Introduction
Tumor blood vessels have emerged as critical targets for therapeutic intervention. Therapeutic strategies include suppressing the growth or perturbing functions of the neovessels that sustain tumor lesions1,2 and leveraging the unique characteristics of tumor blood vessels to enhance the delivery of cytotoxic anticancer agents via specialized drug delivery systems. Therapeutic agents can be directed to malignant cells in the tumor microenvironment by using affinity ligands such as antibodies, peptides, aptamers, or small molecules that engage tumor-associated markers3,4. The accessibility of target receptors from the bloodstream and their adequate expression level are prerequisites for any effective affinity-targeted therapy4. Current affinity-targeting strategies primarily engage the cell surface biomarkers on malignant and tumor endothelial cells. Targeting the extracellular matrix (ECM) in the tumor microenvironment is less common despite its potential advantages. ECM components are estimated to be ~ 10-fold more abundant than cellular receptors and offer a higher capacity for drug delivery5,6. Compared to receptors overexpressed on the surface of malignant cells, ECM molecules provide opportunities for more stable targeting as they are predominantly deposited by genetically stable nonmalignant cells7. To some degree, ECM targeting peptides allow for intracellular delivery due to their ability to exploit the natural cellular uptake pathways associated with cell-matrix interactions, as shown for the PL1 peptide that uses macropinocytosis for cellular entry7. The ECM provides a dynamic physical and biochemical microenvironment that actively regulates essential cellular functions such as adhesion, proliferation, and migration, thereby influencing cellular developmental pathways8,9.
Overexpression of alternatively spliced ECM isoforms is associated with cancer, and specific ECM splicing variants have been linked to stromal activation9. Fibronectin Extra Domain-B (Fn-EDB), an acidic 91-amino acid splice variant of fibronectin generated by type III homology repeats, is a key component of the angiogenic signature, distinguished by its overexpression in solid tumors and absence in most adult tissues, except for the female reproductive tract10,11. A comparative analysis of Fn-EDB expression in ~ 18,800 cancer samples and ~ 4,500 normal samples demonstrated upregulation of Fn-EDB in 15 types of cancer, including in grade I to IV malignant gliomas12. In addition, Fn-EDB is a specific marker for tumor neovessels13. To date, several classes of Fn-EDB targeting ligands, including antibodies, peptides, and aptamers, have been applied to deliver therapeutic agents, such as cytokines, cytotoxic agents, chemotherapeutic drugs, and radioisotopes, to Fn-EDB-expressing tumors12,14,15. The Fn-EDB targeting L19 antibody and its derivatives (diabodies, Single-chain variable fragment (scFv), Small immunoprotein (SIP)) have exhibited potential in preclinical and clinical studies in EDB-FN-positive cancer patients using both systemic and intratumoral administration routes15. Daromun, an intralesional immunocytokine combining IL-2 and TNF conjugated to the L19 antibody, has shown effectiveness in local tumor destruction and treating distant disease through an immune-mediated mechanism in early clinical studies16,17,18. In contrast to antibodies, short peptides offer easy synthesis, low immunogenicity, low cost, biocompatibility, and moderate affinity, which helps to circumvent the affinity site barrier6,19,20,21. Peptides, such as linear PL1 and cyclic ZD2, specifically bind to Fn-EDB and have been used to target intracranial glioblastomas and prostate cancer, respectively22,23.
One potential limitation in the application of matrix targeting ligands for tumor delivery is the variability of accessibility in the ECM to circulating ligands. The accessibility, influenced by the degree of tumor blood vessel leakiness, generally correlates with histological grade and malignancy but varies significantly both within individual tumors and across tumor types. Several pharmacological strategies have been employed to increase tumor vascular permeability and improve tumor delivery (e.g., drugs modulating tumor blood pressure, inflammatory cytokines, and bradykinin mediators)24,25. We investigated the pharmacological stimulation of extravasation to enhance tumor ECM exposure to circulating targeting ligands by designing bispecific ligands that simultaneously bind the ECM molecule Tenascin-C and the cell and tissue penetration receptor neuropilin-1 (NRP-1)26,27. The engagement of the peptide with NRP-1 induced extravasation and tissue penetration via a mechanism involving cellular entry and vascular transcytosis through the C-end Rule (CendR) pathway26,28,29,30.
Here, we used peptide-phage display on the recombinant Fn-EDB domain to identify a novel heptameric PL2 peptide that interacts specifically with Fn-EDB. The peptide also engages the tissue penetration receptor NRP-1 via the C-terminal arginine containing motif of the peptide, promoting cellular uptake in vitro and penetration of cell and tissue barriers in vivo. The in vivo phage playoff and systemic PL2-guided iron oxide nanoparticles showed specific accumulation in a panel of tumor xenografts implanted in mice. Our study suggests that PL2-guided agents can be used for detection, imaging, and payload delivery to solid tumors positive for the expression of Fn-EDB and NRP-1.
Materials and methods
Materials
Phosphate-buffered saline (PBS) was purchased from Lonza (Verviers, Belgium). K3[Fe(CN)6], HCl, isopropanol, Triton-X, Tween-20, CHCl3, MeOH, Isopropyl β-D-1-thiogalactopyranoside (IPTG), and dimethylformamide (DMF) were purchased from Sigma-Aldrich (Munich, Germany).
Peptides and proteins
The peptides and proteins used in the study were Cys-5(6)-carboxyfluorescein (FAM)-PL2, biotin-PL2, Cys-FAM peptides, and biotin with 6-aminohexanoic acid spacer, which were purchased from TAG Copenhagen (Denmark). The plasmids pASK75-Fn7B8 and pASK75-Fn789 were kindly provided by Prof. Dr. Arne Skerra31. The gene fragment of Fn-EDB domain was amplified from the plasmids and cloned into a pET28a + plasmid containing a N-terminal His6-tag for expression in E. coli strain BL21 Rosetta™ 2 (DE3) pLysS (Novagen, #70956). Recombinant Fn-EDB was produced as a soluble protein and purified using the HisTrap IMAC HP column (GE Healthcare, #17-0920-05) as previously described4,23. SDS-PAGE and mass spectrometry (MS) analyses were used to determine proteins purity, size, and sequence. The NRP-1 b1b2 domain protein was expressed and purified at the Protein Production and Analysis Facility at the Sanford Burnham Prebys Medical Discovery Institute (La Jolla, CA, US), and the NRP-1 b1 domain protein was expressed and purified in-house. Cloning, expression, purification of proteins (FN-EDB, TNC-C, NRP1, NCL and single chain antibodies FN-EDB-L19), and generation of polyclonal rabbit antibodies are described in Supplementary Information, Materials and Methods section and PL1 study protocols23.
Cell lines and experimental animals
The study utilized human glioblastoma (U87-MG, HTB-14, RRID: CVCL_0022) and prostate carcinoma (PC3, CRL1435, RRID: CVCL_0035) cells purchased from ATCC (VA, USA). Murine wild-type glioblastoma (WT-GBM) cells were kindly provided by Gabriele Bergers (UCSF, USA), and stem cell-like cancer cells P3, P13 were gifted by Rolf Bjerkvig (University of Bergen, Norway). The M21 (RRID: CVCL_D031) melanoma cells were the gift of David Cheresh (USA). The cells and tumors were prepared as described in previous studies4,23,26,28,32,33,34.
