Introduction
In vivo genome editing using tools like CRISPR-Cas9 have demonstrated great potential for correcting disease-causing mutations and ensuring sustained correction outcome in monogenic inherited diseases1,2,3,4. This method can also integrate therapeutic protein-coding genes into safe genomic sites to treat loss-of-function conditions, achieving sustainable protein production and long-term efficacy5,6,7,8,9. Conventional homology-directed repair (HDR) is widely recognized as the most effective knock-in method for treading monogenic diseases. However, its reliance on cell division limits its use to proliferating tissues, excluding those with low cell turnover, such as muscle and neurons3. Furthermore, HDR is often plagued by suboptimal knock-in efficiency, hindering its broader therapeutic potential. To overcome this limitation, we previously developed Homology-independent target integration (HITI), which offers a more versatile solution by leveraging the highly accessible non-homologous end joining (NHEJ) repair machinery5. This method facilitates in vivo knock-in across diverse tissues and organs for both dividing and non-dividing cells, such as liver, heart, muscle, and retina5,6,7,8,9. Building on the foundation of HITI, we have developed derivative methods such as SATI, HITI-TE, and Gene-DUET, which show promise for use in inherited disease models, including Hutchinson-Gilford Progeria Syndrome, Inherited GPI Deficiency, and Spinal Muscular Atrophy7,10,11. Furthermore, NHEJ-mediated knock-in, including HITI, supports the insertion of large DNA fragments (up to ~50 kb), making it an exceptionally powerful tool for in vivo genome editing therapies12. This capacity to target multiple organs and insert substantial DNA sequences is particularly valuable for therapeutic protein production within the body, enabling sustained, in vivo synthesis of therapeutic proteins over the long term. Notably, we recently demonstrated that HITI outperforms HDR in terms of knock-in efficacy for GFP gene at the Alb locus in mouse hepatocyte when using clinically relevant, lipid nanoparticle (LNP)-based deliver systems. This result underscores HITI’s broad potential for expanding the applications of in vivo genome editing13.
Recently, genome editing therapies have predominantly targeted diseases with well-defined causative mutations, which affect only about 0.5% to 1% of the global population. Given that more than 7500 genetic disorders and 290,000 disease-causing mutations are cataloged in the OMIM and HGMD databases, it is not feasible to develop clinical-grade vectors for each specific mutation. This genetic complexity of human diseases limits the broad application of gene editing therapies, except for well-known diseases caused by mutations of a single gene, like sickle cell anemia14. Despite advances in treating monogenic diseases, the potential of genome editing for treating more complex noncommunicable diseases (NCDs) remains largely untapped. NCDs, including cardiovascular diseases, diabetes, cancers, chronic respiratory conditions, and obesity, account for 74% of global deaths. The lack of clear genetic targets for these conditions has constrained the application of in vivo genome editing.
Biologics, which are cutting-edge recombinant protein-based large-molecule drugs, have transformed the treatment of many complex and non-genetic diseases, including autoimmune disorders, cancers, and chronic inflammatory conditions. Despite their groundbreaking impact, these medications often require frequent administration due to their short half-lives and the necessity of maintaining consistent therapeutic levels15. This frequent administration can lead to various challenges, such as patient non-compliance, injection-site reactions, immune responses, high costs, storage issues, and potential long-term safety concerns. Achieving a single, lifelong therapeutic outcome from biologics could address these issues, but such advancements remain elusive due to the current limitations in biologics’ half-life.
Obesity is a prevalent, complex, progressive, and relapsing disease characterized by excessive or abnormal body fat, which negatively impacts health. It is influenced by various environmental and genetic factors such as diet, increases the risk of metabolic conditions like type 2 diabetes and cardiovascular diseases16. Biologic medications, such as Glucagon-Like Peptide-1 (GLP-1) receptor agonists, promote weight loss and improve blood glucose levels. Exendin-4 (Exe4), a synthetic peptide that potently activates GLP-1, is known for its effective weight loss properties17,18,19. However, despite its small size (39 amino acids) and low effective dose, Exe4 has a short half-life, requiring frequent injections similar to other biologics. Discontinuation often leads to weight regain, making long-term maintenance challenging. Achieving sustained in vivo production of Exe4 could offer a lasting defense against a range of complex metabolic diseases.
Here, we present an innovative concept in applying in vivo genome editing to treat diseases without a clear genetic underpinning. We engineered Exe4 as an artificial secretion peptide, achieving consistent secretion through a single in vivo knock-in using the HITI method. This approach demonstrated long-term therapeutic effect in a dietary-induced obesity mouse model, offering a promising one-shot genome-editing therapy for non-genetic and complex diseases.
Methods
Plasmid construction
The Exe4 gene was codon-optimized using the GeneSmart codon optimization tool (https://www.genscript.com/gensmart-free-gene-codon-optimization.html) based on the amino acid sequence in existing publication (Supplementary Table 1). To construct expression vectors for the secretion of Exe4, double-stranded DNAs of Exe4 fused with signal peptides (SPs) and furin-cleavable sequences were synthesized (Azenta) and inserted into a CAG promoter-containing pCAG backbone vector using the In-Fusion HD cloning kit (Takara Bio). The 20 bp genomic sgRNA target sequence with a 3 bp PAM (underlined) used in this study is shown as follows: sgAlb-E14 (GTTGTGATGTGTTTAGGCTAAGG), targeting exon 14 of the mouse Albumin (Alb) locus. Notably, this sgRNA enabled efficient on-target integration with no noticeable off-target editing in the context of in vivo knock-in, as demonstrated in a previous study20. The sgAlb-E14 sequence was inserted into the AflII (NEB) site of the gRNA_Cloning Vector (Addgene, #41824) using the In-Fusion HD cloning kit. The structure of the HITI donor plasmid followed the previous report5. In brief, for the HITI donor vector containing the sgAlb-E14 expression cassette and scExe4 donor fragment, the sgAlb-E14 expression cassette and scExe4 (NGF-FCS2-Exe4) sequence flanked by two sgAlb-E14 gRNA target sequences were prepared by PCR using PrimeSTAR GXL polymerase (Takara Bio). These fragments were subcloned into the MluI (NEB) site of the HITI backbone vector (Addgene, #87116) using the In-Fusion HD cloning kit. The size of scExe4 donor plasmid and P2A-scExe4 fragment to be inserted are 6774 bp and 551 bp, respectively. To construct the SpCas9 expression vector (referred to as the Cas9 plasmid), the Cas9 DNA fragment was amplified from hCas9 (Addgene #41815) and inserted into a CMV promoter-containing pCMV backbone vector using the In-Fusion HD cloning kit. The size of Cas9 plasmid is 7859 bp. For the NanoLuc expression vector (NanoLuc plasmid), CMV DNA fragment was amplified from the Cas9 plasmid and replaced with the PGK promoter in the pNL1.1.PGK[Nluc_PGK] Vector (Promega) using the In-Fusion HD cloning kit. All constructs were confirmed by Sanger sequencing. The related sequences are listed in Supplementary Table 2.
