Introduction
Prostate cancer ranks fifth internationally in terms of cancer-related mortality among males and also as the second most prevalent malignant tumour1. According to the Global Cancer Statistics (GLOBOCAN), approximately 1.5 million new cases of prostate cancer and 397,000 deaths were reported in 20222. However, by 2040, the global prostate cancer incidence is predicted to rise to 2,408,776 cases accounting for additional 579,684 deaths3. The rising incidence of prostate cancer necessitates investigation into its causes and the development of preventative methods. Numerous studies have shown the involvement of different risk factors, such as lifestyle, age, family history, and environmental conditions; however, the exact cause of prostate cancer continues to remain unclear4.These risk factors influence the severity of treatment-related side effects and therapeutic outcomes in addition to the initial onset of prostate cancer. Currently, patients diagnosed with early-stage of PCa tend to receive radical prostatectomy, brachytherapy, external beam radiation therapy (EBRT), continuous monitoring, or a combination of these treatments5. Besides, androgen deprivation therapy (ADT) is considered as the first-line of treatment for individuals with advanced illness that can be provided either alone or in combination with chemotherapy5,6,7. However, most of the patients in due course develop castration-resistant prostate cancer (CRPC) despite undergoing 12–24 months of ADT, with a median survival time period of only 14.5 months8,9,10. Over the last few years, novel drugs such as cabazitaxel, abiraterone, docetaxel, and enzalutamide have been used as first-line treatments for CRPC. However, their efficacy to improve disease outcomes have remained limited11. Therefore, patients receiving these therapies often experience poorer quality of life due to a variety of side effects including night sweats, hot flashes, anaemia, decreased libido, erectile dysfunction, and other challenges12,13. Thus, development of novel treatments for cancer with low toxicity and high bioavailability is crucial for improving therapeutic outcomes while reducing the adverse effects associated with conventional therapies.
Traditional herbal drugs have received notable acceptance as a key alternative and complementary therapy for cancer patients. Previous studies have suggested that the incorporation of herbal products as an adjuvant treatment in combination with traditional therapies can boost the quality of life, reduce side effects, enhance survival, and improve the therapeutic effects14,15,16,17,18,19. Plant-based foods rich in bioactive chemicals such as polyphenols, salicylates, glucosinates, flavonoids, and sterols yields a wide variety of therapeutic benefits on health20,21,22. A number of phytochemicals have shown anticancer effects by blocking the metastasis, invasion, proliferation and migration of cancer cells by targeting one or more signaling pathways which are essential in modulating the cancer cells23,24. Therefore, there is a growing focus on plant-derived products in the search for developing novel therapeutics for prostate cancer. Curcuma amada (Roxb.), commonly known as mango ginger, is a rhizomatous, perennial herb of the Zingiberaceae family. The plant is widely distributed across the humid subtropical regions of South East Asia, Africa and Australia25. The rhizomes of the plant have been extensively utilized in food industry and as alternative therapies to cure several ailments, including bronchitis, colic, cough, indigestion, hiccups, loss of appetite, and constipation26. Several reports are available on the anticancer activities of leaf and rhizome of C. amada. Methanol extract of mango ginger rhizomes and leaves have been observed to cause cell death in breast cancer cell lines27. Another researcher has reported on the cytotoxic effects of fractionated ethanolic and chloroform-methanolic extracts of C. amada rhizomes against the HCT-116 human colorectal cancer cell line28. Preclinical studies have reported supercritical CO2 extract of C. amada exhibits anticancer effect in in vitro cells (U-87MG human glioblastoma multiforme cells) and in nude mouse xenograft models29. However, hardly any research has been done to explain the possible mode of action of C. amada towards PCa modulation, contrary to claims of its anticancer properties. Therefore, it is essential to discover potential active compounds and molecular targets of C. amada rhizome extract in order to better comprehend its anticancer effect against prostate cancer.
Conventional drug discovery typically focuses on identifying single bioactive molecules that target specific proteins or pathways in order to treat diseases. However, it often fails to address the multifactorial mechanisms underlying diseases including cancer30. Network pharmacology represents a combined in-silico strategy for creating “protein-compound/disease-gene” network to disclose the complex mechanisms behind the coexisting curative effects of conventional drugs31,32. As a result, the theoretical framework shifted from a “one-target, one-drug” to a “network-target, multiple-component-therapeutics” approach32. In addition, advanced computational techniques including molecular docking and molecular dynamics (MD) simulation can be used to assess the relationship between the active constituents of C. amada and the primary targets of prostate cancer. These methods may also offer comprehensive understanding of the flexibility and stability of molecular complexes formed over time.
Therefore, the present investigation seeks to determine the phytoconstituents of C. amada with anti-prostate cancer properties and build a network for possible target modulation using network pharmacology. Firstly, the bioactive components present in Curcuma amada were identified through UHPLC-QTOF-HRMS/MS technique. Subsequently, potential molecular targets linked to both the identified phytochemicals and prostate cancer were retrieved from different publicly accessible databases, respectively. Swiss ADME and pkCSM online servers were used to observe the drug-likeness, physicochemical, and pharmacokinetic characteristics of the detected compounds. A protein-protein interaction (PPI) network with overlapping targets was created, and hub targets were identified. The compound-disease target network was built by connecting the predicted targets for prostate cancer with the active constituents of C. amada using Cytoscape software. The Kyoto Encyclopaedia of Genes and Genomes (KEGG) and Gene Ontology (GO) were used for pathway enrichment and biological function analysis. Molecular docking and MD simulation studies were performed to assess the binding mechanism between drug molecules and target proteins encoded by the genes implicated in prostate cancer. The hub targets were then examined for mRNA expression, survival rates, immune cell infiltration patterns, and genetic alterations. Lastly, experimental verification assays, including cell viability, apoptosis, cell cycle analysis, and qRT-PCR analysis were carried out to validate the prediction of network pharmacology. The detailed workflow outlining the investigation is provided in Figure S1.
Methodology
Plant material
The fresh rhizomes of Curcuma amada were obtained from Chakapada, Khordha Odisha (20°11’27.9” N, 85°24’56.9” E). The sample was verified by taxonomist Prof. P.C. Panda and the voucher specimen (2709/CBT/25.11.2023) was deposited at the herbarium of the Centre for Biotechnology, Siksha ‘O’ Anusandhan (Deemed to be University), Bhubaneswar. The rhizomes were washed, sliced, and shade-dried at room temperature before being thoroughly crushed with a mechanical grinder. The powdered rhizome (50 g) was extracted with 150 mL of methanol via soxhlet apparatus. The obtained extract was filtered, then evaporated using a rotary evaporator (Hei-VAP Core HL, Heidolph, Schwabach, Germany) and stored at 4 °C.
UHPLC-QTOF-HRMS/MS analysis
The UHPLC-QTOF-HRMS/MS analysis of rhizome extract of C. amada was performed on an ultra-high performance liquid chromatography coupled with quadrupole time-of-flight mass spectrometer and a dual AJS electrospray ionisation (ESI) source. The mobile phases were composed of 0.1% fluoroacetic acid in water and 100% acetonitrile, which was employed under gradient settings at 0.3 mL/min flow rate during 35-minute run. The hip sampler model G6550A was used to inject the sample, with 3 µL ejected at 100 µL/min. The mass spectrometer was programmed to scan between 120 and 1200 m/z, with MS and MS/MS scan rates of 1.0 spectra per second (temp. set at 40 °C). Mass spectrum for each with high-resolution was obtained in both the positive and negative ionization modes (gas temp. set at 250 °C; dry gas flow rate of 13 L/min. The nebulizer pressure was set to 35 psi. Further, the compounds’ retention times and mass spectrum data were compared to the in-house reference repository in order to make a tentative identification.
Screening of drug-like candidates of Curcuma amada
The structures of the active compounds obtained from UHPLC-QTOF-HRMS/MS analysis were confirmed using PubChem database (https://pubchem.ncbi.nlm.nih.gov/). The physicochemical, pharmacokinetic properties, and drug similarity of the compounds were assessed using SwissADME (http://www.swissadme.ch/) and pkCSM (http://biosig.unimelb.edu.au/pkcsm/) web servers. Further, Protox II online tool (https://tox-new.charite.de/protoxII/) was used to offer data on toxicity class, LD50 values, and several toxicologic endpoints.
Prediction of active compounds-related and prostate cancer-related target genes
The canonical SMILES of the filtered compounds were submitted to the SuperPRED (https://prediction.charite.de/) and Swiss Target Prediction (http://www.swisstargetprediction.ch/) platforms to predict their probable biological targets of C. amada. The targets predicted with Swiss Target Prediction and SuperPRED, which have a probability greater than zero and 50%, respectively, were selected as potential targets for CARE. To ensure consistency and facilitate integration with disease gene datasets, we standardized the downstream analysis of proteins obtained from SwissTargetPrediction and SuperPRED by converting these targets into their corresponding gene names. Furthermore, potential PCa-associated targets were retrieved from multiple databases, including GeneCard (https://www.genecards.org/), Therapeutic Target Database (TTD) (https://idrblab.net/ttd/), and MalaCard (https://www.malacards.org/), ensuring that the search was limited to Homo sapiens. In addition, the search terms included “prostate adenocarcinoma” and “prostate cancer.” Furthermore, the gene names of the relevant targets were normalised using the UniProt (https://www.uniprot.org/) database, retaining exclusively the “human” genes for the study.Following that, drug and disease-related targets were merged to find potential CARE targets against prostate cancer using Venny 2.1.0 after deleting duplicate entries.
Protein‒protein interaction (PPI) analysis, screening of hub genes and network construction
The overlapping targets between active compounds of CARE and prostate cancer were uploaded to the STRING database (https://string-db.org/) to build functional protein association networks. The intersecting genes were selected by limiting the species to “Homo sapiens,” setting the confidence score to greater than 0.7, and removing independent nodes from the network. The PPI results were then imported into Cytoscape v.3.9.1 software to create and visualize the regulatory network, resulting in the identification of hub genes. The CytoHubba plugin was used to filter and select key targets in the network based on degree centrality (DC) values. Furthermore, the chiplotonline (https://www.chiplot.online/) web-based tool was utilized to show the degree of interaction between the hub genes. Furthermore, the CytoNCA plug-in in Cytoscape software was used to create a compound-target-disease network. The network diagram depicted the multi-component (nodes) and multi-targeted interactions (edges) between CARE bioactive phytoconstituents and their predicted targets in the treatment of prostate cancer.
Enrichment analysis
Gene Ontology (GO) enrichment analysis was conducted to identify important GO terms, including biological processes (BP), molecular functions (MF), and cellular components (CC), using the ShinyGO web tool (https://bioinformatics.sdstate.edu/go77/). The target proteins were uploaded in a text format, and the resulting gene networks were subsequently analysed. GO terms with a p-value of ≤ 0.05 were considered significant and were visualized using SR Plot (https://www.bioinformatics.com.cn/en) and ChiPlotonline server. Additionally, KEGG pathway enrichment analysis was carried out using the DAVID v6.8 platform (https://david.ncifcrf.gov/) to explore the involvement of key targets in various signalling pathways. A p-value of < 0.01 and an FDR of < 0.01 were used to define significant KEGG enrichments. The overlapping targets were then input into KEGG Mapper (https://www.genome.jp/kegg/mapper/) to uncover key pathways and investigate the associated molecular mechanisms.