Athymic nude mice (Hsd/Athymic Fox1 nu Harlan) were purchased from Harlan Sprague Dawley (HSD, Indianapolis, IN, USA) and maintained under standard housing conditions of the Animal Facility of the Institute of Biomedicine and Translational Medicine, University of Tartu (Tartu, Estonia). Inclusion and exclusion criteria for animals were based on standard experimental conditions and we selected age-matched male and female nude mice (11–15 weeks old). The orthotopic glioblastoma (GBM) tumor models were established using P13, P3 stem cell-like, and WT-GBM cells. Approximately 2–3 × 10⁵ cells suspended in 3 µL of PBS were intracranially implanted into mice brain 2 mm right and 1 mm anterior to the bregma. For the subcutaneous models, 2−9 × 106 U87-MG GBM and prostate carcinoma (PC3) cells in 100 µl of PBS were subcutaneously (s.c.) implanted in the right flank of 11−15-week-old male and female nude mice. No attrition was noted during the study, as all animal samples were included as per protocol. No blinding was used during the experiment and study did not include treatment groups; power calculations were not applicable. Our experimental design ensures that minimal bias (or noise) by ensuring same sex, similar animal weight at start of experiment, same animal age, same type of stabling, several animals caged together.
Animal experimentation procedures were approved by the Estonian Ministry of Agriculture, Committee of Animal Experimentation, projects #42 and #48. We confirm that all methods were performed in accordance with the relevant guidelines and regulations. We confirm that the study was conducted in accordance with the ARRIVE guidelines35.
T7 phage peptide library biopanning
We employed T7-Select® phage display system (Novagen, EMD Biosciences, MA, USA) to generate NNK-encoded X7 peptide phage libraries with a diversity of ~ 5 × 108 for biopanning on recombinant Fn-EDB. The first round of selection was carried out on Fn-EDB immobilized on a Costar 96-Well enzyme-linked immunosorbent assay (ELISA) plate (#3590, Corning Life Sciences, MA, USA) by coating the plate with 20 µg/ml recombinant Fn-EDB protein in 100 µl of PBS overnight at 4 °C. The plate was then blocked with 1% bovine serum albumin (BSA) in PBS overnight at 4 °C. The phage library solution (5 × 108 pfu in 100 µl of PBS-BSA) was incubated overnight at 4 °C, followed by 6 washes with PBS + BSA + 0.1% Tween 20 to remove non-specifically bound background phages. The bound phages were rescued and amplified in E. coli strain BLT5403 (Novagen, MA, USA)36. The subsequent selection rounds were performed with His6-tagged Fn-EDB protein (30 µg/10 µl beads) immobilized on Ni-NTA Magnetic Agarose Beads (QIAGEN, Hilden, Germany) at room temperature for 1 h in 400 µl of PBS. The Fn-EDB immobilized beads were washed three times with PBS + BSA + 0.1% NP40, followed by incubation with the phage from the previous round (5 × 108 pfu in 100 µl of PBS + BSA + 0.1% NP40) for 1 h at room temperature. The background and weakly bound phages were removed by rinsing six times with PBS + BSA + 0.1% NP40, and the bound phages were eluted with 1 ml of PBS + 50 0mM imidazole + 0.1% NP40. The recovered phages were titered and amplified for a subsequent round of selection. After 6 rounds of selection, peptide-encoding phage DNA from a randomly selected set of 48 clones from round 5 was subjected to Sanger sequencing to obtain information on the displayed peptides36,37.
For cell-free binding studies with individually amplified phage clones, phages were incubated with Fn-EDB coated magnetic beads as described above. Furthermore, the GRPARPAR phage on NRP-1 coated beads was used as a positive control29while the Nucleolin (NCL) and the C-domain of Tenascin C (TNC-C) were used as negative controls. Finally, we used phage displaying heptaglycine peptide (GGGGGGG, G7) or insertless phage clones for negative controls.
In vivo playoff phage auditioning
To assess the systemic homing of peptide-phages to xenograft tumor models, we employed an internally controlled competitive assay that we have termed in vivo peptide-phage playoff23,36,38. Briefly, the candidate Fn-EDB binding peptides, together with previously published tumor-homing peptides and control peptides, were amplified individually and purified by PEG-8000 precipitation, CsCl gradient ultracentrifugation, and dialysis to minimize amplification biases. The equimolar pooled peptide-phages were intravenously injected into tumor-bearing mice at a concentration of 1 × 1010 pfu in 200 µL of PBS. After 2 h of circulation, the mice were anesthetized by intraperitoneal (i.p.) injection of 350 µL containing 0.1 mg/kg dexmedetomidine and 75 mg/kg ketamine dissolved in saline or 3–4% isoflurane. Following anesthesia, the mice were perfused intracardially with Dulbecco’s Modified Eagle Medium (DMEM; Lonza Ltd, Basel, Switzerland) to remove non-bound phages. Tumors and organs were collected in LB + 1% NP40, and tissue homogenization was carried out to rescue peptide-phages. The lysates were then amplified in E. coli, purified through precipitation with PEG-8000, and the DNA was extracted using a DNA extraction kit (High Pure PCR Template Preparation Kit; Roche, Basel, Switzerland). To evaluate the representation of each phage in the input mixture, tumor, and control organs, we performed next-generation sequencing of phage genomic DNA using the Ion Torrent high-throughput DNA sequencing system (Thermo Fisher Scientific, Waltham, MA, USA), with reads normalized to non-targeted G7 control phage to account for input diversity. The FASTQ data from Ion Torrent were processed using a custom Python (RRID: SCR_024202) script that identified the barcodes and constant flanking residues, and extracted the correct length reads. Table 1 provides a detailed list of the equimolarly pooled phage peptides used in this study.
Peptide binding assay
ELISA plates (Nunc Maxisorp, Thermo Fisher Scientific Inc., MA, USA) were coated with 20 µg of PL2 peptide labeled with FAM in 100 µl of PBS and incubated at 37 °C overnight. The plate wells were blocked with 1% BSA in PBS for 1 h at 37 °C, washed with a blocking solution (PBS containing 1% BSA and 0.1% Tween-20), and incubated with 2 µg of recombinant proteins in PBS per well for 6 h. The wells were washed 3 times with the blocking solution, and the bound protein was detected using an anti-His-tag antibody (Cat #A2-502-100, RRID: AB_11135798, Icosagen, Tartu, Estonia) for 1 h at 37 °C. After washing the wells 3 times with the blocking solution, a horseradish peroxidase-conjugated secondary antibody (Cat# 111-035-008, RRID: AB_2337937, Jackson Immuno Research, Cambridgeshire, UK) was added according to the manufacturer’s instructions. The wells were washed 3 times with the blocking solution, and a peroxidase reaction was initiated by adding 100 µL/well of freshly prepared solution from the TMB Peroxidase EIA Substrate Kit (Bio-Rad, Hercules, CA, USA), followed by a 5-minute incubation at 37 °C. The reaction was stopped with 1 N H2SO4, and the absorbance was measured at 450 nm using a microplate reader (Tecan Austria GmbH, Salzburg, Austria).