Cell lines
Hepa1-6 (mouse hepatocarcinoma cell line) and HepG2 (human hepatocarcinoma cell line) were purchased from KAC and cultured in DMEM (Wako) containing 10% FBS (Biowest). iGL cells, used for the evaluation of glucose-stimulated insulin secretion (GSIS), were purchased from CosmoBio and cultured in RPMI-1640 (Wako) containing 10% FBS. Cell line authentications have been checked by STR analysis. Mycoplasma contamination was checked every 2 months and was found to be negative in all cell lines used.
Animals
All animal procedures were conducted in accordance with the guidelines of the Osaka University Animal Care and Use Committee and received approval from the committee. Mice were housed individually in an approved, ventilation-controlled facility with a 12-h light/dark cycle, at room temperature, and had free access to food and water. Efforts were made to minimize animal use and discomfort. Eight-week-old female Balb/c mice (Oriental Yeast) were utilized for in vivo transfection and knock-in experiments. Female mice were selected to reduce the likelihood of aggression-related stress, which is more frequently observed in group-housed males and may confound metabolic outcomes. Diet-induced obesity (DIO) mice were generated by starting a high-fat diet (HFD-60, lipid 60 kcal%, Oriental Yeast) feeding for C57B6J (B6) mice (Oriental Yeast) at six weeks of age. The pharmacodynamic study was initiated when the mice reached the average body weight around 22 g (around 10 weeks of age). Mice were non-randomly assigned to treatment groups while ensuring that initial body weights were balanced across groups to minimize variability. All animals were maintained under identical housing, handling, and dietary conditions, except for experimental treatments. No animals were excluded from analysis. Blinding was not applied during experiments or data analysis. However, outcome measurements such as ELISA, PCR, and glucose tolerance tests were conducted using objective, quantitative methods with standardized instruments to minimize potential bias. Sample sizes were determined with reference to previously published studies in related fields employing comparable experimental designs and endpoints. Based on this, n = 3–4 mice per group was considered appropriate to detect treatment-associated trends while complying with ethical principles for animal research.
ELISA for Exe4 concentration measurement
Exe4 concentration in the culture medium and mice plasma was measured using the Exendin-4 ELISA kit (Abcam) according to the manufacture’s protocol. Absorbance at 450 nm was measured using a plate reader (Synergy HTX).
Exe4 secretion in hepatocyte-derived cell lines
A series of Exe secretion plasmids containing various SP and FCS (each 1 μg) were transfected into Hepa1-6 and HepG2 cells in 12-well plates using Lipofectamine 3000 (Invitrogen) according to the manufacture’s protocol. One day after transfection, the cells were washed and then incubated for an additional two days. The cell culture medium was subsequently collected, and the Exe4 concentration was measured using the Exendin-4 ELISA kit.
Glucose stimulated insulin secretion (GSIS)
Culture medium containing Exe4 was obtained from Hepa1-6 cells and HepG2 cells transfected with a plasmid expressing NGF-FCS2-Exe4 (referred to as scExe4_Hepa1-6 and scExe4_HepG2, respectively). Additionally, a synthetic Exe4 peptide solution (Wako) was prepared. iGL cells were exposed to different mediums: iGL cells culture medium (Cell only), iGL culture medium supplemented with synthetic Exe4 peptide (Exe4 solution) and with scExe4 peptide secreted from Hepa1-6 and HeG2 cells transfected with the scExe4 expression plasmid (scExe4_Hepa1-6 and scExe4_HepG2, respectively). The Exe4 concentrations in the culture medium for the synthetic Exe4 peptide solution, scExe4_Hepa1-6, and scExe4_HepG2 were 2.5 nM, 2.4 nM, and 2.5 nM, respectively. These media were incubated with iGL cells for 1 h at low (5.6 mM) and high (28 mM) glucose (Wako) concentrations21. The luminescence of Luc-insulin secreted from iGL cells into the culture media was then measured using coelenterazine (CosmoBio) as the substrate. GSIS values were calculated from the luminescence intensity ratio at high (28 mM) to low (5.6 mM) glucose concentrations.
In vitro knock-in of scExe4
Hepa1-6 cells were transfected with scExe4 donor plasmid alone (Donor) or scExe4 donor plasmid with Cas9 plasmid (Donor+Cas9), each 1 μg in 12-well plates using Lipofectamine 3000. The transfected cells were passaged sequentially, and culture medium was collected at passages 2, 5, 8, and 11. The concentration of Exe4 in the culture medium was measured using the Exendin-4 ELISA kit. Genomic DNA from Hepa1-6 at passage 2 and 11 was extracted using DNAeasy Blood & Tissue kit (Qiagen) for further PCR assays, including junction PCR and digital PCR.
Genomic DNA extraction from mouse liver
Liver genomic DNA was extracted 28 weeks post-administration from Balb/c using the All prep DNA/RNA kit, following the manufacture’s protocol.
Junction PCR assay
Genomic DNA extracted from Hepa1-6 cells or mice liver was used as the PCR template for the junction PCR assay. PCR was conducted using PrimeSTAR Max DNA polymerase (Takara Bio) according to the manufacturer’s protocols. The primers employed for junction PCR are outlined in Supplementary Table 3. PCR products were then electrophoresed on a 1% agarose gel, and bands corresponding to the target product size (958 bp for the 5′ junction and 828 bp for the 3′ junction) were identified.
Digital PCR
The QuantStudio Absolute Q Digital PCR System (Thermo Fischer Scientific) was employed for digital PCR analysis. Genomic DNA was digested using MluCI (NEB) and used as the template for the assay. The primers and probes sequences utilized for digital PCR are listed in Supplementary Table 3. The template, primers, and probes were combined with Absolute Q DNA Digital PCR Master Mix (5×) (Applied Biosystems) following the manufacturer’s protocols. The knock-in efficiency was determined using the equation: Knock-in (%) = (Copy number of target)/(Copy number of reference + Copy number of target) × 100.
RT-qPCR
RNA was extracted from various organs (liver, lung, spleen, heart, and kidney) of Balb/c mice from in vivo knock-in experiment using All prep DNA/RNA kit (Qiagen) according to the manufacture’s protocol. The primers used for RT-qPCR are listed in Supplementary Table 3. Reverse transcription from RNA to cDNA was performed using the SuperScript IV First-Strand Synthesis System (Invitrogen). The cDNA samples and primers were combined with SsoAdvanced Universal SYBR Green Supermix (BioRad) and RT-qPCR was conducted using the CFX384 Touch Real-Time PCR System (BioRad). Data analysis was carried out using the ΔΔCt method, and the fold RNA expression levels for Exe4 and Cas9 were calculated by normalizing with the Ct values of the internal control (Gapdh) and the saline-treated control groups.