Molecular docking
Protein preparation is a crucial step that involves refining structures of macromolecule to ensure their compatibility for computational analysis. In this study, X-ray crystallographic structures of seven hub proteins, including BCL2 (PDB ID- 7LH7), CTNNB1 (PDB ID- 1JDH), ESR1 (PDB ID-1XP6), TP53 (PDB ID-1AIE), EGFR (PDB ID-1M17), HIF1A (PDB ID-1H2M), CCND1 (PDB ID-2W96), were retrieved from the RCSB Protein Data Bank (PDB), while PIK3CA and AKT1 was predicted using the I-TASSER web server, since it had missing amino acid residues. Primary structure of PIK3CA and AKT1 was retrieved from the UniProt database with the UniProt ID and “P42336” and “P31749”, respectively. The accuracy of predictions was provided based on the confidence score of the modeling and the best scored model was identified and utilized for model validation. These proteins underwent the following preparation steps prior to docking: (i) removal of water molecules, heteroatoms, and co-crystallized ligands, (ii) addition of polar hydrogen atoms to the target proteins using BIOVIA Discovery Studio Visualizer 2019, (iii) model refinement of side chains using the “Quick and Dirty fixing” feature in SPDBv viewer, (iv) calculation of gasteiger charges using UCSF Chimera software. Ligand 3D spatial data files (SDFs) were traced from the PubChem database and converted to PDB format using BIOVIA Discovery Studio Visualizer. These PDB files for ligands were then imported into the PyRx software using its built-in Open Babel tool, where force fields were applied to optimize the compound geometries. Finally, the protein and ligand PDB files were converted into PDBQT format (containing partial charges and atom types) using the Autodock plug-in in PyRx software.One of the most significant strategies for treating diseases is the binding of ligands to targeted protein positions. Increased toxicity risks and adverse consequences may result from improper binding33.The PrankWeb server (https://prankweb.cz/) was utilized to analyse potential binding sites on the target protein structures. This server employed an advanced machine learning approach to predict ligand binding sites based on protein structure. Following this, an appropriate grid box size and grid point spacing were selected for each protein-ligand interaction, using the 3D coordinate data from the PrankWeb server. The centre (X, Y, and Z) coordinates were modified to provide complete coverage of the protein’s favourable binding regions.Selected hub compounds were further subjected to molecular docking analysis using PyRx virtual screening software34. In this study, molecular docking was performed utilizing the AutoDock Vina plugin. Finally, the compounds exhibiting the strongest binding affinity were selected and visualized via the BIOVIA Discovery Studio.
MD simulation study
The top poses derived from molecular docking investigations on the basis of binding affinity scores were subjected to MD simulations to evaluate the binding mode and stability of the formed complexes at two different time intervals (100 and 200 ns). The simulations were performed using a module developed by SiBioLead LLC that employed GROMACS to analyse the conformational changes in the protein-ligand complexes compared to the unbound apo-protein. Prior to simulation, the complexes were pre-processed with the OPLS/AA force field. The complexes were then placed in a triclinic periodic boundary box filled with a simple point charge (SPC) water solvation model and a 0.15 M NaCl solution to maintain counter ion concentration. Energy minimization was carried out using the steepest descent algorithm. This was followed by NVT/NPT equilibration for 100 ps each to stabilize the system, maintaining a pressure of 1 bar and a temperature of 300 K. Finally, a leap-frog integrator was used to run the system through a 100 and 200 ns production cycle. The peaks developed after conducting MD simulation were further examined using GROMACS’ built-in trajectory analysis tool. The stability of the ligand-protein complexes was evaluated using various parameters derived from the simulation trajectories, such as root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), solvent accessible surface area (SASA), secondary structure count (DSSP) and molecular mechanics energies coupled with Poisson-Boltzmann surface area continuum solvation (MM/PBSA).
Expression analysis of hub genes
Genetic alterations in core targets
cBioPortal (www.cbioportal.org) server was used to evaluate mutations, copy number alteration (CNA), and the gene type summary in prostate adenocarcinoma (PRAD) following the cBioPortal online server.
Gene expression levels and survival analysis of core targets
UALCAN (http://ualcan.path.uab.edu) is an open web tool that employs level 3 RNA-sequencing and clinical data from the TCGA database to analyse 31 cancer types. The “Expression Analysis” component of the UALCAN dataset was used to acquire data with respect to the expression of hub genes. Furthermore, overall survival (OS) and disease-free survival (DFS) analyses based on a median group cutoff were performed on PCa patients through the GEPIA database. The plots indicated the survival curves along with the calculated hazard ratio (HR) and log-rank p-value.
DNA methylation analysis
The UALCAN platform, incorporating data from 93 TCGA datasets, was used to investigate DNA methylation, a significant epigenetic alteration that influences gene expression. The current study focused on the methylation patterns of the promoter region of core genes in order to examine its epigenetic regulation in PRAD cancer type.
Immunohistochemical (IHC) analysis of core targets
RNA sequence analysis and immunohistochemistry analysis offer an extensive array of transcriptomic and proteomic data on human normal tissues or infected tissues. In the current study, a comparison was made between the expression of protein of hub genes in PRAD tissues and normal tissues in HPA. Quality and quantity staining were used to determine the levels of protein expression. The staining intensity was further separated into four ranges based on the proportion of stained cells: none, less than 25%, 25–75%, and greater than 75%.
Immune infiltration levels of core targets
TIMER 2.0 database was utilized to estimate the number of tumour-associated immune cells through the data procured from gene expression from the TCGA-PRAD cohort. Furthermore, a functional correlation heatmap was created using the Chiplot web tool by entering Spearman’s correlation coefficient values obtained from the ‘Gene Module’. Additionally, TISIDB database was used to examine the variations in gene expression among immunological and molecular subtypes (http://cis.hku.hk/TISIDB/)35.
Experimental validation
Cell culture maintenance and cell viability assay
HEK-293 (human embryonic kidney) and PC-3 (prostate adenocarcinoma) cell lines were acquired (NCCS, Pune, India). The cells were cultivated in DMEM with 1% antibiotic-antimycotic solution and 10% FBS added. To ensure optimal growth conditions, they were preserved in a CO2 incubator (37 °C; carbon-dioxide and oxygen concentration of 5% and 18–20%, respectively). The cytotoxic potential of Curcuma amada rhizome extract against PC-3 cells was assessed using the MTT assay. PC-3 cells were first seeded onto 96-well plates at a density of 2 × 104 cells/well and then incubated for 24 h. Following this, the cells were treated with varying concentrations of C. amada rhizome extract (10–200 µg/mL) and allowed to grow for an additional 48 h. After incubation, the medium was removed, and MTT solution (20 µL; 5 mg/mL) was added to each well. The crystals of formazan were then immersed in 100 µL of DMSO and examined at 570 nm. The cells that received only DMEM treatment were regarded as negative controls. The formula used to obtain the percentage of cell viability (%) is given as follows:
$${text{Percentage of cell viability}} (%)={text{Mean absorbance of sample (CARE)}} / {text{Mean absorbance of negative control}} times 100$$
Apoptosis assay
PC-3 cells were cultured for 24 h after being treated with CARE at concentrations of 100 and 200 µg/mL. The cells underwent washing with PBS followed by trypsinization. Subsequently, the cells were reconstituted in a binding buffer that contained 100 µL of staining solution (10 µL of PI and 5 µL of FITC-labelled Annexin-V), and they were allowed to rest at room temperature for 15 min in the dark. Subsequently, flow cytometry was used to determine the proportion of late and early apoptotic cells, viable cells, and necrotic cells.
Cell cycle analysis
Briefly, 6-well culture plates were seeded with PC-3 cells at earlier mentioned conditions. Following that, the cells were treated with CARE at doses of 0, 100, and 200 µg/mL. The cells were incubated for 24 h, then fixed with 100% ethanol, rinsed twice in cold PBS, and stored at − 20 °C. The fixed cells were then immersed in 400 µL of Propidium Iodide/RNase staining buffer and maintained in the dark for another 20 min. Flow cytometry was used to monitor the fluorescence of treated cells.
Gene expression study by qRT-PCR
As previously stated, PC-3 cells were grown for a day to achieve the appropriate cell density for qRT-PCR gene expression study. After incubation, the cells were rinsed with 1 mL of 1X PBS and treated with 100 and 200 µg/mL of CARE in 1 mL of fresh medium for another 24 h. Untreated cells served as the control group. Subsequently, the cells were detached using trypsin-EDTA, centrifuged, and washed twice with PBS. Total RNA was then extracted using the Qiagen RNase kit following the manufacturer’s instructions. cDNA was synthesized using the iScript cDNA synthesis kit (Bio-Rad, CA). Gene-specific primers for AKT1, BCL2, CDK4, MDM2, TP53, and PIK3CA were designed, and amplification was performed using SYBR Premix Ex Taq (Takara Bio, Otsu, Japan) on a Qiagen RotorGene Q 5plex HRM system. The relative gene expression was normalized against β-actin and analyzed using the 2−ΔΔCt method. Primer sequences for the genes examined in this study are provided in Table 1.
Results
Chemical profiling of CARE using UHPLC-QTOF-HRMS/MS analysis
The yield of C. amada rhizome extract after solvent evaporation was calculated as 2.51%. The methanolic extract of C. amada was thoroughly investigated with UHPLC-QTOF-HRMS/MS in both positive and negative ESI modes. This highly precise and sensitive approach allowed the identification of 16 distinctive compounds, with 7 compounds in + ve ionization mode and 9 in -ve ionization mode. Tentative identifications were made by comparing mass spectrum data and retention times to known reference databases including MassBank (https://massbank.eu/MassBank/) and prior literature. Additionally, the fragmentation patterns of the identified compounds were subsequently analyzed using MS/MS to validate the findings. The data provided for the identified compounds includes the molecular formula, retention time, computed and observed m/z, error in parts per million (ppm), MS fragmentation pattern, metabolite classes, names of the compounds which were tentatively discovered (Table 2).
Terpenoids were the primary components of CARE that produced a greater response rate in the negative-ion mode. Six terpenoids were identified using compound database searches in PubChem, mass spectrographic assessment, and related literature. Amongst them, zederone (4) and zedoarondiol (18) were sesquiterpenoids, whereas zerumin A (9), 13-hydroxylabda-8 (17), 14-dien-18-oic acid (13), labda-(17), 12-diene-15,16-dial (14) and coronarin D (15) were diterpenoids. A deprotonated ion was found at m/z 317.2159. Thereafter, a fragment ion was generated at m/z 273.2255 by this ion, showing the loss of a CO2 molecule. Further fragmentation of the ion at m/z 273.2255 led to the production of a product ion at m/z 271.2067, which represents the loss of a hydrogen molecule. These ions suggested that the chemical compound 9 obtained was zerumin A36. Furthermore, the deprotonated ion detected at m/z 301.2035 was determined to be labda-8(17),12-diene-15,16-dial (12). Upon analysis, the MS2 spectrum revealed the loss of a carbonyl group, generating a fragment ion at m/z 273.1581. Following that, the ion at m/z 273.1581 yielded another ion at m/z 258.2323, which corresponded to the elimination of a methyl molecule37,38. Consequently, MS analysis of compound 4 in positive ionization mode showed an m/z value of 247 for [M + H] +, indicating an empirical molecular formula of C15H18O3. A fragment ion at m/z 229, corresponding to a loss of 18 Da, suggested the elimination of H2O from the parent ion. The ion at m/z 229 further produced four major fragment ions at m/z 189, 183, 128, and 105, with mass differences of 40, 6, 55, and 23 Da, respectively. The 40 Da loss suggests the removal of a C3H4 group, possibly caused by cleavage in the aliphatic side chain or within the furan ring structure. Additionally, the 55 Da mass losses may represent the elimination of a C4H7 group, respectively. The 23 Da difference observed in the fragment ion at m/z 105 could result from the loss of a CH3 group (15 Da) and an oxygen-containing fragment, such as OH (17 Da), helping to stabilize the structure. The compound was identified as zederone39. Similarly, MS analysis of compound 16 revealed an [M + H] + ion with an m/z value of 253 in the positive ionization mode, suggesting an empirical molecular formula of C25H39NO2. The first fragmentation step involved the loss of 18 Da (H2O), resulting in a fragment ion at m/z 235. This was followed by the elimination of 46 Da (CO2), producing an ion at m/z 189. Subsequently, the loss of 14 Da (CH2) generated an ion at m/z 175, likely due to the cleavage of a methylene group in the aliphatic chain. A further loss of 28 Da (CO) yielded an ion at m/z 147, followed by another 14 Da loss (CH2), resulting in an ion at m/z 133, consistent with additional aliphatic chain cleavage. Finally, the elimination of 26 Da (C2H2) produced a fragment ion at m/z 107, likely corresponding to the removal of a terminal acetylene group. This was identified as zedoarondiol39.
Phenolic acids were identified as the second most abundant class. Compound 1 was characterized as vanillic acid, based on its fragment ions at m/z 151 and 123, indicating the loss of CH3 and CO2, respectively. Both ions further generated a common fragment at m/z 107, resulting from the elimination of HCO2 and CH4, respectively40. Compound 3 was tentatively assigned in the negative ion mode with precursor ion at m/z 179.0347. MS/MS analysis, supported by previous literature, confirmed caffeic acid, as its product ions at m/z 143 and m/z 133 corresponded to the sequential loss of two H2O molecules (36 Da) and HCOOH (46 Da) from the parent ion41. For compound 5, typical fragment ions were observed at m/z 119.0513, 103.0109, 93.0356, 61.9910 corresponding to the loss of CO2, O, and CH = CH. Similarly, compound 6 produced characteristic fragment ions at m/z 153.0556, 139.0393, 125.0594, 111.0445, 95.0499, and 79.0551. These ions were generated through sequential losses of H2O, CO2, CH3, CH2, CH2O, and O. Based on these fragmentation patterns, compound 5 and 6 were identified as 4-hydroxycinnamic acid and syringic acid, respectively42,43. The MS/MS analysis of compound 2 revealed fragment ions at m/z 93.0347 and 75.0138, resulting from the loss of CO and H2O, respectively. The molecule was determined to be benzoic acid based on its fragmentation pattern43,44. MS analysis of compound 10 in positive ionization mode revealed a parent ion at m/z 369.1340 [M + H] +, which corresponded to the chemical formula C21H20O6. Fragmentation of this ion produced two main fragment ions at m/z 285.1137 and 177.0555, with mass losses of 80 Da and 192 Da, respectively. These losses were attributed to the elimination of C4H4O2 and C11H12O3. A comparison with reported data confirmed the compound as curcumin45,46.