Nanoparticle synthesis and functionalization
The synthesis and functionalization of iron oxide nanoworms (NWs) and silver nanoparticles (AgNPs) followed4,6,23,26,39,40,41. For NWs, aminated NWs were PEGylated with maleimide-5 K-PEG-NH2 (JenKem Technology, TX, USA), enabling thiol-reactive maleimide groups. Subsequently, Peptides with N-terminal cysteine were conjugated to the PEG-NWs via a thioether bond formed between the thiol group of a cysteine residue on the peptide and the maleimide group on the PEGylated NWs. The concentration of NWs was determined by constructing a calibration curve with iron oxide, and the absorbance of NWs at 400 nm was measured using a NanoDrop 2000c spectrophotometer (Thermo Scientific). For AgNPs, CF647-N-hydroxysuccinimide-dye (NHS-dye) was conjugated to the terminal amine group of PEG, and biotinylated peptides were coated on the surface of the AgNPs through NeutrAvidin (NA; Sigma-Aldrich, USA). The nanoparticles were characterized using transmission electron microscopy (TEM, Tecnai 10, Philips, Netherlands) to image, and dynamic light scattering (DLS, Zetasizer Nano ZS, Malvern Instruments, UK) to assess the zeta potential, polydispersity, and the size, as described previously.
Cell binding and internalization assay
The U87-MG, PPC1, and M21 cells were cultured on coverslips and treated with CF555-labeled PL2 AgNPs or non-targeted control AgNPs at 37 °C for 1 h, as previously described7,26,28. After removing unbound particles with culture medium, cells were treated with an etching solution (10 mM working concentration in PBS) made by diluting 0.2 M stock solutions of Na2S2O3 and K3Fe (CN)6 in a 1: 1 ratio for 3 min, followed by washing with PBS. Cells were fixed with methanol at −20 °C for 1−2 min, and thereafter the nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI, Molecular Probes) at 1 µg/mL. Finally, the coverslips were mounted on microscopy slides using Fluoromount-G medium (Electron Microscopy Sciences) for confocal imaging.
Tumor-targeted delivery and biodistribution studies
FAM-labeled PL2 peptide-conjugated NW or control FAM-NW (7.5 mg/kg) in PBS were administered via tail vein injection to subcutaneous U87-MG, PC3, and orthotropic WT-GBM tumor-bearing mice. Five hours after circulation, the tumors and organs were collected via cardiac perfusion of mice under deep anesthesia with 20 ml PBS/DMEM. Macroscopic images of tissues were taken using an Illuminatool Bright Light System LT-9900 (Lightools Research, Encinitas, CA, USA) before snap-freezing. The frozen tissues were then cryosectioned with Leica CM1520 (Leica Camera AG, Germany) into 8–10 μm sections and mounted on Superfrost + slides (Thermo Fisher Scientific, MA, USA). The tissue sections were equilibrated at room temperature and fixed with 4% paraformaldehyde/-20 °C methanol. Tissue staining was performed using primary antibodies, including rabbit anti-fluorescein IgG fragment (Cat # A889, RRID: AB_221561, Thermo Fisher Scientific, MA, USA), rat anti-mouse CD31 (RRID:
AB_393571, BD Biosciences, CA, USA), and in-house prepared CF647/CF546-labeled single-chain antibodies ScFV L19. Secondary antibodies used were Alexa 488 goat anti-rabbit IgG (Cat # A-11034, RRID: AB_2576217), Alexa 647 goat anti-rat IgG (Cat # A-21247, RRID: AB_141778), and Alexa 546 goat anti-mouse IgG (Cat # A-11003, RRID: AB_2534071) from Invitrogen, CA, USA. The tissue nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI, Molecular Probes) at 1 µg/ml concentration. Coverslips were mounted on glass slides with Fluoromount-G (Electron Microscopy Sciences, PA, USA) and imaged using confocal microscopy (Olympus FV1200MPE, Hamburg, Germany). The resulting images were analyzed using FV10-ASW4.2 viewer/Imaris software/Fiji ImageJ.
Ex vivo clinical tumor dipping assay
In compliance with the Ethics Committee of the University of Tartu, Estonia (permit #243/T27), fresh surgical ovarian carcinoma samples were collected from consenting patients undergoing surgery according to relevant guidelines and regulations accordance with the Declaration of Helsinki42. To perform the dipping assay, the fresh ovarian carcinoma tissues were washed with DMEM, and 1 cm3 explants were incubated at 37 °C with PL2-NW or non-targeted control NWs (40 mg/mL Fe in DMEM supplemented with 1% BSA) for four hours. The explants were then washed with PBS, snap-frozen, cryosectioned at 10 μm, and immunostained with rabbit anti-fluorescein primary antibodies, followed by detection with the Alexa-488 anti-rabbit secondary antibody (Invitrogen, Thermo Fisher Scientific, MA, USA).
Statistical analysis
The statistical analysis was conducted using Prism 6 software (GraphPad Software, Inc, RRID: SCR_002798). For comparisons between two groups, a student’s unpaired t-test was used, while an ANOVA test was applied for comparisons involving multiple groups. For Continuous data, including quantifying FAM signal in tissue sections, the fluorescence signal intensity of antibody-amplified FAM was analyzed from 12 to 20 confocal images using Fiji ImageJ freeware (RRID: SCR_003070) and were analyzed using the student’s unpaired t-test. Data are presented as mean values, with error bars representing ± SEM. The data are presented as mean values with error bars showing ± SEM. The significance level was set at p < 0.05, and the P-values are denoted as *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001. The Samples were processed in a randomized order during analysis to prevent systematic bias. Group sizes were determined based on prior studies and standard practices in the field to ensure robust data collection and reproducibility. A priori power analysis was conducted to ensure the sample size was sufficient to detect biologically relevant differences, adhering to the 3R principles43 and guidelines such as PREPARE44 and ARRIVE35.
Results
Identification of Fn-EDB binding peptides
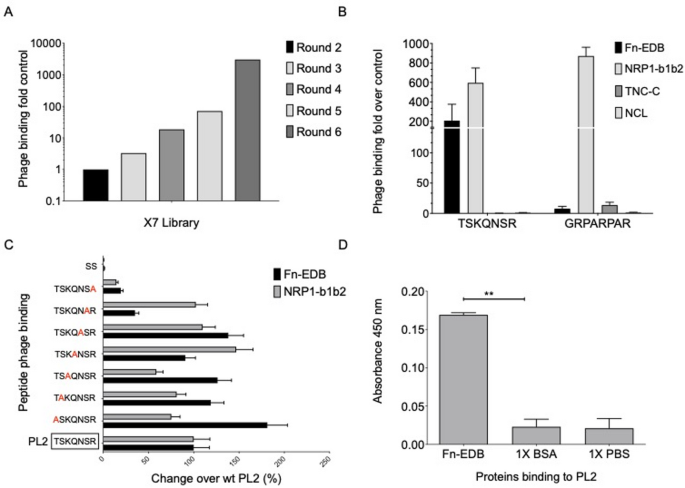
For biopanning, His6-tailed Fn-EDB domain was expressed in E. coli and purified using Ni-NTA chromatography (Supplementary Fig. S1). Subsequently, 6 rounds of selection were performed on immobilized Fn-EDB using X7 peptide T7 phage libraries. The first and fourth rounds of biopanning were performed using Fn-EDB immobilized on polystyrene multiwell plates, and other rounds were performed on Fn-EDB coated on Ni-NTA magnetic beads (Supplementary Fig. S2). We observed enrichment of Fn-EDB binding of the phages through the screening, with round 6 pool showing ~ 3000-fold increased binding over the naive library (Fig. 1A). Sanger sequencing of 48 random phage clones from a selection round 5 resulted in 11 unique peptide-phages that were individually tested for their interaction with Fn-EDB. Among the 11 candidates, 5 clones demonstrated the ability to bind to Fn-EDB in vitro > 50 fold over control phage displaying heptaglycine (Supplementary Fig. S3). To evaluate the systemic tumor homing of the in vitro selected Fn-EDB binding peptides, in vivo phage playoff auditioning was used38. An equimolar mixture of candidate and control peptide-phage were intravenously injected into a panel of glioma and prostate xenograft tumor-bearing mice and, following phage circulation and removal of blood background by perfusion, representation of peptide-phages in tumors was estimated using high-throughput DNA sequencing. We observed that the phage clone displaying the heptameric TSKQNSR peptide, designated PL2, was overrepresented in tested solid tumor models (Table 1). Interestingly, the C-terminal arginine of the PL2 peptide may engage with the “C-wall” binding pocket on the b1 domain of neuropilin-1 (NRP1)29.