LNP preparation
All lipids used in this study were purchased from commercial vendors: DLin-MC3-DMA from Cayman Chemicals, Cholesterol from Sigma-Aldrich and DSPC and PEG2000-DMG from Avanti Polar Lipids. NanoLuc-LNP, Donor-LNP, and Cas9-LNP were prepared using the ethanol dilution method22. This involved adding a lipid-ethanol solution to a plasmid solution in 25 mM citrate buffer (pH 4.0), followed by buffer replacement with PBS through diafiltration using Amicon Ultra Centrifuga Filter (10 kDa MWCO, Millipore). All procedures were conducted at ambient temperature. To generate both scExe4 and Cas9 plasmid-loaded LNPs, the molecular ratio of DLin-MC3-DMA/Cholesterol/DSPC/PEG2000-DMG was 50/38.5/10/1.5, following a previously established protocol23,24,25. The LNP samples were diluted with PBS (pH 7.4), and the z-average diameter, polydispersity index, and zeta potential were measured using dynamic light scattering (Zetasizer Nano ZS, Malvern Instruments). The encapsulation efficiency of DNA into the LNP was determined by the Picogreen assay (Thermo Ficher Scientific) following the manufacturer’s protocol. The characterization data for LNPs are summarized in Supplementary Table 4.
In vivo delivery of NanoLuc expressing plasmid DNA
To evaluate DNA delivery efficiency, luciferase (Luc) activity was measured in major organs (liver, lung, spleen, kidney and heart) of 6-8 weeks-old female Balb/c mice. Mice received tail vein injection of saline (Saline), NanoLuc plasmid-loaded LNP (LNP), or hydrodynamics injection of saline solution with NanoLuc plasmid (HD). The dose of plasmid for both LNP and HD was 0.5 μg g−1, with injection volumes of 10 μl g−1 for LNP and 100 μl g−1 for HD. Mice were euthanized 12 h after treatment by CO2 inhalation, and major organs were collected. Organ lysates were prepared by homogenization using Lysis Buffer (Promega) according to manufacturer’s protocol. Luc activity in organ lysates was measured using Nano-Glo Luciferase Assay System (Promega). The ratio of Luc activity (%) corrected by liver Luc activity was calculated for other organs (lung, spleen, heart and kidney) to assess liver specificity of transfection.
In vivo knock-in study with Balb/c mice
Eight-week-old female Balb/c mice were utilized for an in vivo knock-in experiment to investigate the pharmacokinetic profile of Exe4 produced by scExe4 knock-in. The mice were divided into three groups and treated with tail vein injections of either saline (Saline), scExe4 donor plasmid loaded LNP alone (Donor), or scExe4 donor plasmid loaded LNP plus Cas9 plasmid loaded LNP (Donor+Cas9). The dose of both donor plasmid and Cas9 plasmid was 0.5 μg g−1, with an injection volume of 10 μl g−1 for all treatment groups. Body weight was monitored throughout the study. Blood samples were collected by facial vein puncture at each time points to measure Exe4, AST, ALT, and albumin concentrations. Blood samples were centrifuged at 3000 × g for 15 min at ambient temperature to collect supernatant plasma. The concentrations of Exe4 and other hepatic markers in plasma were assessed using commercially available assay kits (Exe4: Abcam, AST and ALT: Sigma-Aldrich, Albumin: Fuji Rebis) following the manufacturer’s protocols, and the absorbance of each sample was measured using a plate reader. At 28 weeks after initial treatment, the mice were euthanized by CO2 inhalation, and the major organs (liver, lung, spleen, kidney and heart) were collected. DNA and RNA were extracted using All prep DNA/RNA kit following the manufacturer’s protocol and used for various PCR-based assays.
Pharmacodynamic study in DIO mice
Female DIO mice with a body weight approximately 22 g received tail vein injections of either saline (Saline), scExe4 donor plasmid loaded LNP only (Donor), or scExe4 donor plasmid loaded LNP plus Cas9 plasmid loaded LNP (Donor+Cas9). The dose for both the donor and Cas9 plasmid was each 1 μg g−1, with an injection volume of 10 μl g−1 for all treatment groups. Additionally, osmotic pump (ALZET) filled with synthetic Exe4 peptide solution (Vehicle: 50% DMSO in 1% BSA aqueous solution) were subcutaneously implanted to DIO mice (Peptide) to mimic daily Exe4 peptide injections. This pump was designed to release synthetic Exe4 peptide solution over 4 weeks. The pumps were implanted twice at 0 and 4 weeks after initial treatment but were removed at 8 weeks after initial treatment to simulate interruption. Weekly food intake and body weight were measured every 2 weeks. Blood and plasma were collected by facial vein puncture to measure plasma Exe4 concentration.
Glucose tolerance test (GTT)
The GTT was performed at 8, 16, and 24 weeks after the initial treatment. After overnight fasting, mice received an intraperitoneal injection of 2 mg glucose g−1. Blood samples were collected from the tail vein, and the blood glucose concentration (mg dl−1) was measured at 0, 30, 60, 90, and 120 min after administration using a LabGluco tester (Research & Innovation Japan). The area under the curve (AUC) of blood glucose concentration was calculated using the conventional trapezoidal method.
Insulin tolerance test (ITT)
The ITT was performed at 8, 16, and 24 weeks after initial treatment. After overnight fasting, mice received an intraperitoneal injection of 0.65 U kg−1 of recombinant insulin (WAKO). Blood samples were collected from the tail vein, and the blood glucose concentration was measured at 0, 30, 60, 90, and 120 min after administration using a LabGluco tester. The blood glucose reduction level was calculated by dividing the blood glucose concentration by the concentration before insulin treatment. AUC of the blood glucose reduction level was calculated using the conventional trapezoidal method.
Oral glucose challenge and GLP-1 measurement
Oral glucose challenge and plasma GLP-1 measurement were conducted 24 weeks after the initial treatment in DIO mice. Following an overnight fast, mice received an oral gavage of glucose at a dose of 2 mg per gram of body weight. Blood samples were collected via facial vein puncture at 0, 30, 60, and 120 min post-administration for plasma GLP-1 concentration analysis. Blood glucose levels were measured at the same time points using a commercially available ELISA kit (IBL Japan). The AUC for plasma GLP-1 concentration was calculated using the conventional trapezoidal method.
Measurement of plasma HbA1c concentration
HbA1c concentration in plasma samples collected from DIO mice at 8, 16, and 24 weeks after the initial treatment of the pharmacodynamic study was determined using a mouse HbA1c ELISA kit (Aviva Systems Biology) according to the manufacturer’s protocols. The absorbance at 450 nm was measured using a plate reader.
Measurement of plasma insulin concentration
Insulin concentration in plasma samples collected from DIO mice at 8, 16, and 24 weeks after the initial treatment of the pharmacodynamic study was determined using a mouse insulin ELISA kit (Mercodia) according to the manufacturer’s protocols. The absorbance at 450 nm was measured using a plate reader.
5′-rapid amplification of cDNA ends (RACE)-based genome-wide off-target analysis
5′-RACE was performed using the SMARTer RACE 5′/3′ Kit (Takara Bio) according to the manufacturer’s instructions. A total 1 μg of RNA was used for the reaction. The scExe4-specific primer used was 5′-GATTACGCCAAGCTTCCTCGGGCACGTTGCTATCAGTGTAGGGTT −3′. PCR products were cloned using the In-Fusion HD Cloning and sequenced with an ABI 3730xl sequencer (Genewiz). Captured exons located to upstream of scExe4 were mapped to the UCSC mouse genome browser (NCBI37/mm9) (https://genome.ucsc.edu/cgi-bin/hgGateway?db=mm9). Data were obtained from liver tissues of Balb/c mice at 28 weeks post-administration. Sanger sequencing of non-specific bands confirmed that they originated from 18S rRNA and did not contain scExe4.