Drug-likeness and ADMET profiling
The compounds retrieved from CARE were initially analyzed using SwissADME, precisely emphasizing their drug-like characteristics. According to the data obtained from the SwissADME server, out of a total of 16 compounds, 15 adhered to Lipinski’s and Egan’s filter requirements, whereas the remaining 1 compound including N-Undecylbenzenesulfonic acid did not conform to these rules.As shown in Table 3, the chosen compounds manifest good bioavailability scores (≥ 0.55). Moreover, the pharmacokinetic and toxicity profiles of the screened compounds were assessed through a comprehensive ADMET analysis. As shown in Table 4, water solubility varied widely, from highly negative (syringic acid: − 6.415) to moderate values (benzoic acid: − 1.738). It was observed that intestinal absorption was high for most compounds (> 90%), with vanillic acid (78.15%) and coronarin D (73.08%) as notable exceptions. Moreover, skin permeability was generally low, as indicated by negative log Kpvalues, while Caco-2 permeability also exhibited negative values, signifying limited absorption across the intestinal epithelium. However, zedoarondiol exhibited both P-glycoprotein substrate and inhibitor activity. Blood-brain barrier (BBB), central nervous system (CNS) permeability and volume of distribution (VDss) were predominantly minimal for all compounds. In terms of metabolism, none of the compounds were substrates for CYP2D6, minimizing the risk of variability due to genetic polymorphisms in this enzyme. However, syringic acid, retinol, zerumin A, zedoarondiol, and curcumin were identified as substrates for CYP3A4. Notably, zedoarondiol was both a substrate and an inhibitor of CYP3A4, suggesting the possibility of self-inhibition and altered pharmacokinetics. Additionally, total clearance values were low across compounds, suggesting slow systemic elimination. Moreover, toxicity analysis revealed no AMES toxicity, hERG I or hERG II inhibition. However, syringic acid, curcumin, and retinol demonstrated hepatotoxicity, while syringic acid and zerumin A were found to possess skin sensitization potential.
Acquisition of targets associated with active components of C. amada and prostate cancer
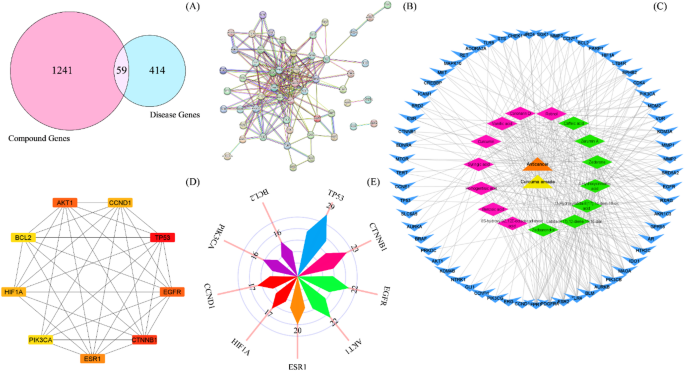
A total of 6,058 potential target genes associated with the active compounds in Curcuma amada were identified using the Swiss Target Prediction (1,700 genes) and SuperPRED (4,358 genes) databases. These targets were then input into the UniProt database to normalize gene names and eliminate duplicate gene entries that were identified across datasets due to overlaps across databases and different naming conventions (e.g., synonyms, isoforms). These duplicates were removed to ensure a non-redundant gene list resulting in 1241 unique target genes. Similarly, 414 prostate cancer-related genes were obtained by combining data from the GeneCards (95 genes), MalaCards (366 genes), and Therapeutic Targets Database (TTD) (93 genes) after removing duplicates and data curation. Furthermore, Venny 2.1.0 software was used to examine the two sets of target genes, that enabled the identification of 59 overlapping targets (Fig. 1A) that were used for downstream analysis.

Retrieval of CARE compounds and prostate cancer-related targets, protein-protein interaction, compound-disease network construction and identification of hub genes (A) Intersecting targets for active compounds of Curcuma amada and prostate cancer-related targets. (B) Analysis of the protein-protein interaction (PPI) network of the 59 common targets obtained using the STRING database. Nodes represent core targets, and edges represent different interactions (C) The visualization network of compound-disease-target was built by Cytoscape 3.8.0. It is composed of target nodes (arrow, blue), less significant compound nodes (diamond, pink), high degree compound nodes (diamond, green), 1 disease node (triangle, orange) and 1 C. amada node (triangle, yellow). (D) PPI network of the top 10 hub genes, analyzed by Cytoscape plug-in CytoHubba. (E) Rose plot showing the degree of hub genes emphasizing their centrality in the network.
PPI network, hub genes screening and compound disease target network construction
The shared targets of CARE and prostate cancer were analyzed using STRING database to explore the PPI network (Fig. 1B). The PPI network consisted of 208 edges, 59 nodes, an average node degree of 7, and a mean local clustering coefficient of 0.553. Moreover, the network showed a significantly higher level of interactions than expected, with a PPI enrichment p-value of < 1.0e-16. Furthermore, the topological assessment of the PPI network was performed in the Cytohubba plugin of Cytoscape 3.7.2 software employing degree, MCC and MNC centrality parameters. Based on the intersection of targets of three centrality parameters, AKT1, CCND1, TP53, EGFR, CTNNB1, ESR1, PIK3CA, HIF1A, and BCL2 were considered as hub genes with significant centrality values. These genes were color-coded to reflect their respective node degrees (Fig. 1D), and Fig. 1E further presents the degree centrality values for these top nine genes. Additionally, the compound-disease target network was constructed (Fig. 1C) which comprised of 78 nodes and 328 edges, with an average neighbour size of 8.410. Moreover, network density was found to be 0.055. The compounds exhibiting the highest degrees were recognized as hub compounds (diamond shaped green-colored nodes, as shown in Fig. 1C). Among the bioactive compounds examined, labda-8(17),12-diene-15,16-dial, zedoarondiol, 4-hydroxycinnamic acid, 13-hydroxylabda-8(17),14-dien-18-oic acid, zederone, zerumin A, and caffeic acid exhibited the highest connectivity, with degrees ranging from 21 to 25.
Enrichment analysis
Fifty-nine overlapping genes were investigated to uncover the biological roles and potential mechanisms of CARE in combating prostate cancer through GO and KEGG enrichment analyses. The top 10 targets associated with phytochemicals from the rhizomes of Curcuma amada were significantly linked to various biological processes (BP). These included responses to light and radiation, reactions to abiotic stimuli, cellular responses to oxygen-containing compounds, peptidyl-amino acid modifications, protein phosphorylation, positive regulation of signal transduction, and the regulation of programmed cell death and apoptotic processes (Fig. 2A). In terms of cellular components (CC), the targets were localized to the phosphatidylinositol 3-kinase complex (class IA), chromosome passenger complex, cyclin-dependent protein kinase holoenzyme complex, and other related components. Meanwhile, the molecular functions (MF) of these targets were predominantly related to protein serine kinase activity, protein kinase activity, and protein serine/threonine kinase activity, among others. A total of 346 GO terms under MF, 191 under CC, and 1000 under BP were identified with P value < 0.05 and FDR < 0.01 (Fig. 2B). The KEGG pathway analysis identified associations between the 59 common genes and 158 signalling pathways. However, pathways unrelated to prostate cancer pathogenesis were eliminated to reveal exclusively the related findings. Figure 3C depicts the top 10 most enriched signalling pathways. Among them, the PI3K-AKT signalling pathway was the most significantly enriched and have the potential to play a crucial role in prostate cancer treatment (Figure S2)47.

Pathway enrichment analysis of hub targets. (A) Gene ontology (GO) chord plot of top 10 ranked over-represented GO terms belonging to the biological process. (B) GO functional enrichment analysis of overlapping targets. The bar plot shows significant GO enrichment terms of common targets in three functional groups: biological processes, cellular component and molecular function. The x-axis indicates the gene number and the y-axis indicates the pathway. (C) Bubble plot combined with Sankey diagram depicting highly enriched top 10 KEGG pathways and the hub genes within each pathway on the y-axis and the gene ratio (p-value ≤ 0.01 and FDR ˂ 0.01) on the x-axis.
Validation of key genes in an external dataset
Survival analysis based on expression data and clinical evidence from TCGA revealed that TP53 expression was significantly related to patient survival in PCa. The survival analysis curve (FigureS3B) shows that TP53 possesses a hazard ratio (HR) > 1, signifying that higher expression represents a negative prognostic factor and poor clinical outcome.In contrast, the expression levels of the other eight hub genes (BCL2, EGFR, HIF1A, CTNNB1, CCND1, AKT1, PIK3CA, and ESR1) showed no significant differences in overall survival, indicating their limited effect on patient prognosis in prostate cancer.
Moreover, the rate and types of genetic alteration data for nine hub genes were obtained from cBioPortal (TCGA, PanCancer Atlas) and are represented in Figure S3D (i). A total of 489 samples, accounting for 25.15% of the queried patients, displayed alterations in these hub genes. Among them, TP53, PIK3CA, and CTNNB1 exhibited the highest alteration rates of 15%, 5%, and 2.7%, respectively. The identified genetic alterations included mutations, amplifications, deep deletions, truncating mutations, missense mutations, inframe mutations, and splice mutations. Mutations were the most frequent alteration type, representing 13.7% of the changes, followed by deep deletions (5.93%), amplifications (3.48%), and multiple alterations (2.04%) (FigureS3D (ii)).
Immunohistochemistry (IHC) data for both malignant and normal prostate tissues were obtained from the HPA database in order to assess the protein expression profiles of the hub genes that have been found in prostate cancer. The research revealed that AKT1, CTNNB1, EGFR, and PIK3CA were predominantly located in the cytoplasm. In Figure S3F, representative IHC images are displayed. Interestingly, prostate cancer tissues had substantially greater positive expression rates of these proteins than nearby normal tissues, which is in line with the results of the bioinformatics study.
TIMER 2.0 database was utilised to investigate the fundamental mechanism of the immune microenvironment in PCa. From the FigureS3G, it was established that PIK3CA showed a positive correlation with neutrophil (r = 0.55) and myeloid dendritic cell (r = 0.53). Additionally, CTNNB1 displayed a positive correlation for neutrophils (r = 0.54).
The TISIDB web-based platform was utilized to analyze the relationship between the expression of nine key hub genes and immune subtypes in PRAD. Data obtained from TISIDB indicated a significant correlation between the expression levels of two specific hub genes, EGFR and HIF1A, and multiple immune subtypes in PRAD (Figure S3E (i-ii)).
Additionally, the UALCAN database was used to evaluate the DNA methylation levels of the nine hub genes within normal and cancerous tissues. The hypomethylation of HIF1A (methylation levels between 0.25 and 0.3) suggests potential transcriptional activation, which has been linked to a poor prognosis in prostate cancer due to its role in promoting tumor development and metastasis (Figure S3C). AKT1, BCL2, TP53, and CCND1 have notably low methylation levels (unmethylated, methylation levels < 0.25), which further suggests that these genes are transcriptionally active.