Identification and characterization of the PL2 peptide binding to Fn-EDB and NRP-1. (A) Biopanning of T7 phage-displayed X7 peptide libraries on immobilized Fn-EDB resulted in ~ 3000-fold enrichment in phage binding after the sixth round of selection. (B) The selected PL2 peptide phage bound to immobilized Fn-EDB and NRP-1 b1b2 but not to control protein TNC-C, or Nucleolin. Phage binding is expressed as fold over control phage displaying heptaglycine (G7) peptide. (C) Alanine substitutions were introduced at each position of the PL2 peptide, and changes in binding were verified by measuring the percentage of changes in binding to immobilized Fn-EDB and NRP-1. The first peptide from the bottom without red in the box represents the parental PL2 peptide. (D) The interaction of synthetic FAM-PL2 peptide immobilized on an ELISA plate with recombinant His-tagged Fn-EDB was probed and detected using a rabbit anti-His-tag primary antibody and secondary goat anti-rabbit HRP antibody, followed by chromogenic peroxidase reaction. Values are expressed as mean ± standard deviation (SD) from 3 independent experiments; statistical analysis was performed using Student unpaired t-test; all statistical tests were two-sided; *p < 0.05; **p < 0.01.
Next, the binding of the PL2 peptide-displaying phage to recombinant Fn-EDB, NRP1, and control proteins (Tenascin-C C-domain, TNC-C; Nucelolin, NCL) was studied. The PL2 phages demonstrated robust binding to Fn-EDB and b1b2 fragment of NRP1 and no binding to TNC-C, a protein with size and negative surface charge similar to Fn-EDB (Fig. 1B). Interestingly, the PL2 phage exhibited ~ 700-fold higher binding to the recombinant NRP-1 b1b2 domain compared to the heptaglycine control phage, demonstrating a binding capacity comparable to the prototypic NRP-1 binding peptide, RPAPRPAR29. The binding of the PL2 displaying phage to Fn-EDB surpassed that of the previously reported Fn-EDB-binding peptide, ZD222 (Supplementary Fig. S4).
Alanine scanning of PL2 (TSKQNSR) was performed to characterize residue contributions to Fn-EDB and NRP-1 binding, using T7 phage display assays (Materials and Methods, Sect. 2.3). Alanine substitutions were introduced at each position (T1A, S2A, K3A, Q4A, N5A, S6A, R7A) relative to the parental PL2 peptide (TSKQNSR), and changes in binding to immobilized Fn-EDB and NRP-1 were quantified by ELISA (Fig. 1C, Supplementary Table S1). Substitution of the C-terminal arginine (R7A) reduced Fn-EDB binding by ~ 80% and NRP-1 binding by > 90%, confirming its role in the CendR motif for NRP-1 and partial contribution to Fn-EDB affinity. Serine at position 6 (S6A) decreased Fn-EDB binding by ~ 60%, suggesting involvement in hydrogen bonding or structural stabilization. Substitutions at threonine (T1A), lysine (K3A), and asparagine (N5A) enhanced Fn-EDB binding by 20–30%, indicating these residues may modulate affinity when replaced with alanine. NRP-1 binding was minimally affected by T1A, S2A, K3A, Q4A, and N5A (~ 0–10% reduction), but S6A reduced it by ~ 30% (Fig. 1C, Supplementary Table S1). These findings are consistent with the in vitro binding data for candidate peptides (Supplementary Fig. S3).
We then evaluated the interaction of synthetic 5(6)-carboxyfluorescein (FAM)-labeled PL2 peptide with Fn-EDB immobilized on polystyrene ELISA plates. The synthetic PL2 peptide retained its ability to bind Fn-EDB (Fig. 1D).
PL2 AgNPs bind to and are internalized by tumor cells in vitro
We next studied cellular internalization of the PL2-functionalized AgNPs45. Extracellular membrane-bound AgNPs were selectively removed by treatment with a mild, biocompatible redox-based hexacyanoferrate/thiosulfate etching solution, ensuring that only the signal from internalized AgNPs remained detectable40,45. CF555-labeled PL2-AgNPs were incubated with the Fn-EDB and NRP-1-expressing U87-MG glioma cells, NRP-1-positive PPC1 prostate carcinoma cells, and Fn-EDB- and NRP-1-negative M21 melanoma cells4,23,26,28. PL2-AgNPs exhibited robust endocytosis in U87-MG and PPC1 cells following 1-hour incubation, whereas non-functionalized control AgNPs exhibited negligible slight uptake in U87-MG and PPC1 cells, but not in M21 cells (Fig. 2A, B, Supplementary Fig. S5). This difference is likely due to higher baseline endocytic activity in U87-MG and PPC1 cells, which may engage non-specific uptake mechanisms such as macropinocytosis and cell surface properties (e.g., glycocalyx, membrane fluidity), compared to M21 cells, which lack Fn-EDB and NRP-1 expression46,47. In contrast, receptor-negative M21 cells did not internalize PL2-AgNPs nor control AgNPs (Fig. 2A, Supplementary Fig. S5), possibly due to less permissive membrane characteristics. Post-etching, the PL2-AgNP signal showed only a modest decrease in intensity compared to non-etched controls (Fig. 2A, B), confirming that the majority of PL2-AgNPs were taken up by the cells.

Cellular internalization of PL2 peptide-functionalized AgNPs. The PL2 AgNPs and control AgNPs labeled with CF555 fluorophore dye were incubated with PPC1 prostate carcinoma, U87-MG glioma, and M21 melanoma cells for 1 h. After washing and optional etching, the cells were processed for confocal imaging. (A) Confocal images show strong uptake of PL2-AgNPs (red) in NRP-1-positive PPC1 and U87-MG cells but not in NRP-1-negative M21 cells. Cells incubated with control particles are shown in boxes. Scale bar: 20 μm. (B) Quantification of binding and internalization of CF555-labeled particles was done using Fiji ImageJ software. Data represents 3 independent experiments. Error bars indicate mean ± SD (N = 3). Statistical significance was determined using unpaired Student’s t-test (ns, p > 0.05; **p ˂ 0.01; ***p ≤ 0.001; ****p < 0.0001).
