONT “chemistry” necessitates building of a large training set which is prohibitively expensive for strands of ALIEN DNA. We used variable-voltage sequencing with the pore MspA for this work because of its higher experimental customizability compared to commercial devices, as well as the higher information content embedded in variable-voltage signal44 compared to the constant voltage signal.
In variable-voltage nanopore sequencing37,40,44, a voltage waveform is applied instead of a DC-voltage. This stretches the DNA within the nanopore periodically, effectively “flossing” DNA back and forth several times during a typical enzyme step, allowing repeated measurements of each nucleotide.
Previous efforts laid the groundwork for results presented here. We showed P, Z, B, and S produced different ion current signals within limited sequence contexts, suggesting ALIEN DNA composed of P, Z, B, and S might be straightforwardly sequenced without additional pore mutation. We also found that P, Z, B, and S possess acceptable compatibility with the Hel308 helicase commonly used for nanopore sequencing37. While the Hel308 helicase did tend to dissociate from the DNA when translocating over dense tracts of the C-glycoside bases S and Z, helicase processivity was shown to be sufficient for sequencing short synthetic oligos37. In this work, we demonstrate de novo variable-voltage sequencing of DNA containing long, contiguous regions of ALIEN DNA bases. To achieve this, we follow the method which enabled the first sequencing of ACGT DNA45: building empirical reference maps of measured ion currents to DNA sequence for all possible k-mers in a range of sequence contexts.
Results
The ion current measured in variable-voltage nanopore sequencing with MspA is affected by the 6 nucleotides in or adjacent to the pore constriction44,45. This is due to the size of the pore constriction (as it compares to the size of individual bases) compounded with the rapid Brownian motion of the DNA within the pore. The set of bases which affect the ion current is referred to as a k-mer, where the number k is the number of bases which contribute to the ion current. For variable-voltage sequencing ACGT DNA with MspA, we have found that a 6-mer describes nanopore data well, where the central 4 bases of the 6-mer describe the majority of the variation seen in the data44,45. By establishing a reference map for all possible k-mers, the ion current trace of an analyte sequence can be decoded into sequence.
We built a variable-voltage 4-mer (k = 4) model by measuring every possible PZBS 4-mer using a compact de Bruijn sequence (Fig. 2, Supplementary Figs. 1–3, Methods and Supplementary Information section S1), which contains all such 4-mers. Just as the cyclical sequence “AABB” contains all 2-letter combinations of A and B (AA, AB, BB, and, wrapping around, BA), a similar 256-base long de Bruijn sequence can be constructed which contains all 4-letter-long combinations of PZSB. Because phosphoramidite DNA synthesis limits the length of oligonucleotides and previous results suggested Hel308 helicase may have reduced processivity on dense regions of S and Z, we divided this de Bruijn sequence into 12 separately-synthesized strands. We designed and synthesized four additional oligonucleotide strands to allow for limited repeated measurements of some 4-mers. These additional strands helped to ensure internal consistency within the 4-mer sequencing model, i.e., we aligned nanopore signals to DNA sequence ensuring that each instance of a repeated 4-mer appeared identically. All model-training strands were designed with a variable 28 nt region of ALIEN DNA sandwiched between two sections of ACGT-DNA used for ion current calibration and helicase loading (Fig. 1C, Supplementary Fig. 4). A complete list of DNA sequences used in this study and their associated consensus traces are found in Supplementary Table 1 and Supplementary Figs. 5–7 respectively. Model training and subsequent sequencing was performed using the purely ALIEN sections of the DNA strands. A summary of all collected data can be found in Table S2.

Conductance curve consensuses for four of the 16 strands used in training the 4mer model for variable-voltage sequencing of ALIEN DNA. Consensus curves are constructed by hand aligning PZBS 4-mers to the variable voltage conductance curves ensuring consistency across strands, i.e., conductance curves for the 4-mer “PZZZ” should be similar each place it occurs in the training data. Because Hel308 helicase moves DNA through the nanopore in two ~half-nucleotide steps per ATP hydrolysed, each 4-mer is assigned two conductance curves. We have determined previously that these steps correspond to conformational changes of the helicase. Integer nucleotide steps correspond to Hel308 helicase in the open conformation while half-integer steps correspond to the two walker domains in the closed conformation. Sequences displayed are in time-order so appear 3′ to 5′ because Hel308 helicase walks from 3′ to 5′. Consensuses are constructed from 20, 24, 30, and 27 reads, respectively.
To benchmark the performance of our sequencing model, we de novo sequenced four testing strands containing 28 nucleotide pseudorandom sequences of ALIEN DNA using a hidden Markov model solver46,47 developed for variable-voltage nanopore sequencing44 (Fig. 3, Supplementary Information section S2). We achieved a mean, per-base accuracy of 74 ± 1% (s.e.m.) for single passages of single DNA molecules (Fig. 3C), comparable with per-base variable-voltage nanopore sequencing accuracy of standard DNA (79.3 ± 0.3% s.e.m.)44. For comparison, sequencing accuracy on training data is 83.7 ± 0.5% s.e.m (Supplementary Fig. 8), indicating some overfitting of the model to the training data.