On-target and off-target analysis by amplicon sequencing
Liver genomic DNA from in vivo knock-in experiments with Balb/c mice was analyzed for on-target and off-target effects using amplicon sequencing on a MiSeq platform (Illumina). The top 10 off-target sites were predicted using The COSMID Tool (http://crispr.bme.gatech.edu), following a previously established approach20. Primers for amplicon preparation are listed in Supplementary Table 5. Data from amplicon sequencing were processed and analyzed by CRISPResso2 (http://crispresso2.pinellolab.org/submission).
Statistics and reproducibility
GraphPad Prism 10 was used for both statistical analysis (ANOVA) and generation of bar plots. All other graphs were generated in Excel. The sample sizes for both in vitro transfection studies (Hepa1-6 and HepG2 cells) and in vivo studies (Balb/c and DIO mice) were predetermined. Limited material availability necessitated that the off-target analysis and 5′-RACE assay be performed with one sample per group. To ensure reproducibility, 5′-RACE assays were conducted utilizing two distinct primer pairs per group. All specific sample sizes are further detailed within the corresponding figure captions.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Results
Development of the secreted Exe4 peptide-coding gene
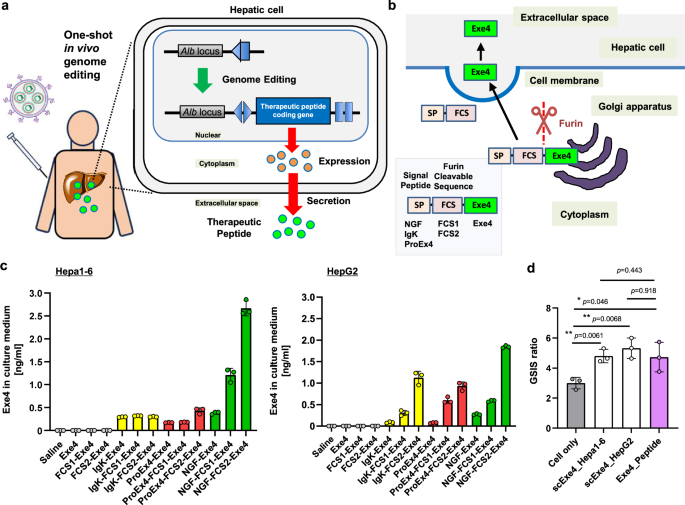
To develop a one-shot in vivo genome editing-based therapy for DIO and pre-diabetes, we targeted the liver due to its large size and ease of transduction, anticipating effective therapeutic peptide/protein production and secretion following successful genome editing (Fig. 1a). We fused a signal peptide (SP) and a furin-cleavable sequence (FCS) to the upstream region of the Exe4 gene to enable Exe4 secretion from the edited liver. The SP promotes protein secretion from cells, while the FCS is cleaved in the Golgi apparatus and on cell membrane, leading to the secretion of bioactive Exe4 (Fig. 1b). To ensure optimal Exe4 secretion from hepatic cells, we engineered a series of secreted Exe4 expression plasmids with various SP (NGF, IgK, and ProEx4) and FCS (FCS1 and FCS2) combinations, and transfected them into hepatic Hepa1-6 and HepG2 cells (Supplementary Table 1)26,27,28,29,30,31,32. Two days later, we quantified the secreted Exe4 protein from the collected culture medium (Fig. 1c). While Exe4 was undetectable in the culture medium after transfection of Exe4 alone, the fusion with FCS and SP effectively enhanced secretion, with the combination of NGF-FCS2 yielding the highest Exe4 secretion in both cell lines. Thus, NGF-FCS2 was selected to engineer the secreted Exe4 in hepatic cells, hereafter referred to as scExe4.

a Scheme depicting the concept of this research, showing genome editing-mediated stable secretion of therapeutic peptides in the liver. b Designs of SP and FCS fused Exe4 (scExe4) and schematic of its secretion from the cell. c Secretion of Exe4 in the culture medium following transfection of a series of scExe4 expression plasmids in Hepa1-6 cells and HepG2 cells. Data are shown as mean ± SD (n = 3 biological replicates). d Glucose stimulated insulin secretion (GSIS) of iGL cells induced by adding cultured medium from scExe4 (NGF-FCS2-Exe4) transfected Hepa1-6 and HepG2 cells. Data are shown as mean ± SD (n = 3 biological replicates). Asterisks (*: p < 0.05, **: p < 0.01) indicate a significant difference among comparing groups using one-way ANOVA. No significant difference (no asterisk; p > 0.05) was observed between the scExe4_Hepa1-6 and Exe4_Peptide groups, nor between the scExe4_HepG2 and Exe4_Peptide.
The bioactivity of scExe4 was evaluated through measuring GSIS using pancreatic iGL cells, which release Gaussia luciferase-fused insulin in response to high glucose levels21. Incubating iGL cells with medium containing secreted Exe4 from scExe4-transfected cells significantly increased GSIS to a level comparable to that of iGL cells treated with commercial synthetic Exe4 peptide solution (Fig. 1d). This indicates that the bioactivity of scExe4 is equivalent to that of synthetic Exe4 peptide.
Secretion of Exe4 peptide via gene knock-in in Hepa1-6 cells
To achieve sustained Exe4 secretion, we targeted the Albumin (Alb) locus due to its hepatocyte-specific high expression levels, ensuring robust expression of the integrated scExe4 gene9,33,34. Employing HITI for in vivo knock-in of large DNA fragments, we designed a guide RNA (gRNA) targeting exon 14 of the Alb gene (sgAlb-E14), which has demonstrated efficient ectopic gene integration via genome editing20. Our dual-plasmid DNA system comprised a Cas9 expression DNA and an sgAlb-E14 expression DNA containing the donor sequence (Fig. 2a). The Cas9 and sgAlb-E14 facilitated the generation of double-strand breaks (DSBs) at the sgAlb-E14 target sites on both the genomic target and the donor DNA (Supplementary Fig. 1). These DSBs initiated repair processes, predominantly through the NHEJ pathway, enabling the integration of the linearized donor DNA at the genomic DSB site with a predetermined orientation5. This resulted in the expression of an Alb-P2A-scExe4 fusion protein, with ribosome skipping at the P2A peptide, facilitating the separation of Albumin and scExe4. The scExe4 is then processed in the Golgi apparatus and on the cell membrane, followed by selective release of Exe4 peptide from the Albumin-expressing hepatocytes (Fig. 2a).

a Schematic representation of HITI-mediated knock-in of the scExe4 expression cassette at the Alb locus, with plasmids for HITI targeting the liver Alb locus and Cas9. Dotted line indicates the site cut by Cas9. b Time course of Exe4 secretion in the culture medium for no transfection (Saline), scExe4 donor plasmid alone (Donor), and scExe4 donor plasmid with Cas9 plasmid (Donor+Cas9). Data are shown as mean ± SD (n = 3 biological replicates). Asterisks (**p < 0.01, ***p < 0.001) indicate a significant difference compared to Donor-treated group using one-way ANOVA. No significant difference (p > 0.05) was observed between Donor and Donor+Cas9 at passage 2. c Knock-in efficiency in Hepa1-6 cells at passage 11 determined by digital PCR. Data are shown as mean ± SD (n = 3 biological replicates).