Molecular docking analysis
Seven hub compounds (4-hydroxycinnamic acid, 13-hydroxylabda-8(17), 14-dien-18-oic acid, labda-8(17), 12-diene-15,16-dial, zederone, zedoarondiol, zerumin A, and caffeic acid) and nine hub proteins encoded by the corresponding genes (AKT1, CCND1, TP53, EGFR, CTNNB1, ESR1, PIK3CA, HIF1A, and BCL2) were subjected to molecular docking in order to evaluate the protein-ligand binding potential. The robustness of this interaction was estimated using a scoring method to predict binding affinity. The greater -ve free energy (ΔG) indicates a favourable binding interaction between protein and ligand. The heatmap depicting the binding energy of the key compounds associated with the hub genes is shown in Fig. 3D. The binding energies of the molecular docking analysis, as displayed in Table 5, were significantly less than − 5 kcal/mol, indicating that the target proteins and the hub compounds of C. amada possessed a strong affinity for binding. The docking scores, amino acid residues involved and nature of the interactions amongst the top seven docked phytocompounds and the target proteins are summarized in Table 5. The findings reveal that zerumin A possesses a favorable docking effect with AKT1, ESR1, and PIK3CA, with respective binding energies of − 9.1, − 8.6, and − 8.2 kcal/mol. Zerumin A established two conventional hydrogen bonds with Lys14 and Arg86 residues at 6.44 and 2.80 Å, respectively. Furthermore, it formed a carbon-hydrogen contact with the Asn53 residue at a distance of 2.46 Å within the binding site of AKT1 protein. In addition, the zerumin A-PIK3CA complex established 1 pi-alkyl and 3 hydrogen bond connections with Pro449, Asn457, Gly460, and Gln1014 residues at distances of 4.31, 2.17, 2.51, and 2.29 Å, respectively. Additionally, Zerumin A formed 2 conventional hydrogen bonds with the residues of Leu387, Arg394 and 1 alkyl bond with Met388 at distances of 2.03, 2.13 Å, and 5.35 respectively, to interact with the ESR1 protein. Furthermore, the relative binding energies of 13-hydroxylabda-8(17) and 14-dien-18-oic acid with AKT1 and PIK3CA are − 8.9 and − 8.1 kcal/mol, respectively. The 13-hydroxylabda-8(17),14-dien-18-oic acid-AKT1 complex was likely to bind to the binding pocket of AKT1 by forming H-bonds with Arg23 and Asn53 (conventional) at a distance of 2.91 and 2.59 Å, respectively. Additionally, 2 hydrogen bond and 1 alkyl interactions with Thr462, Thr679, and Tyr644 residues were formed by the 13-hydroxylabda-8(17),14-dien-18-oic acid-PIK3CA complex at distances of 3.07, 2.63, and 5.04 Å, respectively. Zederone also showed a favourable docking effect with PIK3CA and ESR1, with binding energies of − 8.1 and − 8.5 kcal/mol, respectively. Zederone established one hydrogen bond with Arg818 at a distance of 2.35 Å, along with an alkyl interaction with the same residue at 5.3 Å within the binding pocket of PIK3CA. Additionally, it formed alkyl interactions with His670 and Phe666 at distances of 2.88 Å and 4.86 Å, respectively. However, zederone was likely to bind to the pocket of the ESR1 protein by forming π-π T-shaped bonds with Phe404 at a distance of 5.00 Å, π-alkyl interactions with Leu391 at 4.59 Å, and alkyl interactions with Leu346, Leu387, and Ala350 at distances of 5.20, 4.94, 3.94 and 4.13 Å, respectively. Labda-8(17),12-diene-15,16-dial-ESR1 complex demonstrated favourable binding energy (−8.1 kcal/mol) and bound to the ESR1 binding pocket by forming conventional H-bonds with His524 at a distance of 0.640 Å, π-alkyl interactions with Leu387 and Leu391 at distances of 4.85 Å and 4.57 Å, respectively, and an alkyl bond with Phe404 at 5.41 Å. Zedoarondiol showed a considerable binding affinity (−8.6 kcal/mol) for the AKT1 protein. During the interaction of the ligand zedoarondiol, Arg25 was involved in conventional hydrogen bonding (2.40 Å) at the binding cavity of the protein with no further interactions observed. Figure 3A–C displays the 2D and 3D depictions of the connections amongst the target proteins and top hit docked compounds derived from C. amada.

Molecular docking analysis of core constituents of CARE with hub targets. 3D and 2D interaction of top docked complexes (A) (i) AKT1-ZeruminA (ii) AKT1-labda-8(17),12-diene-15,16-dial (iii) AKT1-13-Hydroxylabda-8(17), 14-dien-18-oic acid; (B) (i) ESR1-ZeruminA (ii) ESR1- Zedoarondiol (iii) ESR1-labda-8(17),12-diene-15,16-dial; (C) (i) PIK3CA-ZeruminA (ii) PIK3CA-13-Hydroxylabda-8(17), 14-dien-18-oic acid (iii) PIK3CA-Zedoarondiol. (D) Heat map showing binding affinities between hub targets with probable drug ligands. The docking score changes from low to high, from blue to pink. Pink numbers on the right of the heat map indicate in terms of better molecular docking. L1: 4-Hydroxycinnamic acid, L2: 13-Hydroxylabda-8(17),14-dien-18-oic acid, L3: Labda-8(17),12-diene-15,16-dial, L4: Zederone, L5: Zedoarondiol, L6: Zerumin A and L7: Caffeic acid.
MD simulation analysis
In addition, the selected docking conformations of target proteins in complex with selected ligands were evaluated by MD simulation at different time scales (100 and 200 ns). The trajectories were assessed through RMSD, RMSF, Rg, SASA, DSSP and MM-PBSA analysis. The findings are presented in Figs. 4 and 5.

Molecular dynamics simulation trajectories of apo-proteins and top-ranked docked complexes. Representative graphs include (A) (i–iii) Root mean square deviations (RMSD) of Cα atoms. (B) (i–iii) Root mean square fluctuation (RMSF) profile of Cα atoms. (C) (i–iii) Radius of gyration (Rg) over 100ns simulation period.

Molecular dynamics simulation trajectories of apo-proteins and top-ranked docked complexes. Representative graphs include (A) (i–iii) Solvent Accessible Surface Area (SASA) and (B) (i–xii) DSSP analysis for the secondary structure fluctuations as function of time at 300 K.
The AKT1 complexes with good docking scores and binding interactions underwent extended 200 ns simulations to capture detailed conformational changes and to assess accurate stability. The RMSD plots for unbound proteins (AKT1, PIK3CA, and ESR1) and their ligand-bound complexes are depicted in Fig. 4A (i-iii). The average RMSD of apo-AKT1 is 0.31 nm. Among the AKT1 complexes, the average RMSD of AKT1-zedoarondiol increased to approximately 0.32 nm from initial value and remained relatively stable at that level till the end of the simulation period. In contrast, AKT1-13-hydroxylabda-8(17),14-dien-18-oic acid complex increased to about 0.3 nm at 40 ns, thereafter decreased. Further, at 60 to 130 ns, the complex maintained the RMSD of about 0.38 nm, then decreased to approximately 0.3 nm and beyond 160 ns it achieved equilibration. Additionally, the AKT1-zerumin A complex exhibited a higher mean RMSD value (0.32 nm) compared to apo-AKT1, implying greater conformational variability during the simulation. The apo-protein conformation exhibited limited level of fluctuations up to 150 ns. Thereafter, the protein attained equilibration. However, the PIK3CA and ESR1 complexes, which demonstrated moderate binding affinities with their corresponding ligands were allowed to simulate for 100 ns. The average RMSD values of PIK3CA complexes were relatively lower compared to apo-PIK3CA, suggesting that compounds binding did not induce notable deviations in PIK3CA conformation. PIK3CA-Zerumin A complex experienced certain deviations throughout the 100ns time. At 10ns and 50ns the RMSD of this complex reached highest to around 0.2 nm. In case of PIK3CA-13-hydroxylabda-8(17),14-dien-18-oic acid complex displayed noticeable rise in average RMSD of about 0.27 nm, then decreased slightly and peaked to the same for 40–60 ns. Afterwards, the protein became equilibrated. Subsequently, the mean RMSD values were measured as 0.29 nm for apo-ESR1, 0.27 nm for ESR1- labda-8(17),12-diene-15,16-dial, 0.27 nm for ESR1-zedoarondiol, and 0.28 nm for ESR1-zerumin A, respectively. The ESR1-Zerumin A complex was stable without any deviation throughout the 100ns time. The ESR1-Zedoarondiol complex displayed noticeable ups and downs upto 60 ns with average RMSD of 0.25 nm. After 65 ns, the complex became equilibrated. In case of ESR1- labda-8(17),12-diene-15,16-dial complex, it is not relatively stable as it experienced noticeable deviations throughout 100 ns.
Unlike RMSD, RMSF was used to evaluate individual residue flexibility rather than positional variations over time (Fig. 4B (i-iii)). The RMSF trajectory, as shown in (Fig. 4B (i)) revealed that the docked complexes of AKT1 along with the undocked AKT1 protein followed a similar pattern with minimal fluctuations throughout 200 ns simulation period. In case of PIK3CA, when bound to 13-hydroxylabda-8(17),14-dien-18-oic acid and zederone, the RMSF plots revealed a similar pattern of fluctuation (Fig. 4B (ii)). The PIK3CA-zederone complex, on the other hand, showed additional variation in two specific regions: residues 171–194 and 257–298. PIK3CA-zerumin A also exhibits greater flexibility at residues 72–93. When ESR1 and labda-8(17)−12-dien-15,16-dial interacted, the RMSF value increased significantly in comparison to apo-ESR1. Furthermore, in ESR1 and labda-8(17)−12-dien-15,16-dial complex, the residues around 326–347 regions showed a considerable increase in protein structure, peaking at approximately 0.51 nm. Similarly, near the end of the simulation, ESR1 bound to zedoarondiol showers a higher number of fluctuations from regions of 512–546 residues with an RMSF value of 0.50–0.53 nm. Besides, no apparent rise in the RMSF value relative to apo-ESR1 was seen during the interaction of ESR1 with zerumin A.
The radius of gyration plots for each structure were recorded during the simulation time to assess structural compression changes. For the first 120 ns, AKT1-13-hydroxylabda-8(17),14-dien-18-oic acid showed observable fluctuations in Rg, with an average of 1.47 nm, after which it increased to approximately 1.5 nm and remained consistent until the simulation terminated, as illustrated in Fig. 4C (i). The AKT1-zerumin A complex with an avergage Rg value of 1.51 nm showed a relatively stable topology without any higher level of deviations up to 150 ns, then displayed a wave-like pattern till the end of the simulation. The AKT1-zedoarondiol complex showed a fluctuated profile up to 90 ns, then the Rg value decreased and exhibited lower level of fluctuation till the end of simulation. The average Rg value for AKT1-zedoarondiol complex (1.46 nm) was little lower than that of apo-AKT1 (1.47 nm). The Rg for the PIK3CA-zederone complex began to fluctuate noticeably at 2.46 nm and lasted up to 65 ns. Following that, the Rg displayed little variations and remained constant. Furthermore, the PIK3CA-13-hydroxylabda-8(17),14-dien-18-oic acid complex showed a similar pattern to apo-PIK3CA after 40 ns, yet initially exhibiting slight instability in Rg values. Throughout the duration, the PIK3CA-zerumin A complex showed a consistent compact structure. This shows that zerumin A and PIK3CA bind compactly and steadily during simulation. The Rg pattern for the ESR1-labda-8(17),12-diene-15,16-dial complex was identical from 50 ns until the end of simulation. On the other hand, the ESR1-Zedoarondiol complex showed a similar pattern to apo-ESR1 after 60 ns, despite initially exhibiting slight instability in Rg values. Additionally, at the beginning of the simulation period, the ESR1-Zerumin complex displayed a larger Rg value of 2.23 nm; however, it did not subsequently exhibit a higher level of deviation.
The SASA plots obtained for the apo-proteins and respective protein-ligand complexes are displayed in Fig. 5A (i-iii). The average SASA values for the AKT1-zedoarondiol (80.84 ± 2.07 nm2) and AKT1-zerumin A (84.02 ± 2.42 nm2) complexes were higher compared to the apo-AKT1 protein (80.82 ± 2.47 nm2). In contrast, the AKT1-13-hydroxylabda-8(17),14-dien-18-oic acid complex displayed a lower SASA (79.96 ± 2.05 nm2) compared to apo-AKT1, suggesting increased structural stability. Similarly, the ESR1-labda-8(17),12-diene-15,16-dial (158.9 ± 2.54 nm2), ESR1-zedoarondiol (160.27 ± 2.34 nm2), and ESR1-zerumin A (159.56 ± 1.89 nm2) complexes exhibited greater SASA values than apo-ESR1 (154.56 ± 2.78 nm2). This implies that ESR1 experiences less stability due to the increased solvent exposure indicated by higher SASA values upon ligand contact. For PIK3CA, the SASA values for the complexes with 13-hydroxylabda-8(17),14-dien-18-oic acid (178.26 ± 3.32 nm2) and zerumin A (177.94 ± 3.12 nm2) were lower than that of the apo-PIK3CA (179.80 ± 2.87 nm2), indicating a more compact structure upon ligand binding. However, the PIK3CA-zederone complex (189.48 ± 2.91 nm2) exhibited a SASA value greater to that of apo-PIK3CA, suggesting greater structural change.