A Three representative variable voltage reads for the ALIEN DNA section of Test sequence 1. Recurring features are labelled by Greek letters. B The variable voltage conductance curve prediction for the true sequence of Test sequence 1 with the same features labelled by Greek letters. Individual de novo base calls for each read are shown below aligned to the true sequence. Because the 4-mer map does not include mixtures of ACGT and PZSB DNA, Sequencing accuracy is only evaluated for the central 24 nucleotides of the 28-base ALIEN DNA region. Thus base-call accuracies are an estimate of bulk sequencing accuracy without edge effects. Sequences displayed are in time-order so appear 3′ to 5′ because Hel308 helicase walks from 3′ to 5′. C Single-read variable-voltage sequencing accuracy and (D) basecall confusion matrix of 4 pseudorandom strands of ALIEN DNA using the k-mer model. We find that B and S are miscalled more often than P and Z. Average per-base sequencing accuracy is 74 ± 1% (s.e.m.) for single passages (reads) of single DNA molecules, substantially better than random accuracy (57.0 ± 0.1% s.e.m., Supplementary Fig. 9) and comparable to per-base variable-voltage sequencing accuracy of standard DNA (79.3 ± 0.3% s.e.m.). Random basecall accuracy exceeds 25% because accuracy is computed by first aligning the called sequence to the true sequence while allowing for gaps, then comparing the number of matches versus errors. Test sequences 1-4 were read 18, 21, 43, and 20 times, respectively.
The breakdown of basecall errors by base shows that P and Z are called at nearly 90% accuracy (Fig. 3D). In contrast, we found that B was miscalled at a higher rate than the other bases (63%), particularly as insertions/deletions.
To further characterize our sequencing models, we estimated the mutual information between the bases and nanopore signal encoded in both the ALIEN and ACGT 4-mer models48 (Supplementary Fig. 10). Mutual information measures the amount of shared information between two sets of data. The question posed here is “How much of the information about the base at location X is encoded in the variable-voltage conductance curve?” Mutual information is encoded in bits (0 or 1) and one needs 2 bits of information to fully specify one of four bases. We find that the estimated mutual information between sequence and signal is comparable between both models, despite the ALIEN model containing orders of magnitude fewer sequence contexts than the ACGT model, suggesting that we have encoded a substantial fraction of the available information about PZBS DNA in the model and that similar, or higher, magnitude sequencing accuracy to ACGT can be obtained with additional training data.
While single-read accuracy is a simple and straight-forward metric for benchmarking sequencing performance, in practice, nanopore reads of the same sequence are often combined into a more accurate consensus sequence. Here we implement a multiple sequence alignment consensus technique for ALIEN nanopore reads which increases the accuracy over single-read sequencing (Fig. 4, Supplementary Information section S3, Supplementary Figs. 11–13).

A An example subset of a multiple sequence alignment for test sequence 1 showing how the consensus sequence can be determined by combining multiple single-molecule reads. B The basecall probability at each sequence position displayed as a stacked bar plot colored by base identity. Sequencing accuracy for each strand is determined by alignment of the final consensus basecall to the central 22 nucleotides of the true sequence. Three bases were trimmed off each end to eliminate edge effects so that the measured sequencing accuracy is an estimate of bulk sequencing accuracy. Sequencing errors (mismatches, insertions, and deletions) are marked with a minus sign below the sequence position. Insertion and deletion errors are also shown as gaps in the true sequence and consensus sequence, respectively. Average per-base consensus accuracy is 81 ± 4% (s.e.m.). The per-base consensus accuracy for Test sequences 1-4 are: 78 ± 8% (N = 18 reads), 78 ± 8% (N = 21 reads), 67 ± 9% (N = 43 reads), and 83 ± 7% (N = 20 reads), respectively. Error bars are s.e.m.
Discussion
Because model training was done with a small number of short oligonucleotides, our initial model was built from relatively few unique sequence contexts for each 4-mer (Supplementary Fig. 15). Many 4-mers in our sequencing model are based on a single sequence context. Further, while 4-mers characterize most of the variation observed in variable-voltage conductance curves, we found previously that 6-mers provide a more complete characterization with ACGT DNA. In a 4-mer model, the surrounding sequence context effectively adds increased variance to 4-mer measurements44. Because we’ve only measured most 4-mers in one or two sequence contexts, we anticipate that sequencing accuracy for ALIEN DNA can be improved dramatically by scaling up the training library size 10 or 100 fold. For example, the 6-mer ACGT model parameterized 46 = 4096 unique ACGT 6-mers using two separate kilobase-scale genomic DNA templates for model training measurements44, compared to the 44 = 256 4-mers parametrized here using 12 short oligos. The datasets used to train the ONT machine learning sequencing models are many orders of magnitude larger49. Given how well our limited 4-mer sequencing model performs, we expect that the use of larger ALIEN training libraries will allow the accuracy of ALIEN sequencing to converge with or exceed that of standard nanopore sequencing of four-nucleotide ACGT DNA.