To assess long-term Exe4 secretion by hepatic cells with HITI-mediated knock-in, we transfected Hepa1-6 cells with either HITI donor DNA only (Donor) or the donor plus Cas9 DNAs (Donor+Cas9). Exe4 secretion was sustained in the Donor+Cas9 transfected cells through passage 11, whereas it declined rapidly in the cells transfected with Donor alone. The observed Exe4 secretion in Donor-transfected cells is likely due to leakage expression from the promoter-less donor, as reported previously (Fig. 2b)35. Knock-in was confirmed via junction PCR and digital PCR targeting the precise knock-in site, showing approximately 3% knock-in efficiency in the Donot+Cas9 group (Fig. 2c and Supplementary Fig. 2). These results suggest that the scExe4 knock-in strategy at the Alb locus enables long-term Exe4 secretion in vitro.
LNP-mediated in vivo DNA delivery in the mouse liver
Efficient delivery of donor DNA, along with Cas9 and gRNA, is crucial for successful in vivo HITI. To achieve this, we employed Lipid Nanoparticles (LNPs), which have components similar to Patisiran, a clinically approved product known for effective hepatic transfection23,24. The plasmid DNA encapsulated by LNPs demonstrated optimal characteristics for liver targeting, including particle size of approximately 100 nm, low polydispersity (approximately 0.2), neutral surface charges, and high DNA encapsulation efficiency (Supplementary Table 4)25. To evaluate the in vivo targeting efficiency, we intravenously administered NanoLuc-encoding DNA-loaded LNPs into Balb/c mice. We observed comparable liver luciferase (Luc) activity between LNP and hydrodynamic (HD) injection, a method well-known for its high liver transfection efficiency (Supplementary Fig. 3a)36. Notably, LNP-based delivery method demonstrated significantly less Luc activity in non-liver organs, highlighting their superior liver selectivity (Supplementary Fig. 3b). These results validate LNPs as an effective tool for specifically delivering DNA vector to the liver, supporting their use in further in vivo genome editing experiments.
Long term secretion of Exe4 peptide in the genome-edited mice
To examine the prolonged expression of Exe4 following a single genome editing procedure, we administrated HITI components, including the scExe4 donor with gRNA expression cassette and Cas9-expression DNAs, via intravenous injection of DNA-loaded LNPs into Balb/c mice (Fig. 3a). Plasma Exe4 concentrations in the Donor+Cas9-treated mice remained detectable for over 28 weeks post-injection, indicating continuous secretion achieved through the knock-in approach (Fig. 3b). We also explored the potential for repeated genome editing to further increase plasma Exe4 concentrations for a controllable therapeutic effect. Unlike AAV vectors, which provoke immunological responses after the second administration, LNPs are expected to allow repeated administrations37,38. Following a second administration of Donor+Cas9, plasma Exe4 levels increased and remained elevated for over 28 weeks, doubling the level of Exe4 production and secretion compared to a single administration, suggesting the potential for fine-tuning the regimen dose through repetitive administration (Supplementary Fig. 4).

a Outline of in vivo scExe4 knock-in study treatments with saline control (Saline), scExe4 donor plasmid-loaded LNP only (Donor) and scExe4 donor plasmid-loaded LNP plus Cas9 plasmid-loaded LNP (Donor+Cas9) in Balb/c mice. b Plasma Exe4 concentration profile of in vivo scExe4 knock-in mice. c Junction PCR assay using liver genomic DNA from treated Balb/c mice (n = 3 biological replicates) at 28 weeks after administration. The uncropped gel image is shown in Supplementary Fig. 9b. d Knock-in efficiency determined by digital PCR of liver genomic DNA from in vivo scExe4 knock-in mice at 28 weeks. e Exe4 RNA expression level in various organs from Donor and Donor+Cas9 treated mice at 28 weeks post-treatment. f Cas9 RNA expression level in various organs from Donor and Donor+Cas9 treated mice at 28 weeks post-treatment. g Long-term evaluation of hepatic parameters (plasma AST, ALT, albumin concentration) for in vivo scExe4 knock-in mice over 28 weeks. Each line in (b and g) represents group means (n = 3 biological replicates). Data in (d–f) are shown as mean ± SD (n = 3 biological replicates). Each data point represents an individual mouse.
Junction PCR confirmed the knock-in event in the liver, and digital PCR indicated an efficiency of approximately 1% for a single administration of Donor+Cas9, consistent with previously reported in vivo knock-in efficiencies (Fig. 3c, d)20. Notably, we previously demonstrated that HITI-mediated GFP gene knock-in efficiency, as measured by digital PCR, closely correlates with the percentage of GFP-positive liver cells detected by flow cytometer13. Given that the both GFP and Exe4 knock-ins were performed under same LNP formulations and transfection conditions, digital PCR is expected to provide a reliable measure of Exe4 gene integration.
To assess safety, we performed next-generation sequencing to evaluate the on-target site and the top 10 potential off-target sites in liver genomic DNA from in vivo knock-in mice. The on-target indel efficiency at the Alb locus was 20.2% in the Donor+Cas9-treated group (Supplementary Fig. 5a), confirming successful genome editing via LNP-mediated HITI. However, knockout events occurred at a higher frequency than knock-in events (Fig. 3d and Supplementary Fig. 5a). Notably, indel frequencies at the top 10 off-target sites were minimal, suggesting a low genomic safety risk (Supplementary Fig. 5b). To further evaluate random donor DNA integration, we performed 5′RACE analysis to identify upstream sequences of scExe4 mRNA transcribed from integrated donor DNA in liver samples from knock-in mice (Supplementary Fig. 6). The results indicated minimal off-target effect, consistent with previous reports of pDNA-mediated genome editing using this sgRNA20.
RT-qPCR demonstrated exclusive Exe4 RNA expression in the liver, with no detectable Cas9 RNA (Fig. 3e, f). Plasma levels of hepatotoxic makers such as aspartate aminotransferase (AST) and alanine aminotransferase (ALT), as well as albumin concentration, were comparable among tested groups, indicating no significant safety concerns, despite the higher occurrence of knockout over knock-in events (Fig. 3d, g and Supplementary Fig. 5a). This may be attributed to the target site being located just upstream of the last five base pairs of the open reading frame at the C-terminal end of exon 14 in the Alb gene (Fig. 2a), where indel mutations are unlikely to impair Alb function. Collectively, these findings demonstrate sustained Exe4 expression and secretion by the liver following a single in vivo genome editing at the mouse Alb locus using LNP-mediated HITI, with no discernible side effects.