Additionally, the structural behaviour of the target proteins bound to ligands was studied by observing shifts in the secondary structure of the protein-ligand complex. As shown in Fig. 5B (i-iv), the secondary structure of apo-AKT1 protein and its complexes did not exhibit any notable alterations throughout the simulation. However, following binding with zederone, PIK3CA displayed minimal fluctuation in its overall structure (Fig. 5B (vii). Similarly, ESR1 showed deviations in coils and α-helices after binding with zedoarondiol (Fig. 5B (xi)).
MM-PBSA analysis
The MM/PBSA analysis involved assessing the potential energy (van der waals and electrostatic) and free solvation energy (nonpolar and polar) of each protein‒ligand complex to calculate the total binding energy (ΔG). Table 6 represents the binding energies of the docked complexes along with the values of individual energy parameters. Among the AKT1 complexes, AKT1-Zerumin A displayed the lowest energy state with a total binding energy (ΔG) of − 30.34 kcal/mol, corresponding to the strongest binding affinity. The PIK3CA-zederone complex showed a significant binding affinity with a ΔG of − 18.53 kcal/mol, followed by PIK3CA-13-hydroxylabda-8(17),14-dien-18-oic acid (ΔG = − 18.16 kcal/mol). However, PIK3CA-zerumin A displayed slightly higher energy value (ΔG = − 15.02 kcal/mol), confirming a weaker interaction. Furthermore, ESR1-labda-8(17),12-diene-15,16-dial showed the strongest binding, with a negative ΔG of − 41.3 kcal/mol. Moreover, favourable van der Waals interactions (VdWaals) between the proteins and ligands were observed for all complexes (indicated by the negative values of ΔVdWaals), revealing a major contribution of nonspecific, short-range forces in stabilizing the complexes. The electrostatic interaction energies (ΔEELs) of all the complexes were largely negative and small, indicating the presence of specific attractive forces. However, these forces appeared to be less significant than van der Waals interactions. The polar solvation energy component (ΔEPB) was uniformly positive across all complexes, revealing that solvation forces acted against the binding interaction. Additionally, the nonpolar solvation energy (ΔENPOLAR) contributed favourably to the overall binding energy of the complexes, despite its small magnitude relative to other energy components. This component represents the hydrophobic interactions between the ligand and protein at the interface.
Experimental verification of network pharmacology findings
The cytotoxic effect of CARE towards normal cells (HEK-293) and prostate cancer cells (PC-3) were evaluated using the MTT assay. As displayed in Fig. 6A, CARE showed a reduction in cell viability in the tested PC-3 cell line as the concentration increased from 10 to 200 µg/mL. However, less distinguished toxic effects were observed for normal cell line (HEK-293).

Effect of CARE on viability and apoptosis of PC-3 cells. (A) The line graph shows cell viability (%) of HEK-293 (non-cancerous) and PC-3 (cancerous) cells treated with the indicated concentrations (10–200 µg/mL) of CARE for 24 h, as determined by MTT assay. (B) The percentage of live, early apoptotic, late apoptotic, and necrotic cells at (0–200 µg/mL) of C. amada (C) Analysis of apoptosis by flow cytometry. The dot plots (i) Control, (ii) CARE (100 µg/mL), (iii) CARE (200 µg/mL) show the following populations: necrotic (Q1-upper left), late apoptosis (Q2-upper right), early apoptosis (Q3-lower right), and viable cells (Q4-lower left).
Furthermore, Annexin V-FITC/Propidium Iodide assay combined with flow cytometry was utilized to estimate the proportion of PC-3 cells undergoing apoptosis (Fig. 6C (i-iii)). As shown in Fig. 6B, the proportion of late and early apoptotic cells increased significantly after 24 h of treatment with different concentrations of CARE. The proportion of late apoptotic cells increased substantially with increasing dosages of CARE including 0.57% (100 µg/mL) and 16.58% (200 µg/mL), in contrast to the control group (0.01%). Likewise, Fig. 6B indicates that the proportion of early apoptotic cells increased to 42.52% (100 µg/mL) and 37.13% (200 µg/mL), respectively, in comparison to the control group (0%). Furthermore, CARE treatment at 100 and 200 µg/mL resulted in less than 2% of the population exhibiting necrotic indications.
The influence of CARE on the cell cycle distribution of PC-3 cells was analysed through flow cytometry. Treatment with CARE at 100 and 200 µg/mL induced notable changes in cell cycle phases compared to the control group (Fig. 7). In untreated cells, the distribution across different phases was as follows: sub G0/G1 (1.04%), G0/G1 (69.07%), S (5.05%), and G2/M (23.88%). Following exposure to 100 µg/mL CARE, the proportion of cells in the sub G0/G1 phase declined to 0.64%, while the G0/G1 phase population dropped to 51.18%. Conversely, the percentage of cells in the S phase increased to 7.68%, and those in the G2/M phase rose to 36.94%. At the higher concentration of 200 µg/mL, the G0/G1 phase further declined to 14.16%, while the sub G0/G1 phase increased to 3.19%. Additionally, the S phase proportion decreased to 3.93%, whereas the G2/M phase significantly increased to 68.82%.

Effect of cell cycle distribution of PC-3 cells exposed to different concentrations of C. amada rhizome extract (0–200 µg/mL) for 24 h. (A) (i–iii) Flow cytometric analysis of cell cycle with PI-DNA staining. (B) The percentage of cell cycle distribution in different phases of cell cycle.
Network pharmacology analysis revealed that CARE demonstrates anticancer activity by modulating the PI3K-AKT signaling pathway. Therefore, we examined the effect of CARE on key downstream genes involved in this pathway by quantifying the mRNA expression levels of five core genes (PI3K, AKT1, BCL2, MDM2, and CDK4) in PC-3 cells through qRT-PCR analysis. Treatment with CARE at 100 and 200 µg/mL resulted in significant dose-dependent changes in gene expression when compared to untreated cells (Fig. 8A). Specifically, the pro-survival genes AKT1, BCL2, CDK4, and MDM2, which are critical for cell proliferation and survival, were markedly downregulated. At 100 µg/mL, their expression was reduced to 0.67 ± 0.09, 0.73 ± 0.0, 0.82 ± 0.01, and 0.93 ± 0.07, respectively. This reduction was even more pronounced at 200 µg/mL, with expression levels decreasing to 0.45 ± 0.14, 0.53 ± 0.01, 0.34 ± 0.04, and 0.66 ± 0.16, respectively. These results suggest that CARE effectively hinders cancer cell survival and growth by targeting the PI3K-AKT signalling pathway. In contrast, the tumor suppressor gene TP53, known for its crucial role in promoting apoptosis and regulating the cell cycle, was significantly upregulated in a dose-dependent manner. At 100 µg/mL, its expression increased to 1.94 ± 0.68, and at 200 µg/mL, it further increased to 2.29 ± 0.53, indicating the potential of CARE to enhance apoptosis in PC-3 cells. Additionally, PI3KCA, an upstream regulator of the pathway, exhibited a moderate reduction in expression to 0.94 ± 0.11 at 100 µg/mL and a significant decrease to 0.59 ± 0.24 at 200 µg/mL, further supporting the inhibitory effect of CARE on the activation of the PI3K-AKT pathway. Figure 8B depicts a schematic illustration of the action of C. amada rhizome extract on PC-3 cancer cells, triggering apoptosis while promoting cell cycle arrest.

Effect of CARE in inducing apoptosis and promoting cell cycle arrest in PC-3 cells. (A) Relative mRNA expression of the genes linked to apoptosis following treatment of PC-3 cells with CARE at different concentrations (100 and 200 µg/ml) and control. Statistical significance was measured by one-way analysis of variance followed by Tukey test. nsp > 0.05 * p < 0.05 and **p < 0.01 between the control and different concentrations of CARE. (B) Schematic representation showing the effect of CARE on PC-3 cancer cells by inducing apoptosis while promoting cell cycle arrest.
Discussion
Prostate cancer is an epithelial malignancy, characterized by high heterogeneity in men. It is usually detected at the de novo metastatic stage, as confirmed by a wide range of clinical findings and treatment outcomes48. Although the course of treatment for localized prostate cancer has improved, there continues to be few treatment options available for the advanced stages of the disease. Researchers have demonstrated anti-prostate cancer activities of several natural compounds both in-vivo and in-vitro49,50,51. The anticancer activity of phenolic compounds, flavonoids and curcuminoids extracted from Curcuma amada have been reported in various cancers52,53,54. However, the corresponding mechanism responsible for its anticancer effect against prostate cancer has not been determined. This work is the primary attempt, as far as we are aware, to use network pharmacology, molecular docking, molecular dynamics simulations, and in-vitro studies to explore the molecular mechanisms underlying the therapeutic utilization of isolated substances from C. amada in the treatment of prostate cancer.
The present work used UHPLC-Q-TOF-MS/MS to execute a qualitative examination of the methanolic extract derived from C. amada. The findings of the subsequent analysis showed the presence of a wide variety of phytocompounds. Each phytochemical belonged to different classes including 6 terpenoids, 5 phenolic acids, 1 carboxylic acid, 1 sulfonic acid derivatives, 1 curcuminoid and 2 compounds of other classes. Terpenoids were identified as the most abundant classes of phytoconstituents. The terpenoids included diterpenoids (zerumin A, 13-hydroxylabda-8(17),14-dien-18-oic acid, labda-8(17),12-diene-15,16-dial and coronarin D) and sesquiterpenoids (zederone and zedoarondiol). Phenolic acids were detected as the second major class comprising vanillic acid, caffeic acid, 4-hydroxycinnamic acid, syringic acid and homogentisic acid. The identified carboxylic acid included benzoic acid. Additionally, a curcuminoid (curcumin) and a retinoid (retinol) were identified. Lipid derivative such as 8 S-hydroxy-9Z,12Z-octadecadienoic acid (fatty acid) and sulfonic acid derivative such as N-undecylbenzenesulfonic acid were also detected. The conclusions drawn from this study are in accordance with previous identified components of C. amada including syringic acid, curcumin and caffeic acid52,55,56. Several Curcuma species such as Curcuma kwangsiensis, Curcuma zedoaria, Curcuma mutabilis have been found to contain various diterpenoids57,58,59. Two sesquiterpene compounds including zederone and zedoarondiol had been reported in the rhizome extract of Curcuma cf. amada (En-Lueang)60. Moreover, (E)-labda-8(17),12-diene-15,16-dial had previously been detected in the ethyl acetate extract of C. amada61. The presence of curcumin has been previously reported in C. amada and related species62,63,64. Amongst the series of compounds found in C. amada, many corresponded to the anticancer properties highlighted in previous studies. Curcumin is an essential polyphenol that exhibits significant anticancer effect, promoting cell cycle arrest and apoptosis in a variety of cancer cell lines, involving acute myeloid leukemia (AML) and HCT116 colon cancer cells65,66,67. Homogentisic acid is a potential natural compound that has been shown to significantly inhibit the proliferation of papillary thyroid carcinoma cells in vitro and in vivo through ROS and p21-induced cell cycle arrest68. Coronarin D, a labdane diterpene, has been shown to possess noticeable cytotoxic effects against MOLT-3, HuCCA-1, P388 and HeLa cell lines69.
The expansion of novel therapeutic agents is often hindered by poor pharmacokinetic profiles, including inadequate absorption, low bioavailability, rapid clearance, and unfavourable metabolism, leading to high attrition rates in clinical trials70,71. Therefore, the current study aimed to screen potential compounds with favourable pharmacodynamic and pharmacokinetic profiles using the SwissADME web tool. The results revealed that 15 out of the 16 compounds from CARE adhered to Lipinski’s and Egan’s drug-likeness criteria, indicating their potential as active therapeutic agents.