The high error rate of calling B compared with the other bases remains conspicuous. Future improvements in identifying B will have the largest impact on total ALIEN sequencing performance. The tendency of B to undergo tautomerization with a non-trivial proportion of B bases adopting an enolic form may play a role in its sequencing difficulty50. Other origins of systematic model errors may include errors from the phosphoramidite chemistry used to synthesize the ALIEN oligonucleotides. This synthesis process is known to produce strands with mutations and chemical adducts37 that are known to affect the nanopore signal and could contaminate both the training and testing datasets. Because B is the leading contributor to ALIEN DNA sequencing errors, 2D or 1D2 sequencing strategies51, in which reads of both the primary strand and its complement are combined to yield a higher accuracy read, should deliver significant improvements in sequencing accuracy even at the single-molecule level, because the complementary base S is sequenced with higher accuracy than B.
The DNA oligos used here consisted of short sections of ALIEN DNA sandwiched between adapters made of ACGT DNA. This was primarily a design choice. Nanopore strand sequencing does not read the first or last several bases of each strand. This is not an issue for sequencing because in practice sequencing adapter strands are ligated to either end of the strand of interest. Due to the higher per-base cost of solid phase synthesis for bases PZSB we chose to use sections of ACGT for these unread sections of the DNA. A leading error in solid-phase synthesis is missed base incorporation at the single molecule level. While poly-acrylamide gel electrophoresis (PAGE) can be used to ensure strand purity, this error mode significantly affects yield for strands significantly longer than 100 nucleotides. Thus, all strands used here were 90 nucleotides long.
In previous work, we found that ALIEN DNA bases interacted somewhat differently with the DNA motor enzyme as compared to standard bases ACGT, modifying motor enzyme step kinetics. We analysed our training and test data to determine how enzyme step dwell time and the probability of an enzyme backwards step were affected by ALIEN bases within the enzyme (Supplementary Information section S4). Similar to canonical bases52, we find that ALIEN bases affect these kinetic observables in a sequence-specific manner (Supplementary Fig. 14). For example, we observe that B is associated with a higher rate of backwards stepping when it is 17 nt upstream of the pore constriction. Such sequence-dependent kinetic observables could be integrated into the nanopore sequencing algorithm, resulting in higher basecall accuracy as compared to using the ion current signal alone. As reported previously, Hel308 helicase appeared to have a higher rate of dissociation from the DNA strand in regions dense in C-glycoside bases S and Z, however the 28 nucleotide long region of ALIEN DNA used here was not a significant challenge. Sequencing of longer sections of ALIEN DNA may require further optimization of the Hel308 helicase via mutation or use of another motor enzyme more compatible with PZBS DNA. Sequencing of 28 consecutive PZBS bases shown here would be sufficient for sequencing of ALIEN DNA aptamers.
This demonstration of full-factorial de novo sequencing of a DNA analog containing entirely artificial bases is an important step forward for the many burgeoning fields that use anthropogenic nucleic acid systems. We show that systems which are biomimetic and yet biorthogonal can be easily substituted into nanopore sequencing workflows to generate training data and enable sequencing of new alphabets in short order with minimal additional development. The system used here is not yet optimized for sequencing accuracy, throughput, sample size, or read-length with ALIEN DNA. The ONT nanopore platforms have orders-of-magnitude higher sequencing throughput than the custom nanopore setup used here, and suggest what may be achievable for sequencing of ALIEN DNA. It is conceivable that with a large and complex enough training library, one could generate training data for an entire high-accuracy sequencing model for a new artificial alphabet in a single experiment. However, training a 9-mer sequencing model would require a training library 45 = 1024 times the size of the current training set. Additionally, the efficacy of the ONT helicase to translocate on contiguous sections of ALIEN DNA remains untested. Typical input requirements for experiments reported here are ~1ul of 500 nMolar DNA (0.5pmol of DNA). Again, the commercial ONT devices suggests that through optimization, input requirements for nanopore sequencing of ALIEN DNA can be reduced 100-1000 fold.
Still, this work shows how nanopore systems can be readily adapted to directly sequence entirely anthropogenic informational molecules. This will enable faster and more economical progress in the field of synthetic biology19,53, technology that leverages anthropogenic bases in molecular diagnostics54,55,56, evolvable nucleic acid ligands/catalysts/receptors12,13,15,50, and ultra-dense digital data storage6,57,58,59. It may even help humankind search for alien life, which by theory must be able to support Darwinian evolution, but need not do so with the same biopolymers as used in all (known) life forms here on Earth60.
Methods
Pore establishment
A single M2-NNN-MspA nanopore was established in a 1,2-di-O-phytanyl-sn-glycero-3-phosphocholine (DOPHPC) lipid bilayer using the painting method61. Lipids were purchased from Avanti Polar Lipids. Cis and trans wells contained 400 mM KCl, 10 mM HEPES, buffered to pH 8.00 ± 0.05. The cis well additionally contains 10 mM MgCl2 and 1 mM ATP. With a bilayer established ~1 ul of M2-NNN MspA is added to the cis well and mixed. A single MspA insertion into the bilayer is detected by a jump in the measured ion current to ~145 pA with 180 mV applied. Then remaining MspA in solution is washed out via perfusion of 1 ml of buffer.