Anti-obese effect in DIO mice model
To evaluate the therapeutic effect of in vivo targeted knock-in of scExe4, we conducted a proof-of-concept study using diet-induced non-genetic obese (DIO) mice, which showed obesity and pre-diabetic symptoms through continuous high fat diet (HFD) feeding (Supplementary Fig. 7a). We assessed the therapeutic effects of in vivo genome-editing, including the suppression of body weight gain and improvement of blood glucose profiles (Fig. 4a). B6 mice fed with a normal fat diet (Normal Diet) or HFD (DIO) received the following treatment regimens: saline (Saline), scExe4 donor DNA-loaded LNP (Donor), or scExe4 donor DNA plus Cas9-coding DNA-loaded LNP (Donor+Cas9). Additionally, a comparison group received synthetic Exe4 peptide via a subcutaneously implanted osmotic pump (Peptide), releasing Exe4 peptide into the host circulatory system and then removed at 8 weeks after initial implantation to simulate treatment interruption. Plasma Exe4 concentration sustained in Donor+Cas9-treated mice over 24 weeks, while Exe4 was undetectable in Donor-treated mice from 4 weeks post-injection (Fig. 4b). The Peptide group maintained sustained Exe4 concentration for 8 weeks, but it became undetectable after pump removal.

a Schematic of pharmacodynamic study treatments with saline control (Saline), scExe4 donor plasmid-loaded LNP only (Donor), scExe4 donor plasmid-loaded LNP plus Cas9 plasmid-loaded LNP (Donor+Cas9) and subcutaneous implantation of an osmotic pump filled with synthetic Exe4 peptide solution (Peptide) in the DIO mice model and mice fed with normal diet (Normal Diet). b Plasma Exe4 concentration profile in vivo scExe4 knock-in DIO mice. c Relative weekly food intake normalized to that of Saline. d Relative body weight normalized to that of Saline. e Glucose tolerance test (left panel) and insulin tolerance (right panel) result for DIO mice at 8, 16, and 24 weeks after dosing. f Plasma insulin levels (left panel) and HbA1c concentration (right panel) of DIO mice at 8, 16, and 24 weeks after dosing. c, d Asterisks (****p < 0.0001) denote a significant difference compared to Saline, Donor and Peptide using one-way ANOVA. No significant difference (no asterisk; p > 0.05) was observed between the Normal Diet and Donor+Cas9 groups at 24 weeks (d). e, f Asterisks (***p < 0.001, ****p < 0.0001) indicate significant differences for the Donor+Cas9 and Normal Diet group compared to other groups by one-way ANOVA. No significant difference (no asterisk; p > 0.05) was observed between the Donor+Cas9 and Normal Diet groups. Each line represents the group mean (n = 4 biological replicates), with each data point corresponding to an individual mouse.
Bi-weekly monitoring of absolute food intake and body weight showed a continuous increase in body weight across all DIO mice groups, including those treated with saline or fed a normal diet, indicating a sustained weight gain trajectory throughout the experimental period (Supplementary Fig. 7b, c). While body weight increased over time in both the Donor+Cas9-treated and normal diet-fed mice, the Saline group exhibited a more rapid weight gain, resulting in a gradual decline in the relative body weight of the Donor+Cas9 and normal diet-fed groups (Fig. 4c, d and Supplementary Fig. 7d). At 24 weeks post-administration, the Donor+Cas9-treated mice showed a significant reduction in both food intake and body weight compared to the Saline group, with a 29% decrease in food intake and a 34% decrease in body weight. Notably, the body weight of Donor+Cas9-treated mice was statistically similar to that of mice fed with normal diet (Normal Diet), demonstrating the strongest suppressive effect on body weight gain despite an HFD. Donor alone transiently decreases food intake and body weight, but both rebounded over time. The Peptide-treated group exhibited a 15% reduction in both food intake and body weight at 8 weeks, though these effects were reversed after pump removal.
Taken together, in vivo knock-in of scExe4 effectively mitigates long-term body weight gain in non-genetic DIO mice models, demonstrating the potent metabolic effects of secreted Exe4 against DIO.
Beneficial metabolic effects on pre-diabetic symptoms in edited DIO mice
Beyond anti-obesity effects, we evaluated the impact of in vivo knock-in of scExe4 on pre-diabetic symptoms, including high blood glucose levels and low insulin sensitivity, in DIO mice, which are hallmark features of diabetes (Fig. 4e). By monitoring blood glucose profiles after administering a glucose solution to the mice, we performed glucose tolerance test (GTT). The GTT indicated significantly lower blood glucose AUC in Donor+Cas9 treated mice compared to Saline and Donor groups at 8, 16, and 24 weeks post-injection, with levels comparable to non-treated mice fed with normal fat diet (Normal Diet) at matching time points. Peptide-treated mice showed improvement only when the pump was present, with symptoms relapsing after pump removal. Additionally, monitoring blood glucose levels after intraperitoneal insulin administration in the ITT demonstrated an improved outcome, indicating a restoration of insulin sensitivity in the Donor+Cas9 group.
Lower insulin and HbA1c levels further indicated improved control of blood glucose. The decreased insulin levels suggest efficient insulin utilization and glucose lowering, while HbA1c reflects long-term blood glucose levels. In Donor+Cas9 treated mice, these markers were comparable to those observed in mice fed with normal fat diet (Normal Diet) over 24 weeks, demonstrating a prolonged anti-diabetic effect from in vivo genome editing (Fig. 4f).
To assess the potential impact of genome editing-mediated Exe4 secretion on endogenous hormone regulation, plasma GLP-1 levels were measured following an oral glucose challenge in saline- and Donor+Cas9-treated mice. Since GLP-1 secretion is physiologically triggered by nutrient intake or glucose exposure, we anticipated minimal interference from Exe4. Indeed, plasma GLP-1 concentrations and their corresponding AUC were comparable between the two groups (Supplementary Fig. 8). These findings indicate that continuous Exe4 secretion via genome editing does not substantially suppress endogenous GLP-1 secretion. Overall, a single administration of in vivo genome editing-mediated scExe4 knock-in provides stable, long-term metabolic benefits, effectively restoring blood glucose homeostasis and preventing relapse of pre-diabetic symptoms in non-genetic DIO mice.
Discussion
In this study, leveraging non-viral LNPs and the HITI-mediated genome editing method, we engineered a secretion-enabled Exe4 peptide, scExe4, and developed an in vivo scExe4 peptide production reservoir via knock-in into mouse liver cells. This innovative approach enabled sustained Exe4 secretion into the bloodstream from the liver for up to 28 weeks after a single administration. In a DIO mice model, scExe4 knock-in led to reduced food intake, suppressed body weight gain, and improved blood glucose levels. These results demonstrate the long-term anti-obesity and anti-diabetic effects achieved from a single in vivo genome editing treatment. To the best of our knowledge, this is the first study to demonstrate the application of in vivo genome editing for treatment of diseases not associated with specific genetic lesions or aberrations.
By leveraging the high expression level of the Alb gene in hepatocytes, we targeted the Alb locus for the knock-in of the secretable Exe4. Despite achieving a knock-in efficiency of approximately 1% in vivo (Fig. 3d), sustained plasma Exe4 concentrations and therapeutic effects were observed in DIO mice, owing to the compensatory high gene expression at the Alb gene in hepatocytes. Notably, we recently conducted LNP-mediated knock-in of the GFP gene at the Alb locus, with GFP expression predominantly observed in hepatocytes, further confirming that hepatocytes are primary target for LNP-mediated gene delivery and genome editing13. The in vivo HITI-mediated knock-in efficiency at the Alb locus in this study is comparable to that of other knock-in methods, such as HDR (~2%) and NHEJ (~2%)33,34.