In this study, a PPI network comprising of fifty-nine overlapping targets was constructed in order to evaluate the combined effect of essential hub genes and uncover the molecular mechanisms behind the therapeutic potential of CARE. The top nine hub targets identified in the PPI network were TP53, CTNNB1, EGFR, AKT1, ESR1, HIF1A, CCND1, PIK3CA, and BCL2. The p53 protein, a crucial tumor suppressor that preserves genomic stability and prevents the spread of cancer, is encoded by the TP53 gene. Mutations in TP53 are frequent in cancer and can occur in up to 50% of metastatic prostate cancer cases, resulting in a poor prognosis and resistance to hormonal therapies72,73,74. Additionally, β-catenin hyperactivation and nuclear accumulation are caused by CTNNB1 mutations, which are found in only 3–5% of cases of prostate cancer. According to Akhoundova et al. this leads to aggressive cancer phenotypes, disease advancement, and therapy resistance, especially in mCRPC75. Furthermore, overexpression of EGFR has been associated with the progression of prostate cancer from an androgen-responsive to an androgen-independent state. It is linked to advanced tumour grade, higher stage, increased risk of PSA recurrence, and bone metastases76,77,78. Mutations in PIK3CA activate the PI3K/AKT pathway, promoting tumor metastasis, invasion, and cell growth. AKT1, a crucial isoform in AKT family, supports cancer cell survival and metastasis. Studies have shown that downregulation of microRNA-143-3p enhance prostate cancer proliferation by targeting AKT179. ESR1 is linked to prostate cancer susceptibility through its regulation of abnormal prostate growth, epithelial cellular proliferation, and apoptosis80,81. Overexpression of HIF1A has been documented in both primary and metastatic prostate cancer, as well as other common cancer types82. Evidence indicates that CCND1 expression is reduced in prostate tumour samples, and its inhibition has been shown to suppress cancer cell growth, migration, and invasion while enhancing apoptosis83. Furthermore, anti-apoptotic proteins from the BCL-2 group serve as an important role in controlling mitochondrial apoptosis. Raffo et al. showed that bcl-2 overexpression guarded prostate cancer cell lines (LNCaP) against apoptotic stimulus in vitro84. The findings demonstrate the significance of the hub genes that contribute to the development of prostate cancer and imply that targeting these genes could offer new treatment options.
In the present investigation, the CTD network revealed that the compounds most closely linked to target proteins were zedoarondiol, labda-8(17),12-diene-15,16-dial, 13-hydroxylabda-8(17),14-dien-18-oic acid, 4-hydroxycinnamic acid, zederone, zerumin A, and caffeic acid. These findings suggest that these seven compounds are likely the most important constituents of CARE. Zedoarondiol, a sesquiterpene lactone, has been shown to inhibit PDGF-BB-induced VSMC proliferation through AMPK signalling85. Zederone, a germacrene-type sesquiterpene, exhibits strong cytotoxicity against human prostate cancer cells86. Literature reports indicate that zerumin A, (E)-labda-8(17),12-dien-15,16-dial, and zederone effectively inhibit MCF-7 breast cancer cell growth, highlighting their potential as anticancer agents87. Caffeic acid alone (100 µM) or in combination with metformin (10 mM) activates the AMPK pathway, which inhibits the synthesis of unsaturated fatty acids, ultimately reducing cancer cell viability88. Furthermore, 4-hydroxycinnamic acid (HCA), particularly in its derivative form, artepillin C (3,5-diprenyl-4-hydroxycinnamic acid), enhances TRAIL (tumor necrosis factor-related apoptosis-inducing ligand)-induced apoptosis in prostate cancer cells89. This effect positions HCA derivatives as promising chemopreventive agents in prostate cancer management. These findings collectively highlight the multi-component and multi-target pharmacological potential of CARE, underscoring its role as a promising therapeutic agent.
GO enrichment results presented that CARE was strongly linked to several biological processes and targeting these processes holds potential for the expansion of therapeutics against PCa. KEGG enrichment analysis showed that C. amada may alleviate the development of prostate cancer through the action of core targets in multiple signalling pathways. Among them, PI3K-Akt signalling pathway was the most enriched. During cancer development, PI3K/Akt signalling pathway may be triggered by various types of toxic damage or cellular stimuli, influencing vital cellular activities including translation, transcription, growth, proliferation, and survival. Increasing evidence has evaluated the overactivation of PI3K/Akt signalling is in favour of growing malignant cells aggressively by modulating apoptosis, cell proliferation, invasion, angiogenesis, migration, immune evasion, and therapy resistance90,91. Several studies propose that natural products which target PI3K-AKT signalling pathways are likely to prevent prostate cancer progression92,93,94. Therefore, its inhibition can provide a new insight in treatment of this urological cancer.
The immunohistochemistry results revealed the upregulation of AKT1, CTNNB1, EGFR, and PIK3CA in prostate cancer tissues. This observation suggests their potential as key biomarkers for early disease detection and potential as effective therapeutic targets. Furthermore, TIMER analysis revealed that among the studied immune cells, PIK3CA expressed a positive correlation with neutrophils and myeloid dendritic cells. According to Zhao et al. a lower risk of biochemical failure is associated with a higher concentration of neutrophils in prostate tissue95. According to a number of studies, dendritic cells (DC) play an essential role in the anti-tumour responses against prostate cancer. Fridman et al. indicated that prostate cancer patients had an improved prognostic effect when these cells activated an adaptive cytotoxic T lymphocytes (CTL) response through antigen presentation96. Furthermore, less aggressive tumours were seen in patients with high circulatory DC levels97,98.
This investigation highlights the binding efficiency and interactions of the chosen active compounds from C. amada with nine significant target proteins. Polar and hydrophobic amino acids were the primary mediators of the ligand-protein interactions. Hydrophobic interactions, in particular, played a significant part by aligning hydrophobic areas on both ligands and receptors, eliminating water molecules, and reducing destabilizing effects, resulting in more stable ligand-receptor complexes. We further evaluated the binding outcomes of three proteins (AKT1, ESR1, and PIK3CA) and their corresponding ligands using molecular dynamics simulations. The results revealed structural alterations at the atomic level before and after ligand interaction. RMSD, RMSF, Rg, SASA, secondary structure analysis, and MM-PBSA remained the indicators that were the focus of the analysis. RMSD analysis revealed how protein geometry altered during ligand binding, with larger deviations indicating structural instability. The greatest RMSD was displayed by the AKT1-zerumin A complex, suggesting significant structural variation. RMSF, which measures amino acid-specific movements, indicated local fluctuation in the protein. In contrast to the apo-PIK3CA complex, the PIK3CA-zederone complex revealed the highest RMSF value, indicating flexible protein domains. The Rg value, which indicates the compactness of the protein, remained largely consistent across all complexes and apo-proteins, indicating stable folding even in the presence of ligands. Protein folding and interaction stability were also revealed by the SASA analysis, that measures the protein surface area exposed to solvent molecules. SASA changes such as the rise in ESR1-zedoarondiol, demonstrated the structural flexibility of the protein. During simulation, the SASA value of PIK3CA-13-hydroxylabda-8(17), 14-dien-18-oic acid complex dropped, indicating the contraction of protein structures or the closure of internal cavities which restrict water from dispersion. Furthermore, alterations in the secondary structure of protein imposed by ligand binding were examined through the Secondary Structure of Proteins (DSSP) method. This method assisted in determining structural modifications caused by the interaction with the ligands. ESR1-zederone complex was found to have minimal fluctuations in secondary structure elements, particularly in the α-helix and coil.
Although molecular docking provided preliminary insights into the binding orientations and binding affinities of the identified compounds with key target proteins, the docking scores alone are insufficient for accurate prediction of binding free energy99. Therefore, to gain a more reliable estimation of ligand-protein binding affinity, we employed molecular mechanics/poisson–Boltzmann surface area (MM-PBSA) calculations following molecular dynamics (MD) simulations. The MM-PBSA results offer a more rigorous and thermodynamically relevant evaluation of the binding energies100. Among all complexes, the ESR1-labda-8(17),12-diene-15,16-dial complex exhibited the most favorable free energy of binding (ΔG = − 41.3 kcal/mol), followed by ESR1-zedoarondiol (ΔG = − 35.36 kcal/mol), ESR1-zerumin A (ΔG = − 33.42 kcal/mol), AKT1-Zerumin A (ΔG = − 30.34 kcal/mol), PIK3CA-zederone (ΔG = − 18.53 kcal/mol), and PIK3CA-13-hydroxylabda-8(17),14-dien-18-oic acid (ΔG = − 18.16 kcal/mol). These values provide a stronger basis for evaluating the binding affinities and stability of the ligand-protein complexes than docking scores alone.
Furthermore, in vitro investigations on PC-3 cells demonstrated that CARE had an anticancer effect against prostate cancer. The optimal anticancer drug should have a strong ability to eliminate tumour cells and/or prevent tumour growth while having very few harmful effects on healthy tissues101. In our study, the methanolic extract of Curcuma amada (CARE) demonstrated concentration-dependent cytotoxicity against PC-3 prostate cancer cells, indicating its potential to effectively inhibit tumor cell proliferation. Importantly, this cytotoxic effect was minimal in non-cancerous HEK-293 cells, suggesting a selective action toward malignant cells. These findings align with previously reported in-vitro cytotoxicity studies of C. amada extracts against other cancer cell lines27,28. Therefore, the results reveal the appeal of CARE for further exploration in prostate cancer treatment as it may offer a safer therapeutic option with reduced toxicity compared to non-selective treatments.
Apoptosis is a physiological process that preserves tissue homeostasis by eliminating redundant or damaged cells102. The current studies demonstrate induction of apoptosis representing a marked increase in both early and late apoptosis in PC-3 cells after exposed to CARE, with minimal necrotic cells. This finding suggests that CARE exhibits a targeted apoptotic effect on cancer cells rather than causing nonspecific cell damage. The selective apoptotic induction further strengthens our findings that the methanolic C. amada extract could be an effective agent for the treatment of prostate cancer.
Uncontrolled cellular proliferation is one of the hallmarks of all cancers, and blockade of cell cycle has been proposed to be an efficient method for eliminating cancer cells103,104,105,106. Additionally, the loss of cell cycle regulation has been linked to several stages of carcinogenesis. Thus, cell cycle detection may indicate the stage of tumour advancement. Our findings on cell cycle analysis revealed that in PC-3 cells, CARE treatment diminished the proportion of G0/G1-phase cells and significantly accumulated cells in the G2/M phase, indicating that the cells were arrested at the mitotic stage. Cell cycle arrest allows DNA repair to occur, limiting replication of damaged templates. Several species of Curcuma genus have shown cell accumulation in the G2/M phase by preventing the multiplication of cells from S to G2, resulting in cell arrest107,108.
Subsequent qRT-PCR validation revealed that CARE exerts dose-dependent regulatory effects on key genes linked with the PI3K-AKT signaling pathway, resulting in significant changes in gene expression profiles that promote apoptosis and limit cancer cell proliferation. AKT1, BCL2, CDK4, and MDM2 expression levels were specifically decreased in a dose-dependent manner, with notable reductions at 200 µg/mL, suggesting that CARE may interfere with key oncogenic pathways that support tumor growth. The reported outcomes are consistent with prior investigations on natural compounds that target the PI3K-AKT signaling pathway. For instance, curcumin has been shown to promote apoptosis in various tumours by disrupting the PI3K/Akt signalling pathway109,110. CARE acts through dual mechanism simultaneously disrupting oncogenic survival pathways while triggering apoptosis, thereby, offering a targeted approach to impair prostate cancer progression.
However, the current study does have a few limitations. Firstly, the intersecting targets between CARE and prostate cancer were obtained from public databases, which may have introduced bias due to inadequate or inconsistent information in these resources. Furthermore, the study depends primarily upon preclinical (in-vitro and in-silico) methodologies with no clinical or in-vivo validation to demonstrate the efficacy and safety of CARE in treating prostate cancer. Moreover, the results are based on PC-3 cells, which reflect androgen-independent prostate cancer. However, it is uncertain if the findings are applicable to androgen-dependent or other subtypes of prostate cancer. Although the study evaluated the mRNA expression levels of the genes involved in the PI3K-AKT pathway, it did not confirm these results at the protein level using techniques such as western blotting which is essential for verifying physiological implications. Despite these limitations, the current study provides useful insights into the possible use of Curcuma amada rhizome extract as a prospective candidate for the development of novel medications to treat prostate cancer.