DNA preparation
DNA oligos containing AEGIS nucleotides were constructed using AEGIS phosphoramidites (Firebird Biomolecular Sciences LLC. www.firebirdbio.com) synthesized on an ABI 394 instrument. Oligonucleotides were purified on 10% denatured PAGE and then desalted on C18 Sep-Pak cartridge, lyophilized and shipped from Florida to the University of Washington. DNA strands were resuspended in 100 mM KCl to a final concentration of 20 µM. Sequences were then mixed with their complement DNA strand and were heated to 90 °C and slowly cooled by ∼10 °C per minute to 4 °C to form dsDNA constructs shown in Fig. 1C. Once annealed, these constructs were further diluted to a concentration of 1 µM and ~1 µl additions of this strand was added to the cis chamber. A complete list of DNA strands used in this work is given in Table S1.
Nanopore experiment
DNA molecules bound to Hel308 helicase are electrophoreticaly captured by the nanopore. As the 5′ end of the DNA is pulled into the pore, the complementary strand is displaced by the pore leaving only the ssDNA template with Hel308 resting on the pore rim. Hel308 will then translocate from 3′ to 5′ in discrete steps approximately half a nucleotide in length42, drawing the ssDNA back out of the pore. Over time ATP is converted to ADP within the cis well. We perfused new reagents into the cis well every 45 min to maintain consistent [ATP]. Variable voltage sequencing is performed by applying a periodic voltage waveform (200 Hz triangle wave, 100 mV peak-to-peak offset to 150 mV) to repeatedly stretch and relax DNA within the pore vestibule, “flossing” the DNA in the constriction by ±1 nt much faster than a typical Hel308 step (~10 Hz).
Data acquisition
Data was acquired with custom LabVIEW software on an Axopatch 200B amplifier at 50 kHz. During experiments, we applied a 200 Hz, 100 mV peak-to-peak triangle waveform in addition to a constant 150 mV DC offset.
Operating conditions
All experiments were run at 400 mM trans [KCl] and 400 mM cis [KCl] with 10 mM HEPES at pH 8.0 and 10 mM [MgCl2] (cis only) at temperature 37 ± 1 degrees Celsius. Once a single M2-NNN MspA nanopore was established, a buffer with the above conditions along with ATP was perfused to the cis well. ATP was ordered from Sigma Aldrich. Experiments were performed at various ATP concentrations between 100 and 1000 µM, but the majority of data was collected at 200 µM ATP. The perfusion is done to maintain constant concentrations of the reactants/products in the reaction volume. DNA, DTT, and Hel308 were added to final concentrations of 10 nM, 1 mM, and 200 nM, respectively. Reactants/products were re-perfused every 45 min.
Proteins
Hel308 from Thermococcus gammatolerans EJ3 (accession number WP 015858487.1 [https://www.ncbi.nlm.nih.gov/protein/WP_015858487.1/]) and M2-NNN-MspA (accession number CAB56052.1 [https://www.ncbi.nlm.nih.gov/protein/CAB56052.1], with the mutations (D90N/D91N/D93N/D118R/D134R/E139K)) were expressed in E. coli. and purified using a 6xhis-tag.
Validation of sequences containing PZBS bases
Synthesis errors of oligos by solid phase synthesis can manifest in two categories: systematic errors and random single-molecule errors. Systematic errors could include the wrong sequence being made, contaminated phosphoramidites, or sequence biases such as secondary structures which systematically affect synthesis. Random errors can include incomplete deprotection of nucleotides, instances of unwanted chemical adducts which can be attached to bases or chemical degradation products such as abasic lesions. Put simply, systematic errors are those which manifest uniformly across all copies of the strand, while random errors are different strand to strand.
P, Z, S, and B phosphoramidites undergo tight quality control. dP, dZ, and dS phosphoramidites were purchased from Firebird Biomolecular (Purity are 97%, 98%, and 97%, respectively). dB phosphoramidite was purchased from Glen Research (Purity is 99.5%)
Previous Liquid Chromatography-Mass Spec. performed on homopolymer strands used in Thomas et al.37 showed such strands are successfully produced using solid phase synthesis. Leading errors consisted of single cyanoethyl base adducts and single-base deletions, both of which can be assumed to be distributed randomly along the DNA strands. Roughly 30% of such molecules contained either a base deletion or cyanoethyl base adduct somewhere along their 90-nucleotide length. This is supported by observed variations in phosphoramidite synthesized DNA within our lab (including ACGT-DNA purchased from IDT). Further, previously published results verified proper construction of PZSB strands constructed by solid phase synthesis via X-ray crystallography.
All constructs used in this work were PAGE purified after synthesis, showing predominantly full-length constructs so systematic errors resulting in change of strand length can be ruled out. In addition, nanopore reads of the synthesized DNA contained reads of the natural DNA handles at both the 5′ and 3′ ends (Fig. 2). Further, nanopore traces contained the expected number of steps for the number of bases contained in each strand. Training data was also checked for internal consistency. For example, several k-mers (e.g. “BBBZ”, “ZBZP” and “PZSS”) appear multiple times within different sequencing contexts within the training strands and each time produce similar conductance curves. Additionally, the ability of the sequencing model to predict ion currents of previously unmeasured sequences (Fig. 3B) and to sequence with meaningful accuracy is also proof positive that the desired strands were indeed constructed as designed in both the training and test datasets.