Systemic PL2-functionalized nanoparticles accumulate in tumor lesions
Next, we investigated the potential of the PL2 peptide as a systemic tumor-targeting probe. The dextran-coated PEGylated paramagnetic iron oxide nanoworms (NWs) have been used as a theranostic nanosystem suitable for systemic affinity targeting, functioning both as a drug carrier and as a magnetic resonance imaging agent with T2 contrast properties23,39. FAM-labeled PL2 peptide or FAM-Cys control was conjugated to NWs with an average size of 88.9 ± 0.9 nm and zeta potential of 9.5 ± 0.4 mV (Supplementary Fig. S6). Conjugation of PL2 peptides did not significantly alter particle size or surface charge, indicating successful functionalization without aggregation (Supplementary Fig. S6C–D). NWs remained stable in PBS during preparation and imaging, consistent with prior reports on NW stability under physiological conditions23,41,48,49. Based on our prior studies using identical maleimide-thiol coupling chemistry, we estimate a PL2 peptide density of ~ 20–22 nmol/mg of iron (~ 50–80 peptides per NW), with conjugation efficiencies of ~ 80–90%, as reported for similar peptide-NW systems. Functionalization efficiency was assessed indirectly via fluorescence intensity of FAM-labeled PL2 peptides, consistent with these prior calibrations23,26,50.
In vivo homing studies were conducted in mice bearing orthotopic WT-GBM glioma, s.c. U87-MG glioma and s.c. PC3 prostate carcinoma xenograft tumor models, all of which express abundant Fn-EDB and NRP-14,23,26. Mice were intravenously injected with 7.5 mg/kg of NWs. After circulation, mice were perfused to remove free NWs, and tumors along with control organs were collected, sectioned, and imaged with confocal microscopy. PL2-functionalization significantly increased NW accumulation in CD31-positive vascular structures across all tumor models (Fig. 3A−C). In some regions, PL2-NWs extravasated and accumulated within the tumor parenchyma (Fig. 3A−C, arrowheads).
Following 5 h of circulation, PL2-NWs demonstrated enhanced tumor accumulation compared to control NWs, with a ~ 7-fold increase in PC3 tumors, ~ 7.5-fold increase in U87-MG tumors, and a ~ 2-fold increase in WT-GBM tumors (Fig. 3A−C, Supplementary Fig. S7). In contrast, the signal for PL2-functionalized and non-targeted NWs was comparable in control organs, including the liver, kidney, and lung (Fig. 3A−C, Supplementary Fig. S7). PL2-NWs showed minimal accumulation in healthy organs (Supplementary Fig. S7), suggesting low off-target effects, consistent with prior studies on peptide-functionalized NWs23,26. Future studies will assess acute and chronic toxicity, including immune responses, to support further translational applications. Furthermore, PL2-NWs exhibited selective accumulation in glioblastoma lesions, with minimal accumulation in nonmalignant brain in the orthotopic WT-GBM model and in the healthy brain of control mice (Supplementary Fig. S8).

Systemic PL2-NWs accumulate in solid tumors. (A, B, C) PL2-NWs and control FAM-NWs were intravenously injected at 7.5 mg/kg into mice bearing (A) subcutaneous PC3 prostate carcinoma, (B) subcutaneous U87-MG glioblastoma, and (C) orthotopic WT GBM glioblastoma xenografts. After 5 h of circulation, mice were perfused with PBS/DMEM, and organs (including tumors and control tissue) were snap-frozen. Cryosections of tissues were immunostained with rabbit anti-FAM (green) and rat anti-CD31 (red) antibodies, counterstained with DAPI (blue), and analyzed using confocal microscopy. The boxes represent mice injected with control non-targeted FAM-NWs. The arrows indicate PL2-NWs (green) located along the CD31-positive tumor blood vessels, and the arrowheads indicate extravasated PL2-NWs in the tumor parenchyma. Bar charts show quantification of tissue homing of FAM signal using Fiji ImageJ software (A-C). Error bars indicate mean ± SEM (N = 3–6 mice per group). Scale bars: 100 μm. P-values were determined using Student unpaired t-test, two-tailed; ns, not significant (p > 0.05); *p < 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p < 0.0001.
Macroscopic fluorescence imaging showed accumulation of the PL2-NWs, but not control NWs, in U87-MG tumors collected from mice (Fig. 4A), while no signal was detected in the control organs (liver, lung, heart, brain, kidney, and spleen) (Fig. 4A and D). The tumor homing pattern of the PL2-NWs was also studied by confocal imaging of U87-MG tumor tissue sections after staining with Fn-EDB- (ScFV L19) and NRP-1-specific antibodies. Fn-EDB and NRP-1 were upregulated in the tumors (Fig. 4B, C, E and F), and the PL2-NW signal showed extensive overlap with the receptors (Fig. 4B and C, arrows).

PL2-NWs accumulate in glioblastoma lesions and colocalize with Fn-EDB and NRP-1. (A–C, upper row) PL2-functionalized NWs and (D–F, lower row) control NWs were injected i.v. at 7.5 mg/kg into mice bearing s.c. U87-MG glioblastoma xenografts. After 5 h of circulation, mice were perfused with PBS/DMEM, and tissues were harvested for ex vivo macroscopic imaging and fluorescence microscopy. (A, D) Ex vivo macroscopic Illumatool images of PL2- and control NWs in the green channel. The superimposed images of white light and green fluorescence channels are representative of three independent experiments. (B, C, E, F) Confocal microscopy images of NWs, Fn-EDB, and NRP-1 in tumor cryosections. PL2-NWs colocalized with Fn-EDB and NRP-1 (arrows), whereas (E, F) FAM-NWs showed only background accumulation. Tumor tissues were stained with rabbit anti-FAM (green), anti-Fn-EDB ScFv L19 (red), and rabbit anti-NRP-1 (red) antibodies. PL2-NWs (B, C) colocalize with Fn-EDB and NRP-1 (indicated by arrows), whereas control NWs (E, F) exhibit only background accumulation. Scale bar, 100 μm; the images are representative of 3 independent experiments.
PL2-NWs bind to surgical explants of human clinical cancers
To evaluate the potential translational relevance of the PL2 peptide, we examined the binding and penetration capabilities of PL2-targeted NWs in fresh surgical explants of human ovarian carcinoma tissue. We first established that in these tumors, Fn-EDB were highly overexpressed (Supplementary Fig. S9). Previous studies have consistently demonstrated the overexpression of NRP-1in ovarian cancers, linking it to malignancy and poor prognosis51,52. Next, we performed an ex vivo tumor binding/penetration assay (“tumor dipping assay”)37,53 and observed that PL2-NWs demonstrated a 9-fold increase of binding and penetration in tumor tissue compared to the control NWs (Fig. 5A and B). To quantify PL2-NW tissue penetration, we performed 3D confocal analysis of ovarian carcinoma explants from three patients. PL2-NW signal (FAM channel) was segmented into discrete surfaces (n = 974) using Imaris. Across all samples, nanoworms penetrated to a maximum depth of 33.37 μm, with a mean depth of 17.43 ± 0.17 μm. The average surface area was 537.53 ± 13.40 μm2, and the volume was 278.32 ± 6.78 μm3, confirming penetration beyond the sub-surface regions of clinical tumor tissue (Fig. 5A, Supplementary Table S4). In contrast, control NWs exhibited only a background fluorescence signal. These findings suggest that the PL2-NW platform holds promise as a targeted drug delivery system for ovarian carcinoma and, potentially, other solid clinical tumors.