Conclusion
The current investigation elucidated the pharmacological mechanism of CARE in treating PCa through a comprehensive, multi-target, multi-component, and multi-pathway approach, integrating metabolomics, network pharmacology, bioinformatics, and experimental studies. Terpenoids were found to be the most prevalent class among the several phytocompounds identified in the UHPLC-Q-TOF-MS/MS analysis of C. amada rhizome extract. Furthermore, 15 of the 16 compounds from C. amada showed favourable pharmacodynamic and pharmacokinetic characteristics. CTD network analysis identified zedoarondiol, labda-8(17),12-diene-15,16-dial, 13-hydroxylabda-8(17),14-dien-18-oic acid, 4-hydroxycinnamic acid, zederone, zerumin A, and caffeic acid as the principal compounds of CARE with high degree values. In addition, TP53, CTNNB1, EGFR, AKT1, ESR1, HIF1A, CCND1, PIK3CA, and BCL2 genes were identified as significant molecular targets in prostate cancer. According to this study, CARE may be able to alter genes involved in the PI3K-AKT signalling pathway, highlighting its potential as prostate cancer treatment option. The bioactive substances found in CARE had low binding energy values and a strong binding affinity for the nine major targets. These interactions were further strengthened via MD simulation, which revealed that the ligand binding did not cause any evident conformational changes in the protein targets. Furthermore, subsequent in-vitro studies suggested that CARE has the potential to suppress cell proliferation, promote cell cycle arrest, and trigger apoptotic death of PC-3 cells, confirming its anticancer action against prostate cancer.
Data availability
The data presented in this study are available in this article.
References
-
Belkahla, S., Nahvi, I., Biswas, S. & Nahvi, I. Ben amor, N. Advances and development of prostate cancer, treatment, and strategies: A systemic review. Front. Cell. Dev. Biol. 10, 991330 (2022).
-
Bray, F. et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 74 (3), 229–263 (2024).
-
Chu, F. et al. Global burden of prostate cancer: age-period-cohort analysis from 1990 to 2021 and projections until 2040. World J. Surg. Oncol. 23 (1), 98 (2025).
-
Gandaglia, G. et al. Epidemiology and prevention of prostate cancer. Eur. Urol. Oncol. 4 (6), 877–892 (2021).
-
Gogola, S. et al. Anti-cancer stem-cell-targeted therapies in prostate cancer. Cancers 15 (5), 1621 (2023).
-
Loblaw, D. A. et al. Initial hormonal management of androgen-sensitive metastatic, recurrent, or progressive prostate cancer: 2007 update of an American society of clinical oncology practice guideline. J. Clin. Oncol. 25 (12), 1596–1605 (2007).
-
Cornford, P. et al. EAU-EANM-ESTRO-ESUR-SIOG guidelines on prostate cancer. Part II—2020 update: treatment of relapsing and metastatic prostate cancer. Eur. Urol. 79 (2), 263–282 (2021).
-
Wen, S., Niu, Y., Lee, S. O. & Chang, C. Androgen receptor (AR) positive vs negative roles in prostate cancer cell deaths including apoptosis, anoikis, entosis, necrosis and autophagic cell death. Cancer Treat. Rev. 40 (1), 31–40 (2014).
-
Klotz, L. et al. Nadir testosterone within first year of androgen-deprivation therapy (ADT) predicts for time to castration-resistant progression: a secondary analysis of the PR-7 trial of intermittent versus continuous ADT. J. Clin. Oncol. 33 (10), 1151–1156 (2015).
-
Pyo, Y. & Kwon, K. H. Current state of prostate cancer treatment and future strategies. J. Mens Health. 20 (11), 9–18 (2024).
-
Li, Y., Pan, J. & Gou, M. The anti-proliferation, cycle arrest and apoptotic inducing activity of peperomin E on prostate cancer PC-3 cell line. Molecules 24 (8), 1472 (2019).
-
Rashid, M. H. & Chaudhary, U. B. Intermittent androgen deprivation therapy for prostate cancer. Oncologist 9 (3), 295–301 (2004).
-
Reiss, A. B. et al. Androgen deprivation therapy for prostate cancer: focus on cognitive function and mood. Medicina 60 (1), 77 (2023).
-
Peyromaure, M. et al. Management of prostate cancer in china: a multicenter report of 6 institutions. J. Urol. 174 (5), 1794–1797 (2005).
-
Yin, S. Y., Wei, W. C., Jian, F. Y. & Yang, N. S. Therapeutic applications of herbal medicines for cancer patients. Evid. Based Complement. Alternat. Med. 2013, 302426 (2013).
-
Liu, Z. L. et al. Traditional Chinese medicinal herbs combined with epidermal growth factor receptor tyrosine kinase inhibitor for advanced non-small cell lung cancer: a systematic review and meta-analysis. J. Integr. Med. 12 (4), 346–358 (2014).
-
Fu, B. et al. Multi-component herbal products in the prevention and treatment of chemotherapy-associated toxicity and side effects: a review on experimental and clinical evidences. Front. Pharmacol. 9, 1394 (2018).
-
Wang, Z. et al. An update on Chinese herbal medicines as adjuvant treatment of anticancer therapeutics. Biosci. Trends. 12 (3), 220–239 (2018).
-
Zhang, X., Qiu, H., Li, C., Cai, P. & Qi, F. The positive role of traditional Chinese medicine as an adjunctive therapy for cancer. Biosci. Trends. 15 (5), 283–298 (2021).
-
Hooper, L. & Cassidy, A. A review of the health care potential of bioactive compounds. J. Sci. Food Agric. 86 (12), 1805–1813 (2006).
-
Signorelli, P. et al. Natural grape extracts regulate colon cancer cells malignancy. Nutr. Cancer. 67 (3), 494–503 (2015).
-
Esmeeta, A. et al. Plant-derived bioactive compounds in colon cancer treatment: an updated review. Biomed. Pharmacother. 153, 113384 (2022).
-
Singh, B. N., Singh, H. B., Singh, A., Naqvi, A. H. & Singh, B. R. Dietary phytochemicals alter epigenetic events and signaling pathways for Inhibition of metastasis cascade: phytoblockers of metastasis cascade. Cancer Metastasis Rev. 33, 41–85 (2014).
-
Akhtar, M. F. et al. Anticancer natural medicines: an overview of cell signaling and other targets of anticancer phytochemicals. Eur. J. Pharmacol. 888, 173488 (2020).
-
Sasikumar, B. Genetic resources of Curcuma: diversity, characterization and utilization. Plant. Genet. Res. 3 (2), 230–251 (2005).
-
Chen, Y., Shukurova, M. K., Asikin, Y., Kusano, M. & Watanabe, K. N. Characterization of volatile organic compounds in Mango ginger (Curcuma amadaRoxb.) from Myanmar. Metabolites 11 (1), 21 (2020).
-
Jambunathan, S., Bangarusamy, D., Padma, P. R. & &Sundaravadivelu, S. Cytotoxic activity of the methanolic extract of leaves and rhizomes of Curcuma amadaRoxb against breast cancer cell lines. Asian Pac. J. Trop. Med. 7, S405–S409 (2014).
-
Patil, V. S., Varashree, B. S., Pai, K., Hari, G. & Keerthi, P. Cytotoxic effects of Chloroform-methanol extract, protein extract and various fractions of ethanolic extract of Curcuma amada rhizome on HCT116 colon cancer cell line. Res. J. Pharm. Technol. 15 (11), 4955–4958 (2022).
-
Ramachandran, C. et al. In vivo antitumor effect of supercritical CO2 extract of mango ginger (Curcuma amadaRoxb) in U-87MG human glioblastoma nude mice xenografts. J. Evid. Based Complement. Alternat. Med. 22(2), 260–267 (2017).
-
Schenone, M., Dančík, V., Wagner, B. K. & Clemons, P. A. Target identification and mechanism of action in chemical biology and drug discovery. Nat. Chem. Biol. 9 (4), 232–240 (2013).
-
Chandran, U., Mehendale, N., Patil, S., Chaguturu, R. & Patwardhan, B. Network pharmacology. Innov. Appr. Drug Discov. 35, 127 (2016).
-
Noor, F. et al. Network Pharmacology approach for medicinal plants: review and assessment. Pharmaceuticals 15 (5), 572 (2022).
-
Molla, M. H. R. et al. Integrative ligand-based pharmacophore modeling, virtual screening, and molecular Docking simulation approaches identified potential lead compounds against pancreatic cancer by targeting FAK1. Pharmaceuticals 16 (1), 120 (2023).
-
Naveed, M. et al. Computational biology assisted exploration of phytochemicals derived natural inhibitors to block BZLF1 gene activation of Epstein–Bar virus in host. Sci. Rep. 14 (1), 31664 (2024).
-
Ru, B. et al. TISIDB: an integrated repository portal for tumor–immune system interactions. Bioinformatics 35 (20), 4200–4202 (2019).
-
Malek, S. N. A. et al. Phytochemical and cytotoxic investigations of Curcuma Mangga rhizomes. Molecules 8, 4539–4548 (2011).
-
Sirat, H. M., Masri, D. & Rahman, A. A. The distribution of labdane diterpenes in the Zingiberaceae of Malaysia. Phytochemistry 36, 699–701 (1994).
-
Sy, L. K. & Brown, G. D. Labdane diterpenoids from Alpinia chinensis. J. Nat. Prod. 60, 904–908 (1997).
-
Hao, M. et al. Metabolic profiling analysis of three processed rhizomes of Curcuma wenyujin YH Chen et C. Ling by ultra-performance liquid chromatography/time-of-flight mass spectrometry. Pharmacogn. Mag. 15, 60 (2019).
-
Kumar, S., Singh, A. & Kumar, B. Identification and characterization of phenolics and terpenoids from ethanolic extracts of Phyllanthus species by HPLC-ESI-QTOF-MS/MS. J. Pharm. Anal. 7 (4), 214–222 (2017).
-
Lin, H. et al. Comparative analysis of chemical constituents of Moringa oleifera leaves from China and India by ultra-performance liquid chromatography coupled with quadrupole-time-of-flight mass spectrometry. Molecules 24 (5), 942 (2019).
-
Yuan, H. E., Zhou, X. D., Meng, L. J., Qin, F. M. & Zhou, G. X. Chemical constituents from Commelina communis. Zhongguo Zhong Yao Za zhi = ZhongguoZhongyaoZazhi = China J. Chin. Materia Med. 38 (19), 3304–3308 (2013).
-
Zhang, X. et al. Simultaneous qualitative and quantitative study of main compounds in Commelina communis linn. By UHPLC–Q-TOF-MS-MS and HPLC–ESI-MS-MS. J. Chromatogr. Sci. 56 (7), 582–594 (2018).
-
Yang, Q., Ye, G. & Zhao, W. M. Chemical constituents of Commelina communis Linn. Biochem. Syst. Ecol. 35 (9), 621–623 (2007).
-
He, X. G. L. L. Z., Lian, L. Z. & &Lindenmaier, M. Liquid chromatography–electrospray mass spectrometric analysis of curcuminoids and sesquiterpenoids in turmeric (Curcuma longa). J. Chromatogr. A. .818, 127–132 (1998).
-
Shen, Y., Han, C., Chen, X., Hou, X. & Long, Z. Simultaneous determination of three curcuminoids in Curcuma wenyujinY.H.chen et c.ling. By liquid chromatography tandem mass spectrometry combined with pressurized liquid extraction. J. Pharm. Biomed. Anal. 81–82, 146–150 (2013).
-
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53 (D1), D672–D677 (2025).
-
Malihi, P. D. et al. Clonal diversity revealed by morphoproteomic and copy number profiles of single prostate cancer cells at diagnosis. Converg Sci. Phys. Oncol. 4 (1), 015003 (2018).
-
Kallifatidis, G., Hoy, J. J. & Lokeshwar, B. L. Bioactive natural products for chemoprevention and treatment of castration-resistant prostate cancer. Semin Cancer Biol. 40–41, 160–169 (2016).
-
Taylor, W. F. & &Jabbarzadeh, E. The use of natural products to target cancer stem cells. Am. J. Cancer Res. 7, 1588–1605 (2017).
-
Salehi, B. et al. Phytochemicals in prostate cancer: from bioactive molecules to upcoming therapeutic agents. Nutrients 11, 1483 (2019).
-
Siddaraju, M. N. Antiulcer And Anticancer Bioactive Compounds From Ginger (Zingiber officinale) And Mango Ginger (Curcuma amada) (Doctoral dissertation, University of Mysore) (2008).
-
Ramachandran, C., Lollett, I. V., Escalon, E., Quirin, K. W. & Melnick, S. J. Anticancer potential and mechanism of action of Mango ginger (Curcuma amadaRoxb.) supercritical CO2 extract in human glioblastoma cells. J. Evid. Based Complement. Alternat Med. 20 (2), 109–119 (2015).
-
Noor, A. et al. Curcuminoids as cell signaling pathway modulators: a potential strategy for cancer prevention. Curr. Med. Chem. 31 (21), 3093–3117 (2024).
-
Gupta, A. P., Gupta, M. M. & Kumar, S. Simultaneous determination of curcuminoids in Curcuma samples using high performance thin layer chromatography. J. Liq Chromatogr. R Technol. 22 (10), 1561–1569 (1999).