Chemical names.
B:isoguanine 6-amino-9[(1′-β-D-2′-deoxyribofuranosyl)-4-hydroxy-5-(hydroxymethyl)-oxolan-2-yl]-1H-purin-2-one
P: 2-amino-8-(1′-β-D-2′-deoxyribofuranosyl)-imidazo-[1,2a]-1,3,5-triazin-[8H]-4-one
S: 3-methyl-6-amino-5-(1′-β-D-2′-deoxyribofuranosyl)-pyrimidin-2-one
Z: 6-amino-3-(1′-β-D-2′-deoxyribofuranosyl)-5-nitro-1H-pyridin-2-one
Change point detection
A key requirement of enzyme-actuated nanopore sequencing is the segmentation of a conductance trace into segments corresponding to the discrete steps taken by the motor enzyme. Here we use the change-point detection algorithm62 previously demonstrated44 with slight improvements. Previously, change point detection was conducted on a model composed of 5 principal components of periodic functions that represent the raw variable-voltage data of a single read44. In this work, we first subtract the mean cycle ion current for each read individually and use this signal representation solely for the purposes of change point detection. This helps to calibrate out pore-to-pore and read-to-read variations in pore ion current, removing significant systematic errors. This works because the important feature of the data is the sequence-dependent change in pore conductance. Ion current through the nanopore changes non-linearly with applied voltage in ways that depend on the capacitance of the setup. This is known to be variable for example, due to changes in bilayer area/thickness, subtraction of the event-average cycle ion current removes all elements of the non-linear conductance change and isolates the part which we care about: the sequence-dependent change in conductance at various applied voltages.
We construct a model of the variable-voltage data based on principal component vectors, but model the mean cycle normalized signal instead of the raw signal. The mean cycle current subtraction allows us to reduce our model complexity to 2 principal components (Supplementary Fig. 1) as compared to the 5 used in Noakes et al. 44.
Capacitance compensation
Because of the applied voltage waveform in variable-voltage sequencing, we must subtract out the large charging and discharging ion current due to the large capacitance of the lipid bilayer membrane that supports the nanopore if we hope to extract information from the signal. We use the identical computational strategy detailed in ref. 44, which assumes that the non-capacitive component of the positive-going and negative-going ion currents should be identical when variable voltage is applied as a triangle wave. This is because the capacitive current depends only on the rate of change of the applied voltage which, with a triangle wave, has the same magnitude but opposite sign. Thus the nanopore conductances at a given voltage can be calculated as the mean of the up-going and down-going currents.
Conductance normalization
As in ref. 44, we perform a normalization step to remove the remaining DNA-position independent contribution to each state’s conductance curve. This nuisance component is predominately caused by two factors: the intrinsically non-ohmic character of the nanopore conductance when translocating a charged molecule, and the effects of voltage-dependent DNA stretching on the number of bases sensed by the pore constriction. To address the first factor, we find the average conductance at each voltage over each state in a read and subtract this from each state’s conductance curve. The second factor can be subtracted to first order by subtracting a linear component from each curve segment. We find the magnitude of the linear voltage response as a function of conductance by finding the average linear component for each curve averaged over all curve segments in a read. This average slope is independent of the DNA sequence and can be interpreted as the pore’s average change in conductance as a function of applied voltage. We then subtract this line from each state’s conductance curve. The resulting curve segment called the “normalized conductance” contains primarily the DNA-position dependent portion of the nanopore’s conductance curve. This process is originally described in ref. 44.
Principal component reduction
As in ref. 44, we reduce the dimensionality of the conductance curves through principal component reduction, using linear combinations of the top 3 principal component vectors to characterize a conductance curve. This allows for more efficient computation during sequencing and better estimates of feature covariances while retaining 98% of the variance between conductance curves. Because the 3 top principal component vectors roughly correspond to the basic shape characteristics of a conductance curve, they generalize satisfactorily to ALIEN sequencing.
Noise measurement
Noise is calculated by taking the standard deviation of the conductance measurements at each voltage sampled (Eq. 1). The mean of this is taken over all voltages and serves as the holistic noise measurement of the state. The standard error of the measurement noise is computed in Eq. 2, where N is defined as the total number of conductance measurements across all sampled voltages. This is converted into stiffness (Eq. 3) for inclusion into the 4×4 stiffness matrix, which characterizes the full state certainty.
$${sigma }_{G}={{rm{mean}}}({sigma }_{{G}_{V1}},{sigma }_{{G}_{V2}},ldots,{sigma }_{{G}_{V101}})$$
(1)
$${sigma }_{{sigma }_{G}}=,frac{{sigma }_{G}}{sqrt{2N-2}}$$
(2)
$${K}_{{sigma }_{G}}=,{sigma }_{{sigma }_{G}}^{-2}$$
(3)
Filtering
Data is next filtered to 1) remove spurious “bad levels” and 2) consolidate over-segmented data and enzyme backsteps. We determine the optimal ordering of observed conductance states through a two-stage filtering process as described previously44 with slight modifications.