A key consideration in this approach is the impact of continuous Exe4 secretion on endogenous hormone regulation, particularly GLP-1 and glucagon. Our findings indicate that GLP-1 secretion remains comparable between saline- and Donor+Cas9-treated DIO mice, suggesting that genome editing-mediated Exe4 secretion does not disrupt endogenous GLP-1 regulation (Supplementary Fig. 8). Given the antagonistic relationship between glucagon and GLP-1, it is also critical to assess whether sustained Exe4 secretion affects glucagon levels. Notably, previous studies have reported that intravenous Exenatide (Exendin-4) infusion does not significantly alter fasting glucagon secretion in humans39, supporting the expectation that genome editing-mediated Exe4 secretion would similarly exert minimal disruption.
A higher knock-in rate is expected to increase the number of Exe4-expressing cells in the liver, potentially increasing the amount of Exe4 secretion. However, an excessive increase in Exe4 plasma concentration may not be necessary if it surpasses the effective therapeutic range, which varies among patients. Elevated Exe4 levels could also heighten the risk of adverse events, making it essential to carefully balance knock-in efficiency with both efficacy and safety considerations in humans. Further investigations are needed to comprehensively assess the functional and safety aspects of long-term Exe4 secretion. In particular, preclinical studies should assess gastric emptying and conditioned taste aversion to better understand potential physiological effects and safety implications. Additionally, Exe4 has been shown to be less effective in reducing body weight in leptin-deficient ob/ob mice, highlighting the need for further evaluation in diverse obesity models40. Expanding safety assessments to additional animal species, including non-human primates, will be crucial for determining the feasibility of clinical translation.
In comparison to existing GLP-1RAs, our findings align with previous studies demonstrating their anti-obesity and glucose-regulation effects17. The efficacy of a single genome-editing treatment was comparable to that of continuous Exendin-4 peptide infusion via an osmotic pump (Peptide group) over the pump implantation period (Fig. 4). Given that the Peptide group mimics conventional peptide therapeutics, we anticipate that genome editing-mediated Exe4 secretion could provide similar efficacy to existing GLP-1RA treatments. Exendin-4, like other GLP-1RAs, requires frequent administration due to its short plasma half-life. However, genome editing-induced Exe4 secretion in the liver led to sustained plasma Exe4 levels, highlighting its potential for long-term clinical benefits, such as improved treatment adherence and prevention of relapse.
Biologics, including recombinant protein- or peptide-based drugs, offer targeted treatment options for various complex and non-genetic diseases. However, most biologics require frequent injections, posing risks of treatment interruption due to patient non-compliance and limited drug supply, which can lead to relapse and worsening of symptoms. This challenge could be overcome by engineer an in vivo reservoir of biologic through a single genome editing treatment.
Further improvements in knock-in and secretion efficiencies would represent an advance in treating a broad range of non-genetic, polygenic and environmentally related complex diseases, including those for which biologics already used, such as inflammatory diseases. A single administration of a genome editing regimen to enable the sustainable production of therapeutic peptides and monoclonal antibodies has the potential to revolutionize treatment paradigms. One promising optimization strategy involves the use of mRNA-encapsulated LNPs, such as Cas9 mRNA, instead of Cas9 plasmid DNA. This approach could theoretically prevent Cas9 DNA integration into the host genome, thereby reducing the risk of unintended off-target effects and further enhancing the safety of LNP-based in vivo genome editing. Optimizing in vivo delivery and genome editing methods, including the use of DSB-free large DNA knock-in technologies like PASTE and dCas9-SSAP editor, could help bridge the gap between pre-clinical development and clinical translation41,42,43,44,45,46.
Data availability
All numerical source data underlying Figs. 1–4 and Supplementary Figs. 3–5, 7, 8 are available in Supplementary Data 1. All amplicon sequencing data are publicly accessible from the National Center for Biotechnology Information BioProject database under accession number PRJNA1242089. Other data used in this study will be made available to qualified researchers upon reasonable request for research purposes, and may be subject to a data use agreement. Inquiries should be directed to the corresponding author.
References
-
Zheng, Y. et al. Precise genome-editing in human diseases: mechanisms, strategies and applications. Signal Transduct. Target. Ther. 9, 47–67 (2024).
-
Anzalone, A. V., Koblan, L. W. & Liu, D. R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 38, 824–844 (2020).
-
Chen, X. et al. Recent advances in CRISPR-Cas9-based genome insertion technologies. Mol. Ther.: Nucleic Acids 35, 1–13 (2024).
-
Pacesa, M., Palea, O. & Jinek, M. Past, present, and future of CRISPR genome editing technologies. Cell 187, 1076–1100 (2024).
-
Suzuki, K. et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 540, 144–149 (2016).
-
Tornabene, P. et al. Therapeutic homology-independent targeted integration in retina and liver. Nat. Commun. 13, 1963–1977 (2022).
-
Suzuki, K. et al. Precise in vivo genome editing via single homology arm donor mediated intron-targeting gene integration for genetic disease correction. Cell Res. 29, 804–819 (2019).
-
Stephenson, A. A. et al. CRISPR-Cas9 homology-independent targeted integration of exons 1-19 restores full-length dystrophin in mice. Mol. Ther.: Methods Clin. Dev. 30, 486–499 (2023).
-
Chen, X. et al. Long-term correction of hemophilia B through CRISPR/Cas9 induced homology-independent targeted integration. J. Genet. Genomics 49, 1114–1126 (2022).
-
Kuwayama, R. et al. Establishment of mouse model of inherited PIGO deficiency and therapeutic potential of AAV-based gene therapy. Nat. Commun. 13, 3107 (2022).
-
Hatanaka, F. et al. Therapeutic strategy for spinal muscular atrophy by combining gene supplementation and genome editing. Nat. Commun. 15, 6191 (2024).
-
Suzuki, K. & Izpisua Belmonte, J. C. In vivo genome editing via the HITI method as a tool for gene therapy. J. Hum. Genet. 63, 157–164 (2018).
-
Hirose, J. et al. Lipid nanoparticles enable efficient in vivo DNA knock-in via HITI-mediated genome editing. Biomolecules 14, 1558 (2024).
-
Parums, D. V. First regulatory approvals for CRISPR-Cas9 therapeutic gene editing for sickle cell disease and transfusion-dependent β-Thalassemia. Med. Sci. Monit. 30, e944204 (2024).
-
Senior, M. Fresh from the biotech pipeline: fewer approvals, but biologics gain share. Nat. Biotechnol. 41, 174–182 (2023).
-
Müller, T. D., Blüher, M., Tschöp, M. H. & DiMarchi, R. D. Anti-obesity drug discovery: advances and challenges. Nat. Rev. Drug Discov. 21, 201–223 (2022).