Confocal fluorescence imaging of PL2-NW binding to human ovarian carcinoma cancer explants. Fresh surgical explants of human ovarian carcinoma were incubated with PL2-NWs and control NWs at 37 °C for 4 h. The tissue sections were then stained with rabbit anti-FAM primary antibodies (green) and Alexa Fluor 488-conjugated anti-rabbit secondary antibodies. The nuclei were counterstained with DAPI (blue). The arrows indicate PL2-NW signals at the tumor rim and extravasation into the tissue. (B) Quantification of FAM fluorescence signal from the confocal images using Fiji ImageJ. Error bars represent mean ± SEM (N = 3). Statistical analysis was performed using Student’s t-test; ** p < 0.01.
Discussion
We report the development of a homing peptide named PL2, which targets the tumor-associated ECM protein Fn-EDB, a stable and abundant antigen overexpressed in many solid tumors. In addition to binding Fn-EDB, PL2 engages the cell and tissue penetration receptor NRP-1, enabling nanoparticles to internalize into cultured cells, home to solid tumors in vivo, and penetrate clinical tumor samples ex vivo. These findings support the potential applications of PL2 in targeting solid tumors enriched for Fn-EDB and NRP-1. In addition, PL2 may hold relevance for other Fn-EDB–positive pathological conditions, such as endometriosis, based on previous evidence of Fn-EDB expression and successful targeting of human endometriotic lesions using the PL1 peptide and the radiolabeled scFv probe [^111In]In-FnBPA554,55. However, dedicated experimental validation in endometriosis models is required54.
PL2 represents a novel linear heptapeptide that uniquely combines Fn-EDB targeting with NRP-1-mediated tissue penetration in a single, unmodified sequence. Unlike iRGD, which requires proteolytic activation53 or PL3, which bind alternative ECM components (TNC-C)26. Whereas PL1 binds to both FnEDB and TNC-C23. PL2 inherently engages both targets through a minimal CendR motif. This dual-binding capability emerged directly from phage biopanning without structural optimization. Compared to previously reported peptides, PL2 offers simplified synthesis and robust tumor accumulation, as demonstrated in multiple xenograft models and ex vivo human tissue. A comparative summary of PL2 versus related peptides is provided in Supplementary Table S2.
ECM-reactive affinity ligands have been used to deliver anticancer payloads such as cytokines/growth factors, proapoptotic peptides, cell-permeable cytotoxic compounds, and imaging agents to tumors4,14,56,57. However, ECM-targeting compounds, including those targeting Fn-EDB, rely on passive delivery through the enhanced permeability and retention (EPR) effect, which is subject to significant inter- and intratumoral variability58,59. Additionally, within the tumor microenvironment, ECM-targeting ligands generally exhibit limited cellular uptake, necessitating the use of cleavable linkers (e.g., disulfides or hydrazones) to release drug payloads into the extracellular milieu57,60,61. In this study, we employed cell-free phage biopanning to identify peptides that bind Fn-EDB. Unlike other selected peptides, PL2 peptide possessed the C-terminal arginine residue forming a minimal C-end Rule (CendR) element known to mediate binding a cellular pleiotropic multiligand receptor NRP-129. Although the PL2 peptide-phage ranked fifth among 11 tested phages in the cell-free Fn-EDB binding assay, it outperformed all others in an in vivo playoff assay. The functionality of the CendR element in PL2 was demonstrated in in vitro studies, where AgNPs functionalized with the PL2 peptide exhibited strong NRP-1-dependent cellular binding and internalization, consistent with our pervious finding for the PL3 peptide26. In vivo, functionalizing nanoparticles with the PL2 peptide significantly enhanced their tropism to prostate carcinoma and glioblastoma, with significant accumulation in the extravascular space and colocalization with both Fn-EDB and NRP-1 immunoreactivities. Finally, PL2-targeted nanoparticles effectively bound to and penetrated clinical surgical explants ex vivo, but its translational applications require further validation through efficacy and safety studies. This finding is not unexpected as small homing peptides typically target evolutionarily conserved binding pockets on their target molecules and the 91-amino acid alternatively spliced EDB domain is conserved across multiple species, including mice, rats, rabbits, dogs, monkeys, and humans11,23. Additionally, CendR peptides consistently bind to the b1 domain of NRP-1 across these species, from mouse to human. These studies, in the context of well-established knowledge of the upregulation of Fn-EDB and NRP-1 in various primary solid tumors and metastatic lesions, highlight the potential of PL2-based nanoparticle targeting strategy for application across a range of solid tumor types12,62. Although our studies show that the PL2 peptide appears a promising targeting agent for nanoparticles, there is still room for improvement and opportunities to broaden the range of applications.
Short homing peptides with a high degree of conformational freedom and small number of contact residues have typically moderate affinity yet are capable of high avidity interactions with their targets due to cooperative multivalent binding when displayed on nanoparticles6,20,21. This is illustrated by complement factor C1q-IgG interactions, where the dissociation constant (Kd) improves dramatically with increasing valency—100 µM for monomers, 1 µM for dimers, and 3 nM for tetramers63. Interestingly, our alanine scan revealed that at certain positions, particularly the N > A substitution at position 5, alanine substitution enhanced peptide phage binding to recombinant receptor molecules. In follow-up studies to improve the affinity of the PL2 peptide, it will be valuable to conduct secondary screens using constrained peptide libraries, created by randomizing nonessential amino acids of the PL2 peptide and/or adding additional flanking amino acids to provide stabilizing interactions. Another potential avenue for improving the PL2 peptide involves enhancing its tumor specificity. While NRP-1 is overexpressed in solid tumors, it is present at lower levels in the vascular beds of normal organs, particularly the lungs, which could result in off-target accumulation. To address this, further improvement of the PL2 peptide could include capping the C-terminal arginine with additional amino acids that are engineered or screened to be cleaved by tumor-expressed extracellular proteases. Using a similar approach, we recently reported the development of a urokinase-type plasminogen activator-dependent CendR peptide to mitigate background accumulation in nontarget tissues27. Such continuation studies could further facilitate the applications of PL2 as a cancer targeting agent.
Many homing peptides, by binding to functionally important binding pockets on target molecules, trigger biological responses through mechanisms such as eliciting conformational changes or competing with natural ligands for receptor binding. Examples include the antitumor effects of LyP-164, tumor penetration induction and immunomodulation of iRGD30,65wound healing promotion by CAR66and suppression of choroidal neovascularization by PL326,67. Similarly, in addition to its use in affinity targeting, the PL2 peptide may possess inherent biological activity by disrupting interactions between Fn-EDB and integrins or other matrix components involved in adhesion, migration, and survival of malignant or tumor-promoting cells, potentially suppressing tumor growth, metastasis, and angiogenesis. If the biological activity of PL2 is confirmed, its improved variants, developed using the strategies outlined above, and proteolysis-resistant PL2 derivatives incorporating nonproteinogenic amino acids, will be advantageous for further applications. This study establishes PL2’s targeting and penetration capabilities, laying the foundation for therapeutic applications. Future studies will evaluate PL2-NWs loaded with cytotoxic agents, such as doxorubicin/ paclitaxel, to assess tumor growth inhibition and survival benefits, building on prior work with peptide-functionalized NWs 68.