The removal filter finds and removes observed states that are not informative of the DNA sequence. They may occur due to transient noise spikes, mid-read gating of the nanopore, enzyme-specific transients (“flickers”), and over-segmented states. The removal filter of44 assigns a bad state probability to each state using a 12-feature Support Vector Machine (SVM). Input features 1–9 are the principal component coefficients of the evaluated state, the previous state, and the subsequence state. Feature 10 is the value of the single conductance measurement in a state that maximally deviates from the mean conductance of the read. Feature 11 characterizes the deviation between the principal component reduced curve and the original conductance curve. Feature 12 is the score of the best match through alignment of the evaluated state with the ACGT 6-mer model. All of these features, with the exception of feature 12, generalize to measurements of ALIEN DNA, so we use the removal filter with feature 12 disabled and tune the sensitivity threshold accordingly.
The recombination filter finds and combines states that represent repeated measurements of the same DNA position within a read. This filter fixes errors due to over-segmentation arising during change point detection, resulting in consecutive states of the same DNA position, as well as repeated states due to the enzyme stepping backward (towards 3′) along the DNA. Because change-point detection and enzyme stepping behavior are similar in ALIEN sequencing, we use the recombination filter as-is.
Read polishing
The final quality control step in read processing is to polish the reads by hand to remove any remaining “bad”, “hold”, or backstep states missed by the previous filtering process. We also trim states at both ends of the read to match the bounds of the consensus trace, typically only redundant calibration region states caused by homopolymer T’s at the 5′ end and redundant states caused by GA repeats at the 3′ end (Supplementary Fig. 4).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The nanopore data generated in this study have been deposited in the figshare database under accession code https://doi.org/10.6084/m9.figshare.28199681.v263.
Code availability
The analysis code underlying this article are available in figshare and can be accessed at https://doi.org/10.6084/m9.figshare.28199681.v263.
References
-
Benner, S. A. Rethinking nucleic acids from their origins to their applications. Philos. Trans. R. Soc. B: Biol. Sci. 378, 20220027 (2023).
-
Kool, E. T., Morales, J. C. & Guckian, K. M. Mimicking the Structure and Function of DNA: Insights into DNA Stability and Replication. Angew. Chem. Int. Ed. 39, 990–1009 (2000).
-
Henry, A. A. & Romesberg, F. E. Romesberg, Beyond A, C, G and T: augmenting nature’s alphabet. Curr. Opin. Chem. Biol. 7, 727–733 (2003).
-
Geyer, C. R., Battersby, T. R. & Benner, S. A. Nucleobase pairing in expanded Watson-Crick-like genetic information systems. Structure 11, 1485–1498 (2003).
-
Hirao, I. et al. An unnatural base pair for incorporating amino acid analogs into proteins. Nat. Biotechnol. 20, 177–182 (2002).
-
Tabatabaei, S. K. et al. Expanding the Molecular Alphabet of DNA-Based Data Storage Systems with Neural Network Nanopore Readout Processing. Nano Lett. 22, 1905–1914 (2022).
-
Liu, H. et al. A Four-Base Paired Genetic Helix with Expanded Size. Science 302, 868–871 (2003).
-
Hoshika, S., Shukla, M. S., Benner, S. A. & Georgiadis, M. M. Visualizing “Alternative Isoinformational Engineered” DNA in A- and B-Forms at High Resolution. J. Am. Chem. Soc. 144, 15603–15611 (2022).
-
Hoshika, S. et al. Hachimoji DNA and RNA: A genetic system with eight building blocks. Science 363, 884–887 (2019).
-
Shukla, M. S., Hoshika, S., Benner, S. A. & Georgiadis, M. M. Crystal structures of ‘ALternative Isoinformational ENgineered’ DNA in B-form. Philos. Trans. R. Soc. B: Biol. Sci. 378, 20220028 (2023).
-
Zhang, L. et al. An Aptamer-Nanotrain Assembled from Six-Letter DNA Delivers Doxorubicin Selectively to Liver Cancer Cells. Angew. Chem. Int. Ed. 59, 663–668 (2020).
-
Jerome, C. A., Hoshika, S., Bradley, K. M., Benner, S. A. & Biondi, E. In vitro evolution of ribonucleases from expanded genetic alphabets. Proc. Natl. Acad. Sci. 119, e2208261119 (2022).
-
Biondi, E. et al. Laboratory evolution of artificially expanded DNA gives redesignable aptamers that target the toxic form of anthrax protective antigen. Nucleic Acids Res. 44, 9565–9577 (2016).
-
Biondi, E. & Benner, S. A. Artificially Expanded Genetic Information Systems for New Aptamer Technologies. Biomedicines 6, 53 (2018).
-
Zhang, L. et al. Aptamers against Cells Overexpressing Glypican|3 from Expanded Genetic Systems Combined with Cell Engineering and Laboratory Evolution. Angew. Chem. Int. Ed. 55, 12372–12375 (2016).