-
Tatarkiewicz, K., Sablan, E. J., Polizzi, C. J., Villescaz, C. & Parkes, D. G. Long-term metabolic benefits of exenatide in mice are mediated solely via the known glucagon-like peptide 1 receptor. Am. J. Physiol., Regul., Integr. Comp. Physiol. 306, 490–498 (2014).
-
Su, N. et al. Exenatide in obese or overweight patients without diabetes: a systematic review and meta-analyses of randomized controlled trials. Int. J. Cardiol. 219, 293–300 (2016).
-
Knop, F. P., Brønden, A. & Vilsbøll, T. Exenatide: pharmacokinetics, clinical use, and future directions. Expert Opin. Pharmacother. 18, 555–571 (2017).
-
Zhang, J. P. et al. Curing hemophilia A by NHEJ-mediated ectopic F8 insertion in the mouse. Genome Biol. 20, 276 (2019).
-
Suzuki, T., Kanamori, T. & Inouye, S. Quantitative visualization of synchronized insulin secretion from 3D-cultured cells. Biochem. Biophys. Res. Commun. 486, 886–892 (2017).
-
Charcosset, C., Juban, A., Valour, J. P. & Urbaniak, S. Preparation of liposomes at large scale using the ethanol injection method. Chem. Eng. Res. Des. 94, 508–515 (2015).
-
Holm, A., Løvendorf, M. B. & Kauppinen, S. Development of siRNA therapeutics for the treatment of liver diseases. Methods Mol. Biol. 2282, 57–75 (2021).
-
Kulkarni, J. A. et al. Design of lipid nanoparticles for in vitro and in vivo delivery of plasmid DNA. Nanomed.: Nanotechnol., Biol. Med. 13, 1377–1387 (2017).
-
Böttger, R. et al. Lipid based nanoparticle technologies for liver targeting. Adv. Drug Deliv. Rev. 154, 79–101 (2019).
-
Samson, S. L. et al. Gene therapy for diabetes: metabolic effects of helper-dependent adenoviral exendin 4 expression in diet-induced obesity mouse model. Mol. Ther. 16, 1805–1812 (2008).
-
Parsons, G. B. et al. Ectopic expression of glucagon-like peptide 1 for gene therapy of type II diabetes. Gene Ther. 14, 36–48 (2007).
-
Choi, S. H. & Lee, H. C. Long-term, antidiabetogenic effects of GLP-1 gene therapy using double-stranded, adeno-associated viral vector. Gene Ther. 18, 155–163 (2011).
-
Oh, S., Lee, M., Ko, K. S., Choi, S. & Kim, S. W. GLP-1 gene delivery for the treatment of type 2 diabetes. Mol. Ther. 7, 478–483 (2003).
-
DiPasquale, G. et al. Sustained Exendin-4 secretion through gene therapy targeting salivary glands in two different rodent models of obesity/type 2 diabetes. PLoS ONE 7, e40074 (2012).
-
Beutler, A. S. et al. Retrovirus-mediated expression of artificial beta-endorphin precursor in primary fibroblasts. J. Neurochem. 64, 475–481 (1995).
-
Chirorini, J. A., DiPasquale, G. & Mnnucci, E. AAV Mediated Exendin-4 Gene Transfer to Salivary Glands to Protect Subjects from Diabetes or Obesity. Patent US20190298785A1, filing in 26 April 2019 (2021).
-
Barzel, A. et al. Promoterless gene targeting without nucleases ameliorated hemophilia B in mice. Nature 517, 360–364 (2015).
-
Caneva, A. D. et al. Coupling AAV-mediated promoterless gene targeting to SaCas9 nuclease to efficiently correct liver metabolic diseases. JCI Insight 4, e128863 (2019).
-
Tsunekawa, Y. et al. Developing a de novo targeted knock-in method based on in utero electroporation into the mammalian brain. Development 143, 3216–3222 (2016).
-
Yan, S. et al. High levels of gene expression in the hepatocytes of adult mice, neonatal mice and tree shrews via retro-orbital sinus hydrodynamic injections of naked plasmid DNA. J. Control. Release 161, 763–771 (2012).
-
Moreno, A. M. et al. Immune-orthogonal orthologues of AAV capsids and of Cas9 circumvent the immune response to the administration of gene therapy. Nat. Biomed. Bioeng. 3, 806–816 (2019).
-
Finn, J. D. et al. A single administration of CRISPR/Cas9 lipid nanoparticles achieves robust and persistent in vivo genome editing. Cell Rep. 22, 2227–2235 (2018).
-
Degn, K. B. et al. Effect of intravenous infusion of exenatide (synthetic exendin-4) on glucose-dependent insulin secretion and couterregulation during hypoglycemia. Diabetes 53, 2397–2403 (2004).
-
Xu, F. et al. Short-term GLP-1 receptor agonist exenatide ameliorates intramyocellular lipid deposition without weight loss in ob/ob mice. Int. J. Obes. 44, 937–947 (2020).
-
Lampe, G. D. et al. Targeted DNA integration in human cells without double-strand breaks using CRISPR-associated transposases. Nat. Biotechnol. 42, 87–98 (2024).
-
Awan, M. J. A., Mahmood, M. A., Naqvi, R. Z. & Mansoor, S. PASTE: a high-throughput method for large DNA insertions. Trends Plant Sci. 28, 509–511 (2023).
-
Wang, C. et al. dCas9-based gene editing for cleavage-free genomic knock-in of long sequences. Nat. Cell Biol. 24, 268–278 (2022).
-
Durrant, M. G. et al. Bridge RNAs direct programmable recombination of target and donor DNA. Nature 630, 984–993 (2024).
-
Hiraizumi, M. et al. Structural mechanism of bridge RNA-guided recombination. Nature 630, 994–1002 (2024).
-
Kawamata, M., Suzuki, H., Kimura, R. & Suzuki, A. Optimization of Cas9 activity through the addition of cytosine extensions to single-guide RNAs. Nat. Biomed. Eng. 7, 672–692 (2023).
Acknowledgements
We are grateful to Shinji Sakai, Shinji Deguchi, Kana Furukawa for animal care; Takashi Enjoji for digital PCR; Yun Xia and Ximing Gong for editing manuscript. This work was supported by the Japan Society for the Promotion of Science KAKENHI (21H04811 and 22K19405), AMED (JP23ek0109521), Daiichi Sankyo Foundation of Life Science, The NOVARTIS Foundation (Japan) for the Promotion of Science and G-7 Scholarship Foundation. S.Y. was supported by Osaka University Honors Program for Graduate Schools in Science, Engineering and Informatics. A part of this work was conducted in Advanced Research Infrastructure for Materials and Nanotechnology Open Facilities in Osaka University, supported by ARIM Program of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan, Grant Number JPMXP1224 OS1059.
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Medicine thanks Tamer Coskun and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. [Peer review reports are available.]
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hirose, J., Aizawa, E., Yamamoto, S. et al. Targeted in vivo gene integration of a secretion-enabled GLP-1 receptor agonist reverses diet-induced non-genetic obesity and pre-diabetes. Commun Med 5, 269 (2025). https://doi.org/10.1038/s43856-025-00959-8
-
Received:
-
Accepted:
-
Published:
-
DOI: https://doi.org/10.1038/s43856-025-00959-8