In conclusion, our study presents the development of the PL2 peptide, which holds potential for applications in targeted drug delivery and molecular imaging. The dual targeting of Fn-EDB and NRP-1 by the PL2 peptide offers a promising strategy for advancing targeted cancer therapies. The use of PL2 nanoparticles to tumor tissues shows promise for the development of novel cancer treatments. Further studies are needed to optimize the peptide and assess its clinical efficacy in targeting solid tumors as well as safety.
Data availability
The datasets generated and/or analysed during the current study are included in this article and its supplementary information files. No additional data are required to be deposited in public repositories. Any additional data supporting the findings of this study are available from the corresponding author upon reasonable request.
References
-
Guelfi, S., Hodivala-Dilke, K. & Bergers, G. Targeting the tumour vasculature: from vessel destruction to promotion. Nat. Rev. Cancer 24, 1456 (2024).
-
Forster, J., Harriss-Phillips, W., Douglass, M. & Bezak, E. A review of the development of tumor vasculature and its effects on the tumor microenvironment. Hypoxia 5, 21–32 (2017).
-
Ruoslahti, E., Bhatia, S. N. & Sailor, M. J. Targeting of drugs and nanoparticles to tumors. J. Cell Biol. 188, 759–768 (2010).
-
Lingasamy, P. Development of Multitargeted Tumor Penetrating Peptides (University of Tartu, 2020). http://hdl.handle.net/10062/70738.
-
Jailkhani, N. et al. Proteomic profiling of extracellular matrix components from patient metastases identifies consistently elevated proteins for developing nanobodies that target primary tumors and metastases. Cancer Res. 83, 2052 (2023).
-
Lingasamy, P. & Teesalu, T. Homing peptides for Cancer therapy. Adv. Exp. Med. Biol. 1295, 29–48 (2021).
-
Lingasamy, P. et al. PL1 peptide engages acidic surfaces on Tumor-Associated fibronectin and Tenascin isoforms to trigger cellular uptake. Pharmaceutics 13, 1998 (2021).
-
Muncie, J. M. & Weaver, V. M. The physical and biochemical properties of the extracellular matrix regulate cell fate. Curr. Top. Dev. Biol. 130, 1 (2018).
-
Rekad, Z., Izzi, V., Lamba, R., Ciais, D. & Van Obberghen-Schilling, E. The alternative matrisome: alternative splicing of ECM proteins in development, homeostasis and tumor progression. Matrix Biol. 111, 26–52 (2022).
-
Khan, Z. A. et al. EDB fibronectin and angiogenesis – a novel mechanistic pathway. Angiogenesis 8, 183–196 (2005).
-
Zardi, L. et al. Transformed human cells produce a new fibronectin isoform by Preferential alternative splicing of a previously unobserved exon. EMBO J. 6, 2337–2342 (1987).
-
Saw, P. E. et al. Extra-domain B of fibronectin as an alternative target for drug delivery and a cancer diagnostic and prognostic biomarker for malignant glioma. Theranostics 11, 941–957 (2021).
-
Castellani, P. et al. The fibronectin isoform containing the ed-b oncofetal domain: a marker of angiogenesis. Int. J. Cancer. 59, 612–618 (1994).
-
Teesalu, T. & Lingasamy, P. Bi-Specific extracellular matrix binding peptides and methods of use thereof. PCT Patent No WO 2020161602A1 (2020).
-
Lieverse, R. I. Y. et al. Human fibronectin extra domain B as a biomarker for targeted therapy in cancer. Mol. Oncol. 14, 1555–1568 (2020).
-
Miura, J. T. & Zager, J. S. Neo-DREAM study investigating Daromun for the treatment of clinical stage IIIB/C melanoma. Future Oncol. 15, 3665–3674 (2019).
-
Flatz, L. et al. Intratumoral administration of Daromun in non-melanoma skin cancer: preliminary results from a phase 2 non-randomized controlled trial. J. Eur. Acad. Dermatol. Venereol. https://doi.org/10.1111/JDV.20163 (2024).
-
Danielli, R. et al. Intralesional administration of L19-IL2/L19-TNF in stage III or stage IVM1a melanoma patients: results of a phase II study. Cancer Immunol. Immunother. 64, 999–1009 (2015).
-
Albericio, F. & Kruger, H. G. Therapeutic peptides. Future Med. Chem. 4, 1527–1531 (2012).
-
Zhao, N., Qin, Y., Liu, H. & Cheng, Z. Tumor-Targeting peptides: ligands for molecular imaging and therapy. Anticancer Agents Med. Chem. 18, 74–86 (2017).
-
Fosgerau, K. & Hoffmann, T. Peptide therapeutics: current status and future directions. Drug Discov Today. 20, 122–128 (2015).
-
Han, Z. et al. EDB fibronectin specific peptide for prostate Cancer targeting. Bioconjug. Chem. 26, 830–838 (2015).
-
Lingasamy, P. et al. Bi-specific tenascin-C and fibronectin targeted peptide for solid tumor delivery. Biomaterials 219, 119373 (2019).
-
Nagamitsu, A., Greish, K. & Maeda, H. Elevating blood pressure as a strategy to increase Tumor-targeted delivery of macromolecular drug SMANCS: cases of advanced solid tumors. Jpn J. Clin. Oncol. 39, 756–766 (2009).
-
Subhan, M. A., Parveen, F., Filipczak, N., Yalamarty, S. S. K. & Torchilin, V. P. Approaches to improve EPR-Based drug delivery for Cancer therapy and diagnosis. J. Personalized Med. 2023. 13, 389 (2023).
-
Lingasamy, P. et al. Tumor-penetrating peptide for systemic targeting of Tenascin-C. Sci. Rep. 10, 5809 (2020).
-
Tobi, A. et al. Protease-activated CendR peptides targeting tenascin-C: mitigating off-target tissue accumulation. Drug Deliv Transl Res. 14, 2945–2961 (2024).
-
Lingasamy, P., Laarmann, A. H. & Teesalu, T. Tumor penetrating Peptide-Functionalized Tenascin-C antibody for glioblastoma targeting. Curr. Cancer Drug Targets. 21, 70–79 (2021).
-
Teesalu, T., Sugahara, K. N., Kotamraju, V. R. & Ruoslahti, E. C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration. Proc. Natl. Acad. Sci. U S A. 106, 16157–16162 (2009).
-
Sugahara, K. N. et al. Tissue-Penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell. 16, 510–520 (2009).
-
Schiefner, A., Gebauer, M. & Skerra, A. Extra-domain B in oncofetal fibronectin structurally promotes fibrillar head-to-tail dimerization of extracellular matrix protein. J. Biol. Chem. 287, 17578–17588 (2012).
-
Bougnaud, S. et al. Molecular crosstalk between tumour and brain parenchyma instructs histopathological features in glioblastoma. Oncotarget 7, 31955–31971 (2016).
-
Talasila, K. M. et al. EGFR wild-type amplification and activation promote invasion and development of glioblastoma independent of angiogenesis. Acta Neuropathol. 125, 683–698 (2013).
-
Keunen, O. et al. Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proc. Natl. Acad. Sci. U S A. 108, 3749–3754 (2011).