-
Shaker, S. et al. Cancer cell target discovery: comparing laboratory evolution of expanded DNA six-nucleotide alphabets with standard four-nucleotide alphabets. Nucleic Acids Res. 53, gkaf072 (2025).
-
Yang, Z., Chen, F., Chamberlin, S. G. & Benner, S. A. Expanded genetic alphabets in the polymerase chain reaction. Angew. Chem. Int. Ed. 49, 177–180 (2010).
-
Yang, Z., Benner, S. A. Compositions for the Multiplexed Detection of Viruses (2020).
-
Zhou, A. X.-Z., Sheng, K., Feldman, A. W. & Romesberg, F. E. Progress toward Eukaryotic Semisynthetic Organisms: Translation of Unnatural Codons. J. Am. Chem. Soc. 141, 20166–20170 (2019).
-
Bain, J. D., Switzer, C., Chamberlin, R. & Benner, S. A. Ribosome-mediated incorporation of a non-standard amino acid into a peptide through expansion of the genetic code. Nature 356, 537–539 (1992).
-
Yang, Z., Hutter, D., Sheng, P., Sismour, A. M. & Benner, S. A. Artificially expanded genetic information system: a new base pair with an alternative hydrogen bonding pattern. Nucleic Acids Res. 34, 6095–6101 (2006).
-
Wang, X. et al. Biophysics of Artificially Expanded Genetic Information Systems. Thermodynamics of DNA Duplexes Containing Matches and Mismatches Involving 2-Amino-3-nitropyridin-6-one (Z) and Imidazo[1,2-a]-1,3,5-triazin-4(8H)one (P). ACS Synth. Biol. 6, 782–792 (2017).
-
Pham, T. M. et al. DNA Structure Design Is Improved Using an Artificially Expanded Alphabet of Base Pairs Including Loop and Mismatch Thermodynamic Parameters. ACS Synth. Biol. 12, 2750–2763 (2023).
-
Yang, Z., Chen, F., Alvarado, J. B. & Benner, S. A. Amplification, Mutation, and Sequencing of a Six-Letter Synthetic Genetic System. J. Am. Chem. Soc. 133, 15105–15112 (2011).
-
Hollenstein, M., Hipolito, C. J., Lam, C. H. & Perrin, D. M. A self-cleaving DNA enzyme modified with amines, guanidines and imidazoles operates independently of divalent metal cations (M 2+). Nucleic Acids Res. 37, 1638–1649 (2009).
-
Gold, L. et al. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLOS ONE 5, e15004 (2010).
-
Manrao, E. A. et al. Reading DNA at single-nucleotide resolution with a mutant MspA nanopore and phi29 DNA polymerase. Nat. Biotechnol. 30, 349–353 (2012).
-
Loman, N. J. & Watson, M. Successful test launch for nanopore sequencing. Nat. Methods 12, 303–304 (2015).
-
Kasianowicz, J. J., Brandin, E., Branton, D. & Deamer, D. W. Characterization of individual polynucleotide molecules using a membrane channel. Proc. Natl. Acad. Sci. 93, 13770–13773 (1996).
-
Laszlo, A. H. et al. Detection and mapping of 5-methylcytosine and 5-hydroxymethylcytosine with nanopore MspA. Proc. Natl. Acad. Sci. USA 110, 18904–18909 (2013).
-
Rand, A. C. et al. Mapping DNA methylation with high-throughput nanopore sequencing. Nat. Methods 14, 411–413 (2017).
-
Schreiber, J. et al. Error rates for nanopore discrimination among cytosine, methylcytosine, and hydroxymethylcytosine along individual DNA strands. Proc. Natl. Acad. Sci. 110, 18910–18915 (2013).
-
Simpson, J. T. et al. Detecting DNA cytosine methylation using nanopore sequencing. Nat. Methods 14, 407–410 (2017).
-
Nookaew, I. et al. Detection and discrimination of DNA Adducts Differing In Size, Regiochemistry, And Functional Group By Nanopore Sequencing. Chem. Res. Toxicol. 33, 2944–2952 (2020).
-
Tavakoli, S. et al. Semi-quantitative detection of pseudouridine modifications and type I/II hypermodifications in human mRNAs using direct long-read sequencing. Nat. Commun. 14, 334 (2023).
-
Brinkerhoff, H., Kang, A. S. W., Liu, J., Aksimentiev, A. & Dekker, C. Multiple rereads of single proteins at single–amino acid resolution using nanopores. Science 374, 1509–1513 (2021).
-
Thomas, C. A. et al. Assessing readability of an 8-Letter Expanded Deoxyribonucleic Acid Alphabet With Nanopores. J. Am. Chem. Soc. 145, 8560–8568 (2023).
-
Kawabe, H. et al. Enzymatic synthesis and nanopore sequencing of 12-letter supernumerary DNA. Nat. Commun. 14, 6820 (2023).
-
Smith, D. C. et al. Nanopores map the acid-base properties of a single site in a single DNA molecule. Nucleic Acids Res. 52, 7429–7436 (2024).