-
Al-Qudah, T. S. et al. Mango ginger (Curcuma amadaRoxb.): A phytochemical mini review. IJCBS 11, 51–57 (2017).
-
Schramm, A. et al. Phytochemical profiling of Curcuma Kwangsiensis rhizome extract, and identification of labdane diterpenoids as positive GABAA receptor modulators. Phytochemistry 96, 318–329 (2013).
-
Hamdi, O. A. A. Chemical Constituents from the Rhizomes of Curcuma zedoaria and Curcuma Purpurascens and Assessment of their Biological Activities (University of Malaya (Malaysia), 2015).
-
Soumya, T., Lakshmipriya, T., Klika, K. D., Jayasree, P. R. & Manish Kumar, P. R. Anticancer potential of rhizome extract and a labdane diterpenoid from Curcuma mutabilis plant endemic to Western Ghats of India. Sci. Rep. 11 (1), 552 (2021).
-
Anuchapreeda, S. et al. Characterization and biological properties of Zederone and Zedoarondiol from rhizomes of En-lueang (Curcuma cf. amada). Nat. Prod. Commun. 13 (12), 1934578X1801301211 (2018).
-
Singh, S., Singh, R., Banerjee, S., Negi, A. S. & Shanker, K. Determination of anti-tubercular agent in Mango ginger (Curcuma amadaRoxb.) by reverse phase HPLC-PDA-MS. Food Chem. 131 (1), 375–379 (2012).
-
Rafi, M. et al. Curcuminoid’s content and fingerprint analysis for authentication and discrimination of Curcuma xanthorrhiza from Curcuma longa by high-performance liquid chromatography-diode array detector. Food Anal. Methods. 8, 2185–2193 (2015).
-
Shirsath, S. R. et al. Intensification of extraction of Curcumin from Curcuma amada using ultrasound assisted approach: effect of different operating parameters. Ultrason. Sonochem. 38, 437–445 (2017).
-
Akter, J., Islam, M. Z., Takara, K., Hossain, M. A. & Sano, A. Isolation and structural Elucidation of antifungal compounds from Ryudai gold (Curcuma longa) against Fusarium solanisensu Lato isolated from American manatee. Comp. Biochem. Physiol. C: Toxicol. Pharmacol. 219, 87–94 (2019).
-
Collett, G. P. & Campbell, F. C. Curcumin induces c-jun N-terminal kinase-dependent apoptosis in HCT116 human colon cancer cells. Carcinogenesis 25 (11), 2183–2189 (2004).
-
Watson, J. L., Hill, R., Lee, P. W., Giacomantonio, C. A. & Hoskin, D. W. Curcumin induces apoptosis in HCT-116 human colon cancer cells in a p21-independent manner. Exp. Mol. Pathol. 84 (3), 230–233 (2008).
-
Zhou, H., Ning, Y., Zeng, G., Zhou, C. & Ding, X. Curcumin promotes cell cycle arrest and apoptosis of acute myeloid leukemia cells by inactivating AKT. Oncol. Rep. 45 (4), 1–1 (2021).
-
Xie, R. et al. Homogentisic acid metabolism inhibits papillary thyroid carcinoma proliferation through ROS and p21-induced cell cycle arrest. Life Sci. 347, 122682 (2024).
-
Chimnoi, N. et al. Phytochemical reinvestigation of labdane-type diterpenes and their cytotoxicity from the rhizomes of Hedychium coronarium. Phytochem Lett. 2 (4), 184–187 (2009).
-
Alavijeh, M. S. & Palmer, A. M. The pivotal role of drug metabolism and pharmacokinetics in the discovery and development of new medicines. Curr. Opin. Investig Drugs. 5, 755–763 (2004).
-
Basavaraj, S. & &Betageri, G. V. Can formulation and drug delivery reduce attrition during drug discovery and development—review of feasibility, benefits and challenges. Acta Pharm. Sin B. .4 (1), 3–17 (2014).
-
Barbieri, C. E. et al. The mutational landscape of prostate cancer. Eur. Urol. 64 (4), 567–576 (2013).
-
Robinson, D. et al. Integrative clinical genomics of advanced prostate cancer. Cell 161 (5), 1215–1228 (2015).
-
Nizialek, E. et al. Genomic profiles and clinical outcomes in primary versus secondary metastatic hormone-sensitive prostate cancer. Prostate 81 (9), 572–579 (2021).
-
Akhoundova, D. et al. Rare histologic transformation of a CTNNB1 (β-catenin) mutated prostate cancer with aggressive clinical course. Diagn. Pathol. 19 (1), 83 (2024).
-
Schlomm, T. et al. Clinical significance of epidermal growth factor receptor protein overexpression and gene copy number gains in prostate cancer. Clin. Cancer Res. 13, 6579–6584 (2007).
-
Day, K. C. et al. HER2 and EGFR overexpression support metastatic progression of prostate cancer to bone. Cancer Res. 77, 74–85 (2017).
-
Nastały, P. et al. EGFR as a stable marker of prostate cancer dissemination to bones. Br. J. Cancer. 123 (12), 1767–1774 (2020).
-
Armstrong, L., Willoughby, C. E. & McKenna, D. J. Targeting of AKT1 by miR-143-3p Suppresses Epithelial-to-Mesenchymal Transition in Prostate Cancer. Cells. 12(18), 2207 (2023).
-
Cunningham, J. M. et al. Evaluation of genetic variations in the androgen and Estrogen metabolic pathways as risk factors for sporadic and Familial prostate cancer. Cancer Epidemiol. Biomarkers Prev. 16, 969–978 (2007).
-
Nicolaiew, N. et al. Association between Estrogen and androgen receptor genes and prostate cancer risk. Eur. J. Endocrinol. 160, 101–106 (2009).
-
Zhong, H., Semenza, G. L., Simons, J. W. & De Marzo, A. M. Up-regulation of hypoxia-inducible factor 1α is an early event in prostate carcinogenesis. Cancer Detect. Prev. 28 (2), 88–93 (2004).
-
Wang, M. et al. MicroRNA-487a‐3p functions as a new tumor suppressor in prostate cancer by targeting CCND1. J. Cell. Physiol. 235 (2), 1588–1600 (2020).
-
Raffo, A. J. et al. Overexpression of bcl-2 protects prostate cancer cells from apoptosis in vitro and confers resistance to androgen depletion in vivo. Cancer Res. 55, 4438–4444 (1995).
-
Lyu, Y. et al. Zedoarondiol inhibits human bronchial smooth muscle cell proliferation through the CAV-1/PDGF signalling pathway. Sci. Rep. 14 (1), 13145 (2024).
-
Winuthayanon, W. et al. Diarylheptanoid phytoestrogens isolated from the medicinal plant Curcuma comosa: biologic actions in vitro and in vivo indicate Estrogen receptor-dependent mechanisms. Environ. Health Perspect. 117 (7), 1155–1161 (2009).
-
Malek, S. N. A. et al. Phytochemical and cytotoxic investigations of Curcuma Mangga rhizomes. Molecules 16 (6), 4539–4548 (2011).
-
Aikins, A. R. et al. Caffeic acid inhibits proliferation, migration, and stemness of DU-145 prostate Cancer cells. Nat. Prod. Commun. 18 (8), 1934578X231193848 (2023).
-
Kłósek, M. et al. Tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in prostate cancer cells after treatment with xanthohumol—A natural compound present in Humulus lupulus L. Int. J. Mol. Sci. 17 (6), 837 (2016).
-
Barra, F. et al. Investigational PI3K/AKT/mTOR inhibitors in development for endometrial cancer. Expert Opin. Investig Drugs. 28 (2), 131–142 (2019).
-
Peng, Y., Wang, Y., Zhou, C., Mei, W. & Zeng, C. PI3K/Akt/mTOR pathway and its role in cancer therapeutics: are we making headway? Front. Oncol. 12, 819128 (2022).
-
Chen, J. et al. The effect of lycopene on the PI3K/Akt signalling pathway in prostate cancer. Anti-Cancer Agents Med. Chem. 14 (6), 800–805 (2014).
-
Lim, W., Park, S., Bazer, F. W. & Song, G. Naringenin-induced apoptotic cell death in prostate cancer cells is mediated via the PI3K/AKT and MAPK signaling pathways. J. Cell. Biochem. 118 (5), 1118–1131 (2017).
-
Zoi, V. et al. The role of Curcumin in cancer: a focus on the PI3K/Akt pathway. Cancers 16 (8), 1554 (2024).
-
Zhao, H. B. et al. Novel immune-related signature for risk stratification and prognosis in prostatic adenocarcinoma. Cancer Sci. 112 (10), 4365–4376 (2021).
-
Fridman, W. H. et al. The immune microenvironment: a major player in human cancers. Int. Arch. Allergy Immunol. 164 (1), 13–26 (2014).
-
Alessandro, S. et al. Characterization of Circulating blood dendritic cell subsets DC123+ (lymphoid) and DC11C+ (myeloid) in prostate adenocarcinoma patients. Prostate 67 (1), 1–7 (2007).
-
Wang, C., Zhang, Y. & Gao, W. Q. The evolving role of immune cells in prostate cancer. Cancer Lett. 525, 9–21 (2022).
-
Chen, Y. C. Beware of docking! Trends Pharmacol. Sci. 36 (2), 78–95 (2015).
-
Wang, E. et al. End-point binding free energy calculation with MM/PBSA and MM/GBSA: strategies and applications in drug design. Chem. Rev. 119 (16), 9478–9508 (2019).
-
Makimoto, A., Fang, J. & Maeda, H. Development of a selective tumor-targeted drug delivery system: Hydroxypropyl-acrylamide polymer-conjugated pirarubicin (P-THP) for pediatric solid tumors. Cancers 13 (15), 3698 (2021).
-
Ergün, S. et al. Beyond death: Unmasking the intricacies of apoptosis Escape. Mol. Diagn. Ther. 86, 1–21 (2024).
-
Collins, K., Jacks, T. & Pavletich, N. P. The cell cycle and cancer. Proc. Nat. Acad. Sci. 94(7), 2776–2778 (1997).
-
McDonald, E. & El-Deiry, W. S. Cell cycle control as a basis for cancer drug development. Int. J. Oncol. 16 (5), 871–957 (2000).
-
Buolamwini, J. K. Cell cycle molecular targets in novel anticancer drug discovery. Curr. Pharm. Des. 6 (4), 379–392 (2000).
-
Adhami, V. M. et al. Sanguinarine causes cell cycle Blockade and apoptosis of human prostate carcinoma cells via modulation of Cyclin kinase inhibitor-cyclin-cyclin-dependent kinase machinery. Mol. Cancer Ther. 3 (8), 933–940 (2004).
-
Hu, A. et al. Curcumin induces G2/M cell cycle arrest and apoptosis of head and neck squamous cell carcinoma in vitro and in vivo through ATM/Chk2/p53-dependent pathway. Oncotarget 8 (31), 50747 (2017).
-
Ali, N. M. et al. Synthetic Curcumin derivative DK1 possessed G2/M arrest and induced apoptosis through accumulation of intracellular ROS in MCF-7 breast cancer cells. Cancer Cell. Int. 17, 1–12 (2017).
-
Shankar, S. & Srivastava, R. K. Involvement of Bcl-2 family members, phosphatidylinositol 3’-kinase/AKT and mitochondrial p53 in Curcumin (diferulolylmethane)-induced apoptosis in prostate cancer. Int. J. Oncol. 30 (4), 905–918 (2007).
-
Jin, H., Qiao, F., Wang, Y., Xu, Y. & Shang, Y. Curcumin inhibits cell proliferation and induces apoptosis of human non-small cell lung cancer cells through the upregulation of miR-192-5p and suppression of PI3K/Akt signaling pathway. Oncol. Rep. 34 (5), 2782–2789 (2015).
Acknowledgements
The authors are grateful to Dr. M. R. Nayak, President, Siksha ‘O’ Anusandhan (Deemed to be University) and Dr. Debajyoti Das, Dean, School of Pharmaceutical Sciences for providing necessary facilities for carrying out the research work.
Funding
Open access funding provided by Siksha ‘O’ Anusandhan (Deemed To Be University)
Ethics declarations
Competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Ethics approval
This study did not involve any ethical issues.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Priyadarshini, A., Mohanty, D., Mohanty, S. et al. Unveiling the anticancer potential of Curcuma amada rhizome extract against prostate cancer through computational and experimental approaches. Sci Rep 15, 24739 (2025). https://doi.org/10.1038/s41598-025-10761-0
-
Received:
-
Accepted:
-
Published:
-
DOI: https://doi.org/10.1038/s41598-025-10761-0