-
Kilkenny, C., Browne, W. J., Cuthill, I. C., Emerson, M. & Altman, D. G. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 8, e1000412 (2010).
-
Teesalu, T., Sugahara, K. N. & Ruoslahti, E. Mapping of vascular ZIP codes by phage display. Methods Enzymol. 503, 35–56 (2012).
-
Ikemoto, H. et al. Hyaluronan-binding peptide for targeting peritoneal carcinomatosis. Tumor Biol. 39, 1010428317701628 (2017).
-
Põšnograjeva, K., Pleiko, K., Haugas, M. & Teesalu, T. New tools for streamlined in vivo homing peptide identification. Methods Mol. Biol. 2383, 385–412 (2022).
-
Park, J. H. et al. Magnetic iron oxide nanoworms for tumor targeting and imaging. Adv. Mater. 20, 1630–1635 (2008).
-
Willmore, A. M. A. et al. Targeted silver nanoparticles for ratiometric cell phenotyping. Nanoscale 8, 9096–9101 (2016).
-
Säälik, P. et al. Peptide-guided nanoparticles for glioblastoma targeting. J. Controlled Release. 308, 109–118 (2019).
-
World Medical Association. Declaration of helsinki: ethical principles for medical research involving human subjects. JAMA 310, 2191–2194 (2013).
-
Balls, M. It’s time to reconsider the principles of humane experimental technique. Altern. Lab. Anim. 48, 40–46 (2020).
-
Smith, A. J., Clutton, R. E., Lilley, E., Hansen, K. E. A. & Brattelid, T. PREPARE: guidelines for planning animal research and testing. Lab. Anim. 52, 135–141 (2018).
-
Braun, G. B. et al. Etchable plasmonic nanoparticle probes to image and quantify cellular internalization. Nat. Mater. 13, 904–911 (2014).
-
Ruoslahti, E. Peptides as targeting elements and tissue penetration devices for nanoparticles. Adv. Mater. 24, 3747–3756 (2012).
-
Wilhelm, S. et al. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 1, 16014 (2016).
-
Park, J. H. et al. Systematic surface engineering of magnetic nanoworms for in vivo tumor targeting. Small 5, 694–700 (2009).
-
Agemy, L. et al. Nanoparticle-induced vascular Blockade in human prostate cancer. Blood 116, 2847–2856 (2010).
-
Sharma, S. et al. Tumor-Penetrating nanosystem strongly suppresses breast tumor growth. Nano Lett. 17, 1356–1364 (2017).
-
Baba, T. et al. Neuropilin-1 promotes unlimited growth of ovarian cancer by evading contact Inhibition. Gynecol. Oncol. 105, 703–711 (2007).
-
Jiang, H. et al. Increased expression of neuropilin 1 is associated with epithelial ovarian carcinoma. Mol. Med. Rep. 12, 2114–2120 (2015).
-
Sugahara, K. N. et al. Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs. Sci. (1979). 328, 1031–1035 (2010).
-
Simón-Gracia, L. et al. Homing peptide-based targeting of tenascin-C and fibronectin in endometriosis. Nanomaterials 11, 1–28 (2021).
-
Trachsel, B. et al. Relaxed fibronectin: a potential novel target for imaging endometriotic lesions. EJNMMI Res. 14, 17 (2024).
-
Yeow, Y. L. et al. Immune-mediated ECM depletion improves tumour perfusion and payload delivery. EMBO Mol. Med 11, (2019).
-
Raavé, R., van Kuppevelt, T. H. & Daamen, W. F. Chemotherapeutic drug delivery by tumoral extracellular matrix targeting. J. Control Release. 274, 1–8 (2018).
-
Maeda, H. Macromolecular therapeutics in cancer treatment: The EPR effect and beyond. J. Controlled Release 164, 138–144. https://doi.org/10.1016/j.jconrel.2012.04.038 (2012).
-
Prabhakar, U. et al. Challenges and key considerations of the enhanced permeability and retention effect for nanomedicine drug delivery in oncology. Cancer Res. 73, 2412–2417 (2013).
-
Dal Corso, A., Cazzamalli, S., Gébleux, R., Mattarella, M. & Neri, D. Protease-Cleavable linkers modulate the anticancer activity of noninternalizing Antibody-Drug conjugates. Bioconjug. Chem. 28, 1826–1833 (2017).
-
Gébleux, R., Wulhfard, S., Casi, G. & Neri, D. Antibody format and drug release rate determine the therapeutic activity of non-internalizing antibody-drug conjugates. Mol. Cancer Ther. 14, 2606 (2015).
-
Locher, R. et al. Abundant in vitro expression of the oncofetal ED-B-containing fibronectin translates into selective pharmacodelivery of 131I-L19SIP in a prostate cancer patient. J. Cancer Res. Clin. Oncol. 140, 35–43 (2014).
-
Male, D., Champion, B. & Cooke, A. Adv. Immunol. Parasitol. 96, 643–644 (1987).
-
Laakkonen, P. et al. Antitumor activity of a homing peptide that targets tumor lymphatics and tumor cells. Proc. Natl. Acad. Sci. U S A. 101, 9381–9386 (2004).
-
Sugahara, K. N. et al. Tumor-penetrating iRGD peptide inhibits metastasis. Mol. Cancer Ther. 14, 120–128 (2015).
-
Maldonado, H. et al. Systemically administered wound-homing peptide accelerates wound healing by modulating syndecan-4 function. Nat. Commun. 14, 1456 (2023).
-
Puranen, J. et al. Intravitreal CendR peptides target laser-induced choroidal neovascularization sites in mice. J. Controlled Release. 360, 810–817 (2023).
-
Simón-Gracia, L. et al. iRGD peptide conjugation potentiates intraperitoneal tumor delivery of Paclitaxel with polymersomes. Biomaterials 104, 247–257 (2016).
Acknowledgements
T.T. was funded by the Estonian Research Council (grants PRG230 and PRG1788), EuronanomedIII projects ECM-CART and iNanoGun, and TRANSCAN3 project ReachGLIO (all coordinated by Estonian Research Council).
Ethics declarations
Competing interests
T.T. and P.L. hold a patent on the PL2 peptide (“Bi-Specific Extracellular Matrix Binding Peptides and Methods of Use Thereof”, WO. patent no. WO 2020/161602 A1), and TT is an inventor of CendR peptides. Other authors declare that they have no competing interests.
Ethics and informed consent
The fresh surgical ovarian carcinoma samples were obtained under protocols approved by the Ethics Committee of the University of Tartu, Estonia (permit #243/T27). Additionally, informed consent was obtained from all the patients, and methods for using human samples were carried out under relevant guidelines and regulations accordance with the Declaration of Helsinki.
Animals and ethics statement
Animal experimentation procedures were approved by the Estonian Ministry of Agriculture, Committee of Animal Experimentation, projects #42 and #48 (IACUC). We confirm that all methods were performed in accordance with the relevant guidelines and regulations. We confirm that the study was conducted in accordance with the ARRIVE guidelines.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lingasamy, P., Tobi, A., Kurm, K. et al. Targeting oncofetal fibronectin and neuropilin-1 in solid tumors with PL2 peptide. Sci Rep 15, 29369 (2025). https://doi.org/10.1038/s41598-025-11299-x
-
Received:
-
Accepted:
-
Published:
-
DOI: https://doi.org/10.1038/s41598-025-11299-x