-
Ledbetter, M. P. et al. Nanopore sequencing of an expanded genetic alphabet reveals high-fidelity replication of a predominantly hydrophobic unnatural base pair. J. Am. Chem. Soc. 142, 2110–2114 (2020).
-
Craig, J. M. et al. Revealing dynamics of helicase translocation on single-stranded DNA using high-resolution nanopore tweezers. Proc. Natl. Acad. Sci. 114, 11932–11937 (2017).
-
Derrington, I. M. et al. Subangstrom single-molecule measurements of motor proteins using a nanopore. Nat. Biotechnol. 33, 1073–1075 (2015).
-
Ying, Y.-L. et al. Nanopore-based technologies beyond DNA sequencing. Nat. Nanotechnol. 17, 1136–1146 (2022).
-
Noakes, M. T. et al. Increasing the accuracy of nanopore DNA sequencing using a time-varying cross membrane voltage. Nat. Biotechnol. 37, 651–656 (2019).
-
Laszlo, A. H. et al. Decoding long nanopore sequencing reads of natural DNA. Nat. Biotechnol. 32, 829–833 (2014).
-
Viterbi, A. Error bounds for convolutional codes and an asymptotically optimum decoding algorithm. IEEE Trans. Inf. Theory 13, 260–269 (1967).
-
Durbin, R., Eddy, S. R., Krogh, A., Mitchison, G. Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids (Cambridge University Press, 1998).
-
Ross, B. C. Mutual Information between Discrete and Continuous Data Sets. PLOS ONE 9, e87357 (2014).
-
Wick, R. R., Judd, L. M. & Holt, K. E. Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol. 20, 129 (2019).
-
Eberlein, L. et al. Tautomeric equilibria of nucleobases in the Hachimoji expanded genetic alphabet. J. Chem. Theory Comput. 16, 2766–2777 (2020).
-
Lin, B., Hui, J. & Mao, H. Nanopore technology and its applications in gene sequencing. Biosensors 11, 214 (2021).
-
Craig, J. M. et al. Determining the effects of DNA sequence on Hel308 helicase translocation along single-stranded DNA using nanopore tweezers. Nucleic Acids Res. 47, 2506–2513 (2019).
-
Malyshev, D. A. et al. A semi-synthetic organism with an expanded genetic alphabet. Nature 509, 385–388 (2014).
-
Glushakova, L. G. et al. Detecting respiratory viral RNA using expanded genetic alphabets and self-avoiding DNA. Anal. Biochem. 489, 62–72 (2015).
-
Glushakova, L. G. et al. High-throughput multiplexed xMAP Luminex array panel for detection of twenty two medically important mosquito-borne arboviruses based on innovations in synthetic biology. J. Virological Methods 214, 60–74 (2015).
-
Kawabe, H. et al. Harnessing Non-standard Nucleic Acids for Highly Sensitive Icosaplex (20-plex) Detection of Microbial Threats for Environmental Surveillance. ACS Synth. Biol.14, 470–484 (2025).
-
Ceze, L., Nivala, J. & Strauss, K. Molecular digital data storage using DNA. Nat. Rev. Genet. 20, 456–466 (2019).
-
Church, G. M., Gao, Y. & Kosuri, S. Next-generation digital information storage in DNA. Science 337, 1628–1628 (2012).
-
Goldman, N. et al. Towards practical, high-capacity, low-maintenance information storage in synthesized DNA. Nature 494, 77–80 (2013).
-
Carr, C. E. et al. Advancing the search for extra-terrestrial genomes in 2016 IEEE Aerospace Conference (2016), pp. 1–15.
-
Laszlo, A. H., Derrington, I. M. & Gundlach, J. H. MspA nanopore as a single-molecule tool: From sequencing to SPRNT. Methods 105, 75–89 (2016).
-
LaMont, C. H. & Wiggins, P. A. The Development of an Information Criterion for Change-Point Analysis. Neural Comput. 28, 594–612 (2016).
-
Thomas, C. A., Laszlo, A. H. Craig, Jonathan M., Sequencing a DNA Analog Composed of Artificial Bases, figshare https://doi.org/10.6084/m9.figshare.28199681.v2 (2025).
Acknowledgements
This research was supported by grants from the National Human Genome Research Institute U24HG011735 (AHL, JHG, SAB), the National Institutes of Health, National Human Genome Research Institute R01HG005115 (AHL & JHG), the National Institutes of Health under the Director’s Award Number R01GM128186 (SH & SAB), the National Science Foundation MCB-1939086 (SH & SAB), and the National Institutes of General Medical Sciences 1R01GM141391-01A1 (SH & SAB).
Ethics declarations
Competing interests
The authors declare the following competing financial interest(s): S.A.B. owns Firebird Biomolecular Sciences, which makes various ALIEN reagents available for sale. S.H. is affiliated with Firebird Biomolecular Sciences. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Lingjun Li, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Thomas, C.A., Brinkerhoff, H., Craig, J.M. et al. Sequencing a DNA analog composed of artificial bases. Nat Commun 16, 7240 (2025). https://doi.org/10.1038/s41467-025-61991-9
-
Received:
-
Accepted:
-
Published:
-
DOI: https://doi.org/10.1038/s41467-025-61